
Current Status of Gene Therapy Research in Polyglutamine Spinocerebellar Ataxias


Abstract
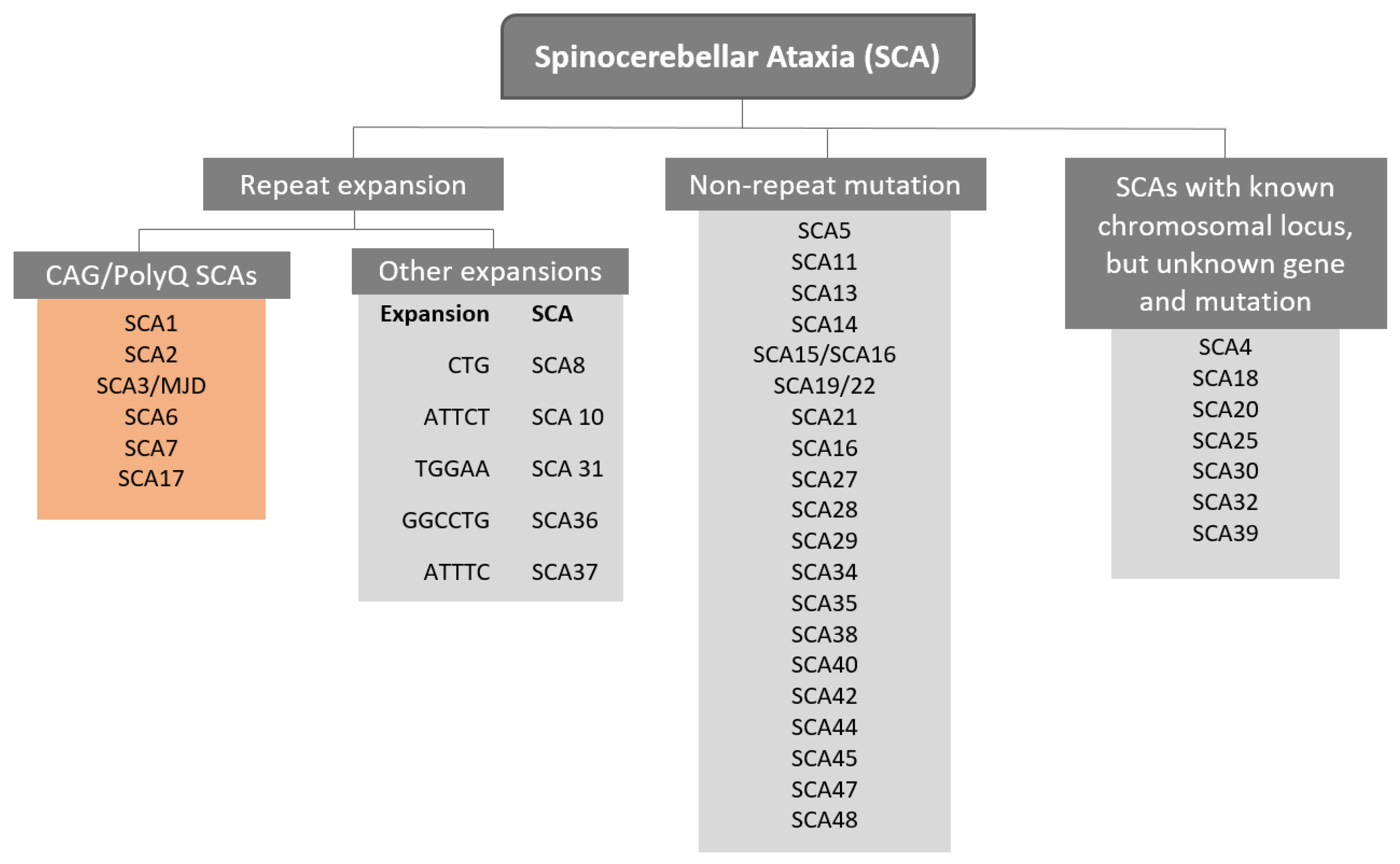
1. Introduction
Therapeutic Advances
2. Gene Therapy Augmentation Strategies
2.1. Strategies Activating Autophagy
2.2. Neuroprotective Strategies

{kind=link}
| Disease | Molecular Target | Gene Delivery System | Strategy | References |
|---|---|---|---|---|
| SCA1 | Homer-3 | AAV vectors | Autophagy | [45] |
| Ataxin-1 like | AAV vectors | Neuroprotection | [47] | |
| SCA3 | CYP46A1 | AAV vectors | Autophagy | [42] |
| Beclin-1 | Lentiviral vector | Autophagy | [44] | |
| Calpastatin | AAV vectors | Proteolytic cleavage | [51] | |
| Wild-type ataxin-3 | Lentiviral vector | Neuroprotection | [48] | |
| Ataxin-2 | Lentiviral vector | Neuroprotection | [49] | |
| CRAG | Lentiviral vector | Neuroprotection | [52] | |
| NPY | AAV vectors | Neuroprotection | [53] |
3. Gene Silencing Strategies
3.1. RNAi-Based Gene Silencing Strategies
3.1.1. Short Hairpin and Small Interfering RNAs Mediated Silencing
| Disease | Target | Allele Specificity | Technology | Experimental Systems | Delivery | References |
|---|---|---|---|---|---|---|
| SCA1 | Ataxin-1 | Non-specific | shRNA | SCA1 transgenic mouse model | AAV-mediated transduction | [66] |
| SCA3/MJD | Mutant Ataxin-3 | Allele-specific | siRNA | HEK 293T cells | Transfection | [73] |
| Mutant Ataxin-3 | Allele-specific | shRNA | LV-induced SCA3/MJD rat model | LV-mediated transduction | [72] | |
| Mutant Ataxin-3 | Allele-specific | siRNA | SCA3/MJD transgenic mouse model | SNALPs-mediated transduction | [76] | |
| Mutant Ataxin-3 | Allele-specific | shRNA | Patient derived fibroblasts | LV-mediated transduction | [79] | |
| Ataxin-3 | Non-specific | shRNA | LV-induced SCA3/MJD rat model | LV-mediated transduction | [48] | |
| SCA7 | Mutant Ataxin-7 | Allele-specific | siRNA | Patient derived fibroblasts | Transfection | [77] |
| Mutant Ataxin-7 | Allele-specific | siRNA | Patient derived fibroblasts | Transfection | [78] | |
| Mutant Ataxin-7 | Allele-specific | shRNA | Patient derived fibroblasts | LV-mediated transduction | [79] |
3.1.2. MicroRNA-Mediated Silencing
| Disease | microRNAs | Target | Experimental System | Delivery | References |
|---|---|---|---|---|---|
| SCA1 | Artificial miRNA | ATXN1 | C2C12 cells, SCA1 transgenic mouse model and non-human primates | AVV-mediated transduction | [67,68,81,83] |
| miR-19, miR-101 and miR-130 mimic | ATXN1 | HEK 293T, HeLa and MCF7 cells | Transfection | [91] | |
| miR-144 mimic | ATXN1 | HEK 293T cells | Transfection | [92] | |
| SCA3/MJD | Artificial miRNA | ATXN3 | SCA3/MJD transgenic mouse model | AAV-mediated transduction | [82,86] |
| miR-25 mimic | ATXN3 | HEK 293T and SH-S5Y5 cells | Transfection | [87] | |
| miR-9, miR-181a and miR-494 mimics | ATXN3 | HEK 293T cells and LV-induced SCA3/MJD mouse model | LV-mediated transduction | [88] | |
| SCA6 | miR-3191-5p | CACNA1A | AAV-induced SCA6 mouse model | AAV-mediated transduction | [89] |
| SCA7 | Artificial miRNA | ATXN7 | SCA7 transgenic mouse model | AAV-mediated transduction | [84,85] |
| miR-124 mimic | Lnc-SCA7 and ataxin-7 | N2a cells | Transfection | [90] |
3.2. Antisense Oligonucleotides
| Disease | Target | Mechanism | Delivery Method | References |
|---|---|---|---|---|
| SCA1 | Ataxin-1 | RNAse-H dependent degradation | ICV | [101] |
| Ataxin-1 | Translation hindrance | ICV | [105] | |
| SCA2 | Ataxin-2 | RNAse-H dependent degradation | ICV | [102] |
| SCA3/MJD | Ataxin-3 | Exon 9 and 10-skipping | ICV | [96] |
| Ataxin-3 | RNAse-H dependent degradation | ICV | [99] | |
| Ataxin-3 | Exon10-skiping | ICV | [98] | |
| Ataxin-3 | Translation hindrance and Exon10-skiping | ICV | [105] | |
| SCA7 | Ataxin-7 | RNAse-H dependent degradation | IVI | [103] |
4. Gene Edition
5. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Klockgether, T.; Mariotti, C.; Paulson, H.L. Spinocerebellar ataxia. Nat. Rev. Dis. Primers 2019, 5, 24. [Google Scholar] [CrossRef]
- Artero Castro, A.; Machuca, C.; Rodriguez Jimenez, F.J.; Jendelova, P.; Erceg, S. Short Review: Investigating ARSACS: Models for understanding cerebellar degeneration. Neuropathol. Appl. Neurobiol. 2019, 45, 531–537. [Google Scholar] [CrossRef]
- Ceylan, A.C.; Acar Arslan, E.; Erdem, H.B.; Kavus, H.; Arslan, M.; Topaloglu, H. Autosomal recessive spinocerebellar ataxia 18 caused by homozygous exon 14 duplication in GRID2 and review of the literature. Acta Neurol. Belg. 2020. [Google Scholar] [CrossRef]
- Zanni, G.; Bertini, E. X-linked ataxias. Handb. Clin. Neurol. 2018, 155, 175–189. [Google Scholar] [CrossRef]
- Genis, D.; Ortega-Cubero, S.; San Nicolás, H.; Corral, J.; Gardenyes, J.; de Jorge, L.; López, E.; Campos, B.; Lorenzo, E.; Tonda, R.; et al. Heterozygous STUB1 mutation causes familial ataxia with cognitive affective syndrome (SCA48). J. Neurol. 2018, 91, e1988–e1998. [Google Scholar] [CrossRef]
- Ashizawa, T.; Oz, G.; Paulson, H.L. Spinocerebellar ataxias: Prospects and challenges for therapy development. Nat. Rev. Neurol. 2018, 14, 590–605. [Google Scholar] [CrossRef]
- Ruano, L.; Melo, C.; Silva, M.C.; Coutinho, P. The global epidemiology of hereditary ataxia and spastic paraplegia: A systematic review of prevalence studies. Neuroepidemiology 2014, 42, 174–183. [Google Scholar] [CrossRef]
- Coutinho, P.; Ruano, L.; Loureiro, J.L.; Cruz, V.T.; Barros, J.; Tuna, A.; Barbot, C.; Guimaraes, J.; Alonso, I.; Silveira, I.; et al. Hereditary ataxia and spastic paraplegia in Portugal: A population-based prevalence study. JAMA Neurol. 2013, 70, 746–755. [Google Scholar] [CrossRef] [PubMed]
- Erichsen, A.K.; Koht, J.; Stray-Pedersen, A.; Abdelnoor, M.; Tallaksen, C.M. Prevalence of hereditary ataxia and spastic paraplegia in southeast Norway: A population-based study. Brain 2009, 132, 1577–1588. [Google Scholar] [CrossRef]
- Tsuji, S.; Onodera, O.; Goto, J.; Nishizawa, M.; Study Group on Ataxic, D. Sporadic ataxias in Japan--a population-based epidemiological study. Cerebellum 2008, 7, 189–197. [Google Scholar] [CrossRef]
- Monin, M.L.; Tezenas du Montcel, S.; Marelli, C.; Cazeneuve, C.; Charles, P.; Tallaksen, C.; Forlani, S.; Stevanin, G.; Brice, A.; Durr, A. Survival and severity in dominant cerebellar ataxias. Ann. Clin. Transl. Neurol. 2015, 2, 202–207. [Google Scholar] [CrossRef]
- La Spada, A.R. Trinucleotide repeat instability: Genetic features and molecular mechanisms. Brain Pathol. 1997, 7, 943–963. [Google Scholar] [CrossRef]
- Buijsen, R.A.M.; Toonen, L.J.A.; Gardiner, S.L.; van Roon-Mom, W.M.C. Genetics, Mechanisms, and Therapeutic Progress in Polyglutamine Spinocerebellar Ataxias. Neurotherapeutics 2019, 16, 263–286. [Google Scholar] [CrossRef]
- Paulson, H.L.; Shakkottai, V.G.; Clark, H.B.; Orr, H.T. Polyglutamine spinocerebellar ataxias—from genes to potential treatments. Nat. Rev. Neurosci. 2017, 18, 613–626. [Google Scholar] [CrossRef]
- Coarelli, G.; Brice, A.; Durr, A. Recent advances in understanding dominant spinocerebellar ataxias from clinical and genetic points of view. F1000Res 2018, 7. [Google Scholar] [CrossRef]
- Bauer, P.O.; Nukina, N. The pathogenic mechanisms of polyglutamine diseases and current therapeutic strategies. J. Neurochem. 2009, 110, 1737–1765. [Google Scholar] [CrossRef]
- Evers, M.M.; Toonen, L.J.; van Roon-Mom, W.M. Ataxin-3 protein and RNA toxicity in spinocerebellar ataxia type 3: Current insights and emerging therapeutic strategies. Mol. Neurobiol. 2014, 49, 1513–1531. [Google Scholar] [CrossRef][Green Version]
- Jimenez-Sanchez, M.; Thomson, F.; Zavodszky, E.; Rubinsztein, D.C. Autophagy and polyglutamine diseases. Prog. Neurobiol. 2012, 97, 67–82. [Google Scholar] [CrossRef]
- Durr, A. Autosomal dominant cerebellar ataxias: Polyglutamine expansions and beyond. Lancet Neurol. 2010, 9, 885–894. [Google Scholar] [CrossRef]
- Matilla-Duenas, A.; Ashizawa, T.; Brice, A.; Magri, S.; McFarland, K.N.; Pandolfo, M.; Pulst, S.M.; Riess, O.; Rubinsztein, D.C.; Schmidt, J.; et al. Consensus paper: Pathological mechanisms underlying neurodegeneration in spinocerebellar ataxias. Cerebellum 2014, 13, 269–302. [Google Scholar] [CrossRef]
- Matos, C.A.; de Almeida, L.P.; Nobrega, C. Machado-Joseph disease/spinocerebellar ataxia type 3: Lessons from disease pathogenesis and clues into therapy. J. Neurochem. 2019, 148, 8–28. [Google Scholar] [CrossRef] [PubMed]
- Katsuno, M.; Watanabe, H.; Yamamoto, M.; Sobue, G. Potential therapeutic targets in polyglutamine-mediated diseases. Expert Rev. Neurother. 2014, 14, 1215–1228. [Google Scholar] [CrossRef]
- Sullivan, R.; Yau, W.Y.; O’Connor, E.; Houlden, H. Spinocerebellar ataxia: An update. J. Neurol. 2019, 266, 533–544. [Google Scholar] [CrossRef]
- Marcelo, A.; Brito, F.; Carmo-Silva, S.; Matos, C.A.; Alves-Cruzeiro, J.; Vasconcelos-Ferreira, A.; Koppenol, R.; Mendonca, L.; de Almeida, L.P.; Nobrega, C. Cordycepin activates autophagy through AMPK phosphorylation to reduce abnormalities in Machado-Joseph disease models. Hum. Mol. Genet. 2019, 28, 51–63. [Google Scholar] [CrossRef]
- Santana, M.M.; Paixao, S.; Cunha-Santos, J.; Silva, T.P.; Trevino-Garcia, A.; Gaspar, L.S.; Nobrega, C.; Nobre, R.J.; Cavadas, C.; Greif, H.; et al. Trehalose alleviates the phenotype of Machado-Joseph disease mouse models. J. Transl. Med. 2020, 18, 161. [Google Scholar] [CrossRef]
- Mendonca, L.S.; Nobrega, C.; Tavino, S.; Brinkhaus, M.; Matos, C.; Tome, S.; Moreira, R.; Henriques, D.; Kaspar, B.K.; Pereira de Almeida, L. Ibuprofen enhances synaptic function and neural progenitors proliferation markers and improves neuropathology and motor coordination in Machado-Joseph disease models. Hum. Mol. Genet. 2019, 28, 3691–3703. [Google Scholar] [CrossRef]
- Nakamura, K.; Mieda, T.; Suto, N.; Matsuura, S.; Hirai, H. Mesenchymal stem cells as a potential therapeutic tool for spinocerebellar ataxia. Cerebellum 2015, 14, 165–170. [Google Scholar] [CrossRef]
- Oliveira Miranda, C.; Marcelo, A.; Silva, T.P.; Barata, J.; Vasconcelos-Ferreira, A.; Pereira, D.; Nóbrega, C.; Duarte, S.; Barros, I.; Alves, J.; et al. Repeated Mesenchymal Stromal Cell Treatment Sustainably Alleviates Machado-Joseph Disease. Mol. Ther. 2018, 26, 2131–2151. [Google Scholar] [CrossRef]
- Cendelin, J. Transplantation and Stem Cell Therapy for Cerebellar Degenerations. Cerebellum 2016, 15, 48–50. [Google Scholar] [CrossRef]
- Chintawar, S.; Hourez, R.; Ravella, A.; Gall, D.; Orduz, D.; Rai, M.; Bishop, D.P.; Geuna, S.; Schiffmann, S.N.; Pandolfo, M. Grafting neural precursor cells promotes functional recovery in an SCA1 mouse model. J. Neurosci. 2009, 29, 13126–13135. [Google Scholar] [CrossRef]
- Mendonca, L.S.; Nobrega, C.; Hirai, H.; Kaspar, B.K.; Pereira de Almeida, L. Transplantation of cerebellar neural stem cells improves motor coordination and neuropathology in Machado-Joseph disease mice. Brain 2015, 138, 320–335. [Google Scholar] [CrossRef] [PubMed]
- Naldini, L. Gene therapy returns to centre stage. Nature 2015, 526, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, K.B.; Buning, H.; Galy, A.; Schambach, A.; Grez, M. Gene therapy on the move. EMBO Mol. Med. 2013, 5, 1642–1661. [Google Scholar] [CrossRef]
- Nóbrega, C.; Mendonça, L.; Matos, C.A. Gene Therapy Strategies: Gene Augmentation. In A Handbook of Gene and Cell Therapy; Springer: Basel, Switzerland, 2020; pp. 117–126. [Google Scholar]
- Gatchel, J.R.; Watase, K.; Thaller, C.; Carson, J.P.; Jafar-Nejad, P.; Shaw, C.; Zu, T.; Orr, H.T.; Zoghbi, H.Y. The insulin-like growth factor pathway is altered in spinocerebellar ataxia type 1 and type 7. Proc. Natl. Acad. Sci. USA 2008, 105, 1291–1296. [Google Scholar] [CrossRef]
- Zarouchlioti, C.; Parfitt, D.A.; Li, W.; Gittings, L.M.; Cheetham, M.E. DNAJ Proteins in neurodegeneration: Essential and protective factors. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373. [Google Scholar] [CrossRef]
- Cortes, C.J.; La Spada, A.R. Autophagy in polyglutamine disease: Imposing order on disorder or contributing to the chaos? Mol. Cell Neurosci. 2015, 66, 53–61. [Google Scholar] [CrossRef]
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef]
- Lee, L.C.; Chen, C.M.; Wang, P.R.; Su, M.T.; Lee-Chen, G.J.; Chang, C.Y. Role of high mobility group box 1 (HMGB1) in SCA17 pathogenesis. PLoS ONE 2014, 9, e115809. [Google Scholar] [CrossRef] [PubMed]
- Nobrega, C.; Mendonca, L.; Marcelo, A.; Lamaziere, A.; Tome, S.; Despres, G.; Matos, C.A.; Mechmet, F.; Langui, D.; den Dunnen, W.; et al. Restoring brain cholesterol turnover improves autophagy and has therapeutic potential in mouse models of spinocerebellar ataxia. Acta Neuropathol. 2019, 138, 837–858. [Google Scholar] [CrossRef]
- Nascimento-Ferreira, I.; Santos-Ferreira, T.; Sousa-Ferreira, L.; Auregan, G.; Onofre, I.; Alves, S.; Dufour, N.; Colomer Gould, V.F.; Koeppen, A.; Déglon, N.; et al. Overexpression of the autophagic beclin-1 protein clears mutant ataxin-3 and alleviates Machado-Joseph disease. Brain J. Neurol. 2011, 134, 1400–1415. [Google Scholar] [CrossRef]
- Nascimento-Ferreira, I.; Nóbrega, C.; Vasconcelos-Ferreira, A.; Onofre, I.; Albuquerque, D.; Aveleira, C.; Hirai, H.; Déglon, N.; Pereira de Almeida, L. Beclin 1 mitigates motor and neuropathological deficits in genetic mouse models of Machado-Joseph disease. Brain J. Neurol. 2013, 136, 2173–2188. [Google Scholar] [CrossRef]
- Ruegsegger, C.; Stucki, D.M.; Steiner, S.; Angliker, N.; Radecke, J.; Keller, E.; Zuber, B.; Ruegg, M.A.; Saxena, S. Impaired mTORC1-Dependent Expression of Homer-3 Influences SCA1 Pathophysiology. Neuron 2016, 89, 129–146. [Google Scholar] [CrossRef]
- Silva, A.; de Almeida, A.V.; Macedo-Ribeiro, S. Polyglutamine expansion diseases: More than simple repeats. J. Struct. Biol. 2018, 201, 139–154. [Google Scholar] [CrossRef]
- Keiser, M.S.; Geoghegan, J.C.; Boudreau, R.L.; Lennox, K.A.; Davidson, B.L. RNAi or overexpression: Alternative therapies for Spinocerebellar Ataxia Type 1. Neurobiol. Dis. 2013, 56, 6–13. [Google Scholar] [CrossRef]
- Alves, S.; Nascimento-Ferreira, I.; Dufour, N.; Hassig, R.; Auregan, G.; Nóbrega, C.; Brouillet, E.; Hantraye, P.; Pedroso de Lima, M.C.; Déglon, N.; et al. Silencing ataxin-3 mitigates degeneration in a rat model of Machado-Joseph disease: No role for wild-type ataxin-3? Hum. Mol. Genet. 2010, 19, 2380–2394. [Google Scholar] [CrossRef] [PubMed]
- Nobrega, C.; Carmo-Silva, S.; Albuquerque, D.; Vasconcelos-Ferreira, A.; Vijayakumar, U.G.; Mendonca, L.; Hirai, H.; de Almeida, L.P. Re-establishing ataxin-2 downregulates translation of mutant ataxin-3 and alleviates Machado-Joseph disease. Brain 2015, 138, 3537–3554. [Google Scholar] [CrossRef] [PubMed]
- Matos, C.A.; Almeida, L.P.; Nobrega, C. Proteolytic Cleavage of Polyglutamine Disease-Causing Proteins: Revisiting the Toxic Fragment Hypothesis. Curr. Pharm. Des. 2017, 23, 753–775. [Google Scholar] [CrossRef] [PubMed]
- Simões, A.T.; Gonçalves, N.; Koeppen, A.; Déglon, N.; Kügler, S.; Duarte, C.B.; Pereira de Almeida, L. Calpastatin-mediated inhibition of calpains in the mouse brain prevents mutant ataxin 3 proteolysis, nuclear localization and aggregation, relieving Machado-Joseph disease. Brain J. Neurol. 2012, 135, 2428–2439. [Google Scholar] [CrossRef]
- Torashima, T.; Koyama, C.; Iizuka, A.; Mitsumura, K.; Takayama, K.; Yanagi, S.; Oue, M.; Yamaguchi, H.; Hirai, H. Lentivector-mediated rescue from cerebellar ataxia in a mouse model of spinocerebellar ataxia. EMBO Rep. 2008, 9, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Duarte-Neves, J.; Gonçalves, N.; Cunha-Santos, J.; Simões, A.T.; den Dunnen, W.F.; Hirai, H.; Kügler, S.; Cavadas, C.; Pereira de Almeida, L. Neuropeptide Y mitigates neuropathology and motor deficits in mouse models of Machado-Joseph disease. Hum. Mol. Genet. 2015, 24, 5451–5463. [Google Scholar] [CrossRef]
- Li, L.B.; Yu, Z.; Teng, X.; Bonini, N.M. RNA toxicity is a component of ataxin-3 degeneration in Drosophila. Nature 2008, 453, 1107–1111. [Google Scholar] [CrossRef]
- Wang, L.C.; Chen, K.Y.; Pan, H.; Wu, C.C.; Chen, P.H.; Liao, Y.T.; Li, C.; Huang, M.L.; Hsiao, K.M. Muscleblind participates in RNA toxicity of expanded CAG and CUG repeats in Caenorhabditis elegans. Cell. Mol. Life Sci. 2011, 68, 1255–1267. [Google Scholar] [CrossRef]
- Hsu, R.J.; Hsiao, K.M.; Lin, M.J.; Li, C.Y.; Wang, L.C.; Chen, L.K.; Pan, H. Long tract of untranslated CAG repeats is deleterious in transgenic mice. PLoS ONE 2011, 6, e16417. [Google Scholar] [CrossRef]
- Mykowska, A.; Sobczak, K.; Wojciechowska, M.; Kozlowski, P.; Krzyzosiak, W.J. CAG repeats mimic CUG repeats in the misregulation of alternative splicing. Nucleic Acids Res. 2011, 39, 8938–8951. [Google Scholar] [CrossRef]
- Tsoi, H.; Lau, T.C.; Tsang, S.Y.; Lau, K.F.; Chan, H.Y. CAG expansion induces nucleolar stress in polyglutamine diseases. Proc. Natl. Acad. Sci. USA 2012, 109, 13428–13433. [Google Scholar] [CrossRef] [PubMed]
- Krol, J.; Fiszer, A.; Mykowska, A.; Sobczak, K.; de Mezer, M.; Krzyzosiak, W.J. Ribonuclease dicer cleaves triplet repeat hairpins into shorter repeats that silence specific targets. Mol. Cell 2007, 25, 575–586. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Setten, R.L.; Rossi, J.J.; Han, S.P. The current state and future directions of RNAi-based therapeutics. Nat. Rev. Drug Discov. 2019, 18, 421–446. [Google Scholar] [CrossRef] [PubMed]
- Matos, C.A.; Carmona, V.; Vijayakumar, U.G.; Lopes, S.; Albuquerque, P.; Conceição, M.; Nobre, R.J.; Nóbrega, C.; de Almeida, L.P. Gene Therapies for Polyglutamine Diseases. Adv. Exp. Med. Biol. 2018, 1049, 395–438. [Google Scholar] [CrossRef]
- Han, H. RNA Interference to Knock Down Gene Expression. Methods Mol. Biol. 2018, 1706, 293–302. [Google Scholar] [CrossRef]
- Nóbrega, C.; Codêsso, J.M.; Mendonça, L.; Pereira de Almeida, L. RNA Interference Therapy for Machado-Joseph Disease: Long-Term Safety Profile of Lentiviral Vectors Encoding Short Hairpin RNAs Targeting Mutant Ataxin-3. Hum. Gene Ther. 2019, 30, 841–854. [Google Scholar] [CrossRef]
- Bobbin, M.L.; Rossi, J.J. RNA Interference (RNAi)-Based Therapeutics: Delivering on the Promise? Annu. Rev. Pharm. Toxicol. 2016, 56, 103–122. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Mao, Q.; Eliason, S.L.; Harper, S.Q.; Martins, I.H.; Orr, H.T.; Paulson, H.L.; Yang, L.; Kotin, R.M.; Davidson, B.L. RNAi suppresses polyglutamine-induced neurodegeneration in a model of spinocerebellar ataxia. Nat. Med. 2004, 10, 816–820. [Google Scholar] [CrossRef]
- Keiser, M.S.; Boudreau, R.L.; Davidson, B.L. Broad therapeutic benefit after RNAi expression vector delivery to deep cerebellar nuclei: Implications for spinocerebellar ataxia type 1 therapy. Mol. Ther. J. Am. Soc. Gene Ther. 2014, 22, 588–595. [Google Scholar] [CrossRef]
- Keiser, M.S.; Kordower, J.H.; Gonzalez-Alegre, P.; Davidson, B.L. Broad distribution of ataxin 1 silencing in rhesus cerebella for spinocerebellar ataxia type 1 therapy. Brain J. Neurol. 2015, 138, 3555–3566. [Google Scholar] [CrossRef]
- Rodrigues, A.J.; Coppola, G.; Santos, C.; Costa Mdo, C.; Ailion, M.; Sequeiros, J.; Geschwind, D.H.; Maciel, P. Functional genomics and biochemical characterization of the C. elegans orthologue of the Machado-Joseph disease protein ataxin-3. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2007, 21, 1126–1136. [Google Scholar] [CrossRef]
- Schmitt, I.; Linden, M.; Khazneh, H.; Evert, B.O.; Breuer, P.; Klockgether, T.; Wuellner, U. Inactivation of the mouse Atxn3 (ataxin-3) gene increases protein ubiquitination. Biochem. Biophys. Res. Commun. 2007, 362, 734–739. [Google Scholar] [CrossRef]
- Zeng, L.; Zhang, D.; McLoughlin, H.S.; Zalon, A.J.; Aravind, L.; Paulson, H.L. Loss of the Spinocerebellar Ataxia type 3 disease protein ATXN3 alters transcription of multiple signal transduction pathways. PLoS ONE 2018, 13, e0204438. [Google Scholar] [CrossRef]
- Alves, S.; Nascimento-Ferreira, I.; Auregan, G.; Hassig, R.; Dufour, N.; Brouillet, E.; Pedroso de Lima, M.C.; Hantraye, P.; Pereira de Almeida, L.; Déglon, N. Allele-specific RNA silencing of mutant ataxin-3 mediates neuroprotection in a rat model of Machado-Joseph disease. PLoS ONE 2008, 3, e3341. [Google Scholar] [CrossRef]
- Li, Y.; Yokota, T.; Matsumura, R.; Taira, K.; Mizusawa, H. Sequence-dependent and independent inhibition specific for mutant ataxin-3 by small interfering RNA. Ann. Neurol. 2004, 56, 124–129. [Google Scholar] [CrossRef]
- Nóbrega, C.; Nascimento-Ferreira, I.; Onofre, I.; Albuquerque, D.; Déglon, N.; de Almeida, L.P. RNA interference mitigates motor and neuropathological deficits in a cerebellar mouse model of Machado-Joseph disease. PLoS ONE 2014, 9, e100086. [Google Scholar] [CrossRef]
- Nóbrega, C.; Nascimento-Ferreira, I.; Onofre, I.; Albuquerque, D.; Hirai, H.; Déglon, N.; de Almeida, L.P. Silencing mutant ataxin-3 rescues motor deficits and neuropathology in Machado-Joseph disease transgenic mice. PLoS ONE 2013, 8, e52396. [Google Scholar] [CrossRef]
- Conceição, M.; Mendonça, L.; Nóbrega, C.; Gomes, C.; Costa, P.; Hirai, H.; Moreira, J.N.; Lima, M.C.; Manjunath, N.; Pereira de Almeida, L. Intravenous administration of brain-targeted stable nucleic acid lipid particles alleviates Machado-Joseph disease neurological phenotype. Biomaterials 2016, 82, 124–137. [Google Scholar] [CrossRef]
- Scholefield, J.; Watson, L.; Smith, D.; Greenberg, J.; Wood, M.J. Allele-specific silencing of mutant Ataxin-7 in SCA7 patient-derived fibroblasts. Eur. J. Hum. Genet. 2014, 22, 1369–1375. [Google Scholar] [CrossRef]
- Fiszer, A.; Wroblewska, J.P.; Nowak, B.M.; Krzyzosiak, W.J. Mutant CAG Repeats Effectively Targeted by RNA Interference in SCA7 Cells. Genes 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Kotowska-Zimmer, A.; Ostrovska, Y.; Olejniczak, M. Universal RNAi Triggers for the Specific Inhibition of Mutant Huntingtin, Atrophin-1, Ataxin-3, and Ataxin-7 Expression. Mol. Ther. Nucleic Acids 2020, 19, 562–571. [Google Scholar] [CrossRef] [PubMed]
- Krauss, S.; Nalavade, R.; Weber, S.; Carter, K.; Evert, B.O. Upregulation of miR-25 and miR-181 Family Members Correlates with Reduced Expression of ATXN3 in Lymphocytes from SCA3 Patients. Microrna (ShariqahUnited Arab Emir.) 2019, 8, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Boudreau, R.L.; Martins, I.; Davidson, B.L. Artificial microRNAs as siRNA shuttles: Improved safety as compared to shRNAs in vitro and in vivo. Mol. Ther. J. Am. Soc. Gene Ther. 2009, 17, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Costa Mdo, C.; Luna-Cancalon, K.; Fischer, S.; Ashraf, N.S.; Ouyang, M.; Dharia, R.M.; Martin-Fishman, L.; Yang, Y.; Shakkottai, V.G.; Davidson, B.L.; et al. Toward RNAi therapy for the polyglutamine disease Machado-Joseph disease. Mol. Ther. J. Am. Soc. Gene Ther. 2013, 21, 1898–1908. [Google Scholar] [CrossRef] [PubMed]
- Keiser, M.S.; Kordasiewicz, H.B.; McBride, J.L. Gene suppression strategies for dominantly inherited neurodegenerative diseases: Lessons from Huntington’s disease and spinocerebellar ataxia. Hum. Mol. Genet. 2016, 25, R53–R64. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, P.S.; Bhattarai, S.; Singh, P.; Boudreau, R.L.; Thompson, S.; Laspada, A.R.; Drack, A.V.; Davidson, B.L. RNA interference-based therapy for spinocerebellar ataxia type 7 retinal degeneration. PLoS ONE 2014, 9, e95362. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, P.S.; Boudreau, R.L.; Schaefer, K.A.; La Spada, A.R.; Davidson, B.L. Nonallele specific silencing of ataxin-7 improves disease phenotypes in a mouse model of SCA7. Mol. Ther. J. Am. Soc. Gene Ther. 2014, 22, 1635–1642. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Lebrón, E.; Costa Mdo, C.; Luna-Cancalon, K.; Peron, T.M.; Fischer, S.; Boudreau, R.L.; Davidson, B.L.; Paulson, H.L. Silencing mutant ATXN3 expression resolves molecular phenotypes in SCA3 transgenic mice. Mol. Ther. J. Am. Soc. Gene Ther. 2013, 21, 1909–1918. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Zhang, L.; Long, Z.; Chen, Z.; Hou, X.; Wang, C.; Peng, H.; Wang, J.; Li, J.; Duan, R.; et al. miR-25 alleviates polyQ-mediated cytotoxicity by silencing ATXN3. FEBS Lett. 2014, 588, 4791–4798. [Google Scholar] [CrossRef] [PubMed]
- Carmona, V.; Cunha-Santos, J.; Onofre, I.; Simões, A.T.; Vijayakumar, U.; Davidson, B.L.; Pereira de Almeida, L. Unravelling Endogenous MicroRNA System Dysfunction as a New Pathophysiological Mechanism in Machado-Joseph Disease. Mol. Ther. J. Am. Soc. Gene Ther. 2017, 25, 1038–1055. [Google Scholar] [CrossRef]
- Miyazaki, Y.; Du, X.; Muramatsu, S.; Gomez, C.M. An miRNA-mediated therapy for SCA6 blocks IRES-driven translation of the CACNA1A second cistron. Sci. Transl. Med. 2016, 8, 347ra394. [Google Scholar] [CrossRef]
- Tan, J.Y.; Vance, K.W.; Varela, M.A.; Sirey, T.; Watson, L.M.; Curtis, H.J.; Marinello, M.; Alves, S.; Steinkraus, B.; Cooper, S.; et al. Cross-talking noncoding RNAs contribute to cell-specific neurodegeneration in SCA7. Nat. Struct. Mol. Biol. 2014, 21, 955–961. [Google Scholar] [CrossRef]
- Lee, Y.; Samaco, R.C.; Gatchel, J.R.; Thaller, C.; Orr, H.T.; Zoghbi, H.Y. miR-19, miR-101 and miR-130 co-regulate ATXN1 levels to potentially modulate SCA1 pathogenesis. Nat. Neurosci. 2008, 11, 1137–1139. [Google Scholar] [CrossRef]
- Persengiev, S.; Kondova, I.; Otting, N.; Koeppen, A.H.; Bontrop, R.E. Genome-wide analysis of miRNA expression reveals a potential role for miR-144 in brain aging and spinocerebellar ataxia pathogenesis. Neurobiol. Aging 2011, 32, 2316.e17–2316.e27. [Google Scholar] [CrossRef]
- Zamecnik, P.C.; Stephenson, M.L. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 280–284. [Google Scholar] [CrossRef]
- Monia, B.P.; Lesnik, E.A.; Gonzalez, C.; Lima, W.F.; McGee, D.; Guinosso, C.J.; Kawasaki, A.M.; Cook, P.D.; Freier, S.M. Evaluation of 2’-modified oligonucleotides containing 2’-deoxy gaps as antisense inhibitors of gene expression. J. Biol. Chem. 1993, 268, 14514–14522. [Google Scholar] [CrossRef]
- Walder, R.Y.; Walder, J.A. Role of RNase H in hybrid-arrested translation by antisense oligonucleotides. Proc. Natl. Acad. Sci. USA 1988, 85, 5011–5015. [Google Scholar] [CrossRef] [PubMed]
- Evers, M.M.; Tran, H.D.; Zalachoras, I.; Pepers, B.A.; Meijer, O.C.; den Dunnen, J.T.; van Ommen, G.J.; Aartsma-Rus, A.; van Roon-Mom, W.M. Ataxin-3 protein modification as a treatment strategy for spinocerebellar ataxia type 3: Removal of the CAG containing exon. Neurobiol. Dis. 2013, 58, 49–56. [Google Scholar] [CrossRef][Green Version]
- Sazani, P.; Kole, R. Therapeutic potential of antisense oligonucleotides as modulators of alternative splicing. J. Clin. Investig. 2003, 112, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Toonen, L.J.A.; Rigo, F.; van Attikum, H.; van Roon-Mom, W.M.C. Antisense Oligonucleotide-Mediated Removal of the Polyglutamine Repeat in Spinocerebellar Ataxia Type 3 Mice. Mol. Ther. Nucleic Acids 2017, 8, 232–242. [Google Scholar] [CrossRef]
- Moore, L.R.; Rajpal, G.; Dillingham, I.T.; Qutob, M.; Blumenstein, K.G.; Gattis, D.; Hung, G.; Kordasiewicz, H.B.; Paulson, H.L.; McLoughlin, H.S. Evaluation of Antisense Oligonucleotides Targeting ATXN3 in SCA3 Mouse Models. Mol. Ther. Nucleic Acids 2017, 7, 200–210. [Google Scholar] [CrossRef]
- McLoughlin, H.S.; Moore, L.R.; Chopra, R.; Komlo, R.; McKenzie, M.; Blumenstein, K.G.; Zhao, H.; Kordasiewicz, H.B.; Shakkottai, V.G.; Paulson, H.L. Oligonucleotide therapy mitigates disease in spinocerebellar ataxia type 3 mice. Ann. Neurol. 2018, 84, 64–77. [Google Scholar] [CrossRef]
- Friedrich, J.; Kordasiewicz, H.B.; O’Callaghan, B.; Handler, H.P.; Wagener, C.; Duvick, L.; Swayze, E.E.; Rainwater, O.; Hofstra, B.; Benneyworth, M.; et al. Antisense oligonucleotide-mediated ataxin-1 reduction prolongs survival in SCA1 mice and reveals disease-associated transcriptome profiles. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Scoles, D.R.; Meera, P.; Schneider, M.D.; Paul, S.; Dansithong, W.; Figueroa, K.P.; Hung, G.; Rigo, F.; Bennett, C.F.; Otis, T.S.; et al. Antisense oligonucleotide therapy for spinocerebellar ataxia type 2. Nature 2017, 544, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Niu, C.; Prakash, T.P.; Kim, A.; Quach, J.L.; Huryn, L.A.; Yang, Y.; Lopez, E.; Jazayeri, A.; Hung, G.; Sopher, B.L.; et al. Antisense oligonucleotides targeting mutant Ataxin-7 restore visual function in a mouse model of spinocerebellar ataxia type 7. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Evers, M.M.; Pepers, B.A.; van Deutekom, J.C.; Mulders, S.A.; den Dunnen, J.T.; Aartsma-Rus, A.; van Ommen, G.J.; van Roon-Mom, W.M. Targeting several CAG expansion diseases by a single antisense oligonucleotide. PLoS ONE 2011, 6, e24308. [Google Scholar] [CrossRef] [PubMed]
- Kourkouta, E.; Weij, R.; González-Barriga, A.; Mulder, M.; Verheul, R.; Bosgra, S.; Groenendaal, B.; Puoliväli, J.; Toivanen, J.; van Deutekom, J.C.T.; et al. Suppression of Mutant Protein Expression in SCA3 and SCA1 Mice Using a CAG Repeat-Targeting Antisense Oligonucleotide. Mol. Ther. Nucleic Acids 2019, 17, 601–614. [Google Scholar] [CrossRef] [PubMed]
- Maeder, M.L.; Gersbach, C.A. Genome-editing Technologies for Gene and Cell Therapy. Mol. Ther. 2016, 24, 430–446. [Google Scholar] [CrossRef]
- Kc, M.; Steer, C.J. A new era of gene editing for the treatment of human diseases. Swiss Med. Wkly. 2019, 149, w20021. [Google Scholar] [CrossRef]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef]
- Ouyang, S.; Xie, Y.; Xiong, Z.; Yang, Y.; Xian, Y.; Ou, Z.; Song, B.; Chen, Y.; Xie, Y.; Li, H.; et al. CRISPR/Cas9-Targeted Deletion of Polyglutamine in Spinocerebellar Ataxia Type 3-Derived Induced Pluripotent Stem Cells. Stem Cells Dev. 2018, 27, 756–770. [Google Scholar] [CrossRef]
- Nishiyama, J.; Mikuni, T.; Yasuda, R. Virus-Mediated Genome Editing via Homology-Directed Repair in Mitotic and Postmitotic Cells in Mammalian Brain. Neuron 2017, 96, 755–768.e5. [Google Scholar] [CrossRef]
- Bak, R.O.; Gomez-Ospina, N.; Porteus, M.H. Gene Editing on Center Stage. Trends Genet. Tig 2018, 34, 600–611. [Google Scholar] [CrossRef]
- Takata, M.; Sasaki, M.S.; Sonoda, E.; Morrison, C.; Hashimoto, M.; Utsumi, H.; Yamaguchi-Iwai, Y.; Shinohara, A.; Takeda, S. Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J. 1998, 17, 5497–5508. [Google Scholar] [CrossRef] [PubMed]
- Segal, D.J.; Beerli, R.R.; Blancafort, P.; Dreier, B.; Effertz, K.; Huber, A.; Koksch, B.; Lund, C.V.; Magnenat, L.; Valente, D.; et al. Evaluation of a modular strategy for the construction of novel polydactyl zinc finger DNA-binding proteins. Biochemistry 2003, 42, 2137–2148. [Google Scholar] [CrossRef] [PubMed]
- Seligman, L.M.; Chisholm, K.M.; Chevalier, B.S.; Chadsey, M.S.; Edwards, S.T.; Savage, J.H.; Veillet, A.L. Mutations altering the cleavage specificity of a homing endonuclease. Nucleic Acids Res. 2002, 30, 3870–3879. [Google Scholar] [CrossRef] [PubMed]
- Boch, J.; Scholze, H.; Schornack, S.; Landgraf, A.; Hahn, S.; Kay, S.; Lahaye, T.; Nickstadt, A.; Bonas, U. Breaking the code of DNA binding specificity of TAL-type III effectors. Science 2009, 326, 1509–1512. [Google Scholar] [CrossRef] [PubMed]
- Doudna, J.A.; Charpentier, E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef]
- Shin, J.W.; Kim, K.H.; Chao, M.J.; Atwal, R.S.; Gillis, T.; MacDonald, M.E.; Gusella, J.F.; Lee, J.M. Permanent inactivation of Huntington’s disease mutation by personalized allele-specific CRISPR/Cas9. Hum. Mol. Genet. 2016, 25, 4566–4576. [Google Scholar] [CrossRef]
- Monteys, A.M.; Ebanks, S.A.; Keiser, M.S.; Davidson, B.L. CRISPR/Cas9 Editing of the Mutant Huntingtin Allele In Vitro and In Vivo. Mol. Ther. J. Am. Soc. Gene Ther. 2017, 25, 12–23. [Google Scholar] [CrossRef] [PubMed]

| PolyQ | Gene | Locus | Protein | Molecular Function | Repeats | ||
|---|---|---|---|---|---|---|---|
| Normal | Intermediate | Disease | |||||
| SCA1 | ATXN1 | 6p22.3 | Ataxin-1 | Transcription Factor Interactor | 9–39 | 40 | 41–83 |
| SCA2 | ATXN2 | 12q24.12 | Ataxin-2 | RNA metabolism | <31 | 31–33 | 34–200 |
| SCA3/MJD | ATXN3 | 14q32.12 | Ataxin-3 | Deubiquitinase | 12–44 | 45–55 | 56–86 |
| SCA6 | CACNA1A | 19p13.13 | Calcium voltage-gated channel subunit alpha 1 G | Channel and Transcription Factor | <18 | 19 | 20–33 |
| SCA7 | ATXN7 | 3p14.1 | Ataxin-7 | Transcription Factor (SAGA Complex) | 4–19 | 28–33 | 34–460 |
| SCA17 | TBP | 6q27 | TATA box-binding protein | Transcription Factor | 25–40 | - | 41–66 |
| Pathophysiological Features | PolyQ SCAs Therapeutic Targets |
|---|---|
| Mutant RNA and protein derived toxicity | Disease-causing gene, mutant RNA |
| Impairments in protein degradation and aggregate clearance | Autophagy and ubiquitin-protease system, mutant protein aggregation |
| Formation of toxic protein fragments | Mutant protein cleavage and proteolytic enzymes |
| Deficient neuronal survival: | |
| Disfuncional cellular structures | Endoplasmic reticulum stress, mitochondrial functioning |
| Failure of cellular processes | Neuroprotective pathways, inflammation, excitotoxicity, synaptic signaling, calcium homeostasis, post transcription modifications, translation |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Afonso-Reis, R.; Afonso, I.T.; Nóbrega, C. Current Status of Gene Therapy Research in Polyglutamine Spinocerebellar Ataxias. Int. J. Mol. Sci. 2021, 22, 4249. https://doi.org/10.3390/ijms22084249
Afonso-Reis R, Afonso IT, Nóbrega C. Current Status of Gene Therapy Research in Polyglutamine Spinocerebellar Ataxias. International Journal of Molecular Sciences. 2021; 22(8):4249. https://doi.org/10.3390/ijms22084249
Chicago/Turabian StyleAfonso-Reis, Ricardo, Inês T. Afonso, and Clévio Nóbrega. 2021. "Current Status of Gene Therapy Research in Polyglutamine Spinocerebellar Ataxias" International Journal of Molecular Sciences 22, no. 8: 4249. https://doi.org/10.3390/ijms22084249
APA StyleAfonso-Reis, R., Afonso, I. T., & Nóbrega, C. (2021). Current Status of Gene Therapy Research in Polyglutamine Spinocerebellar Ataxias. International Journal of Molecular Sciences, 22(8), 4249. https://doi.org/10.3390/ijms22084249