
Lipopolysaccharide-Enhanced Responses against Aryl Hydrocarbon Receptor in FcgRIIb-Deficient Macrophages, a Profound Impact of an Environmental Toxin on a Lupus-Like Mouse Model

 , , and
, , and 
Abstract
1. Introduction
2. Results
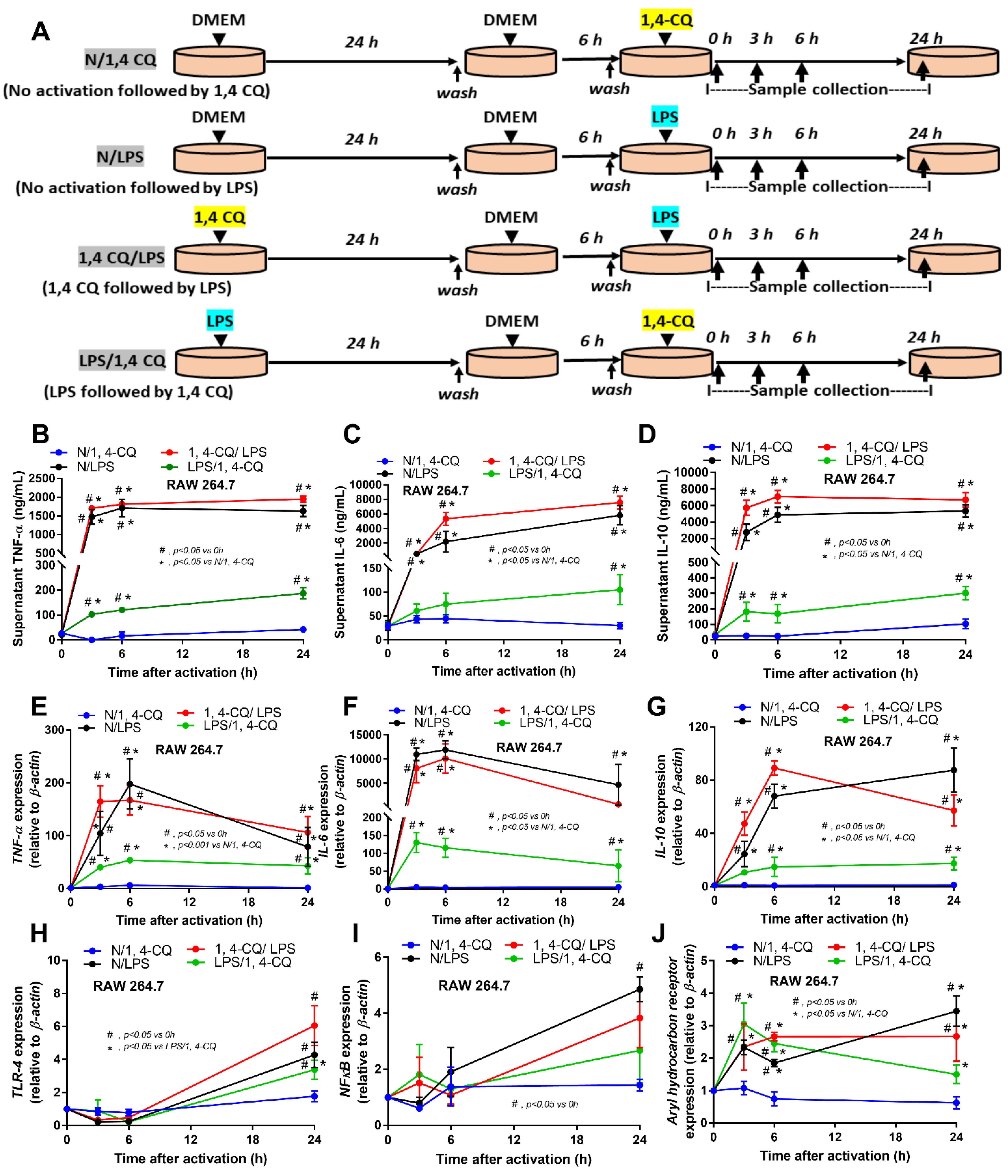
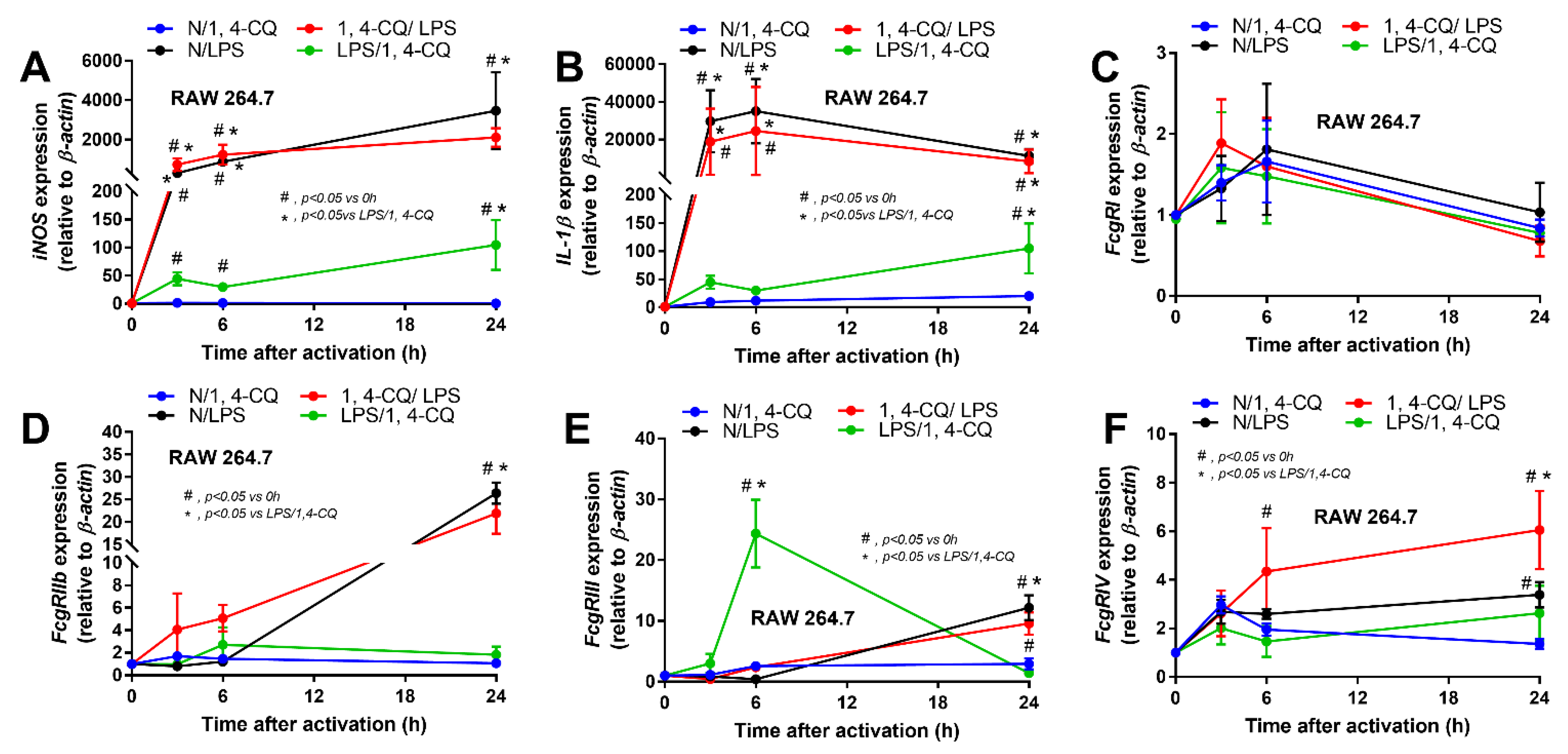
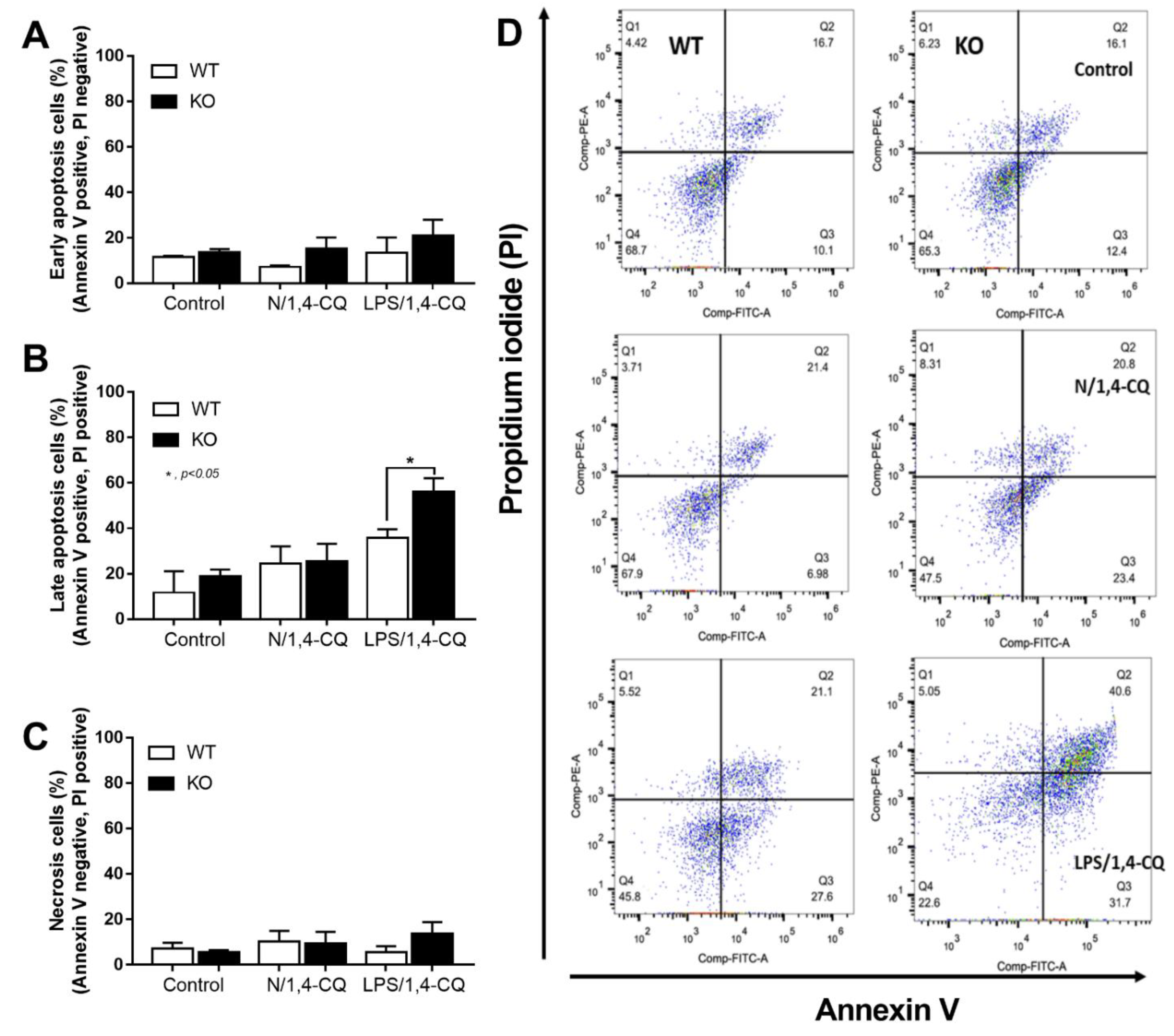
2.1. Pre-Treatment with LPS Enhanced Macrophage Responses toward Ahr Activator, But Not Vice Versa, in the Macrophage Cell Line
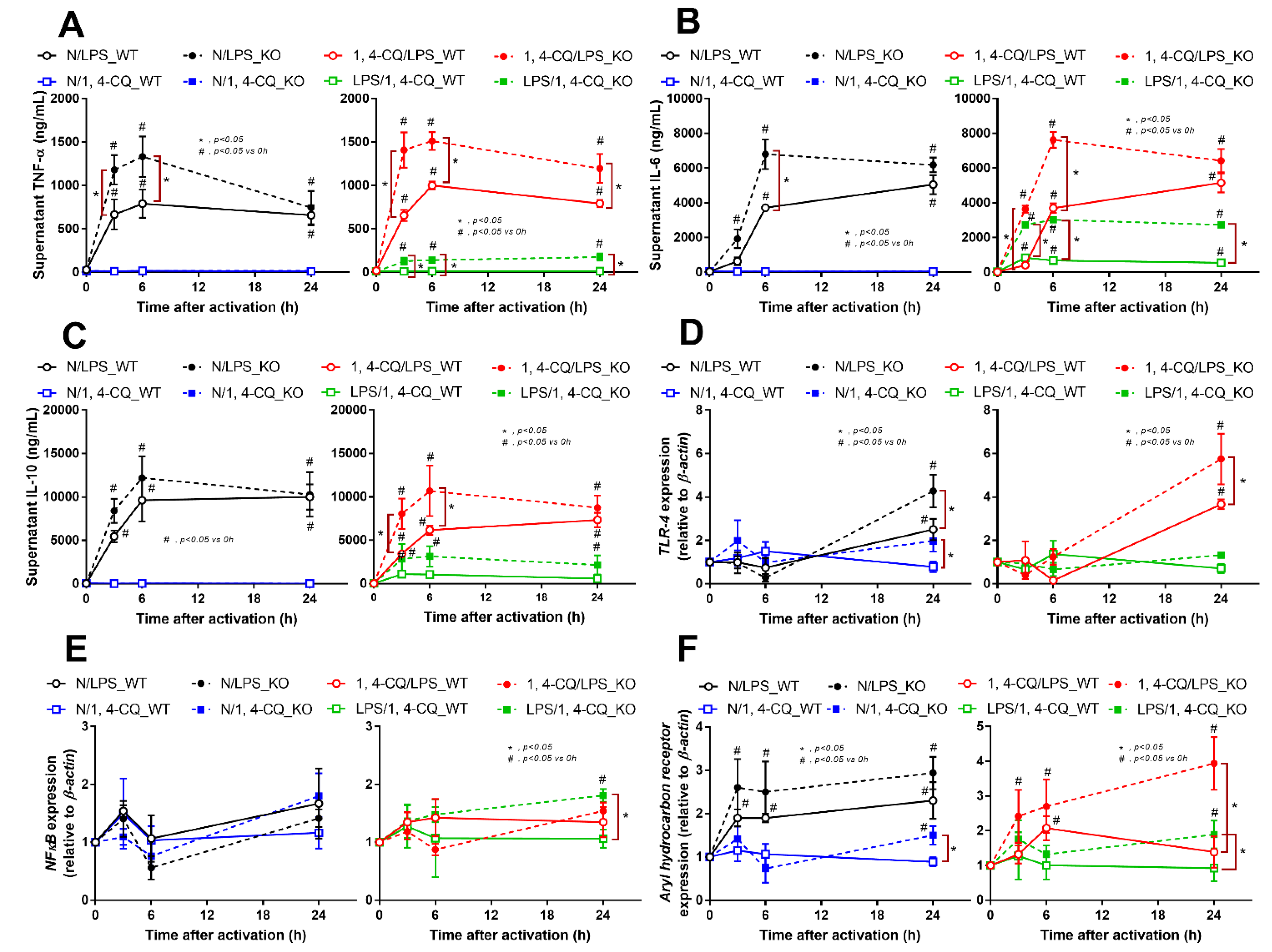
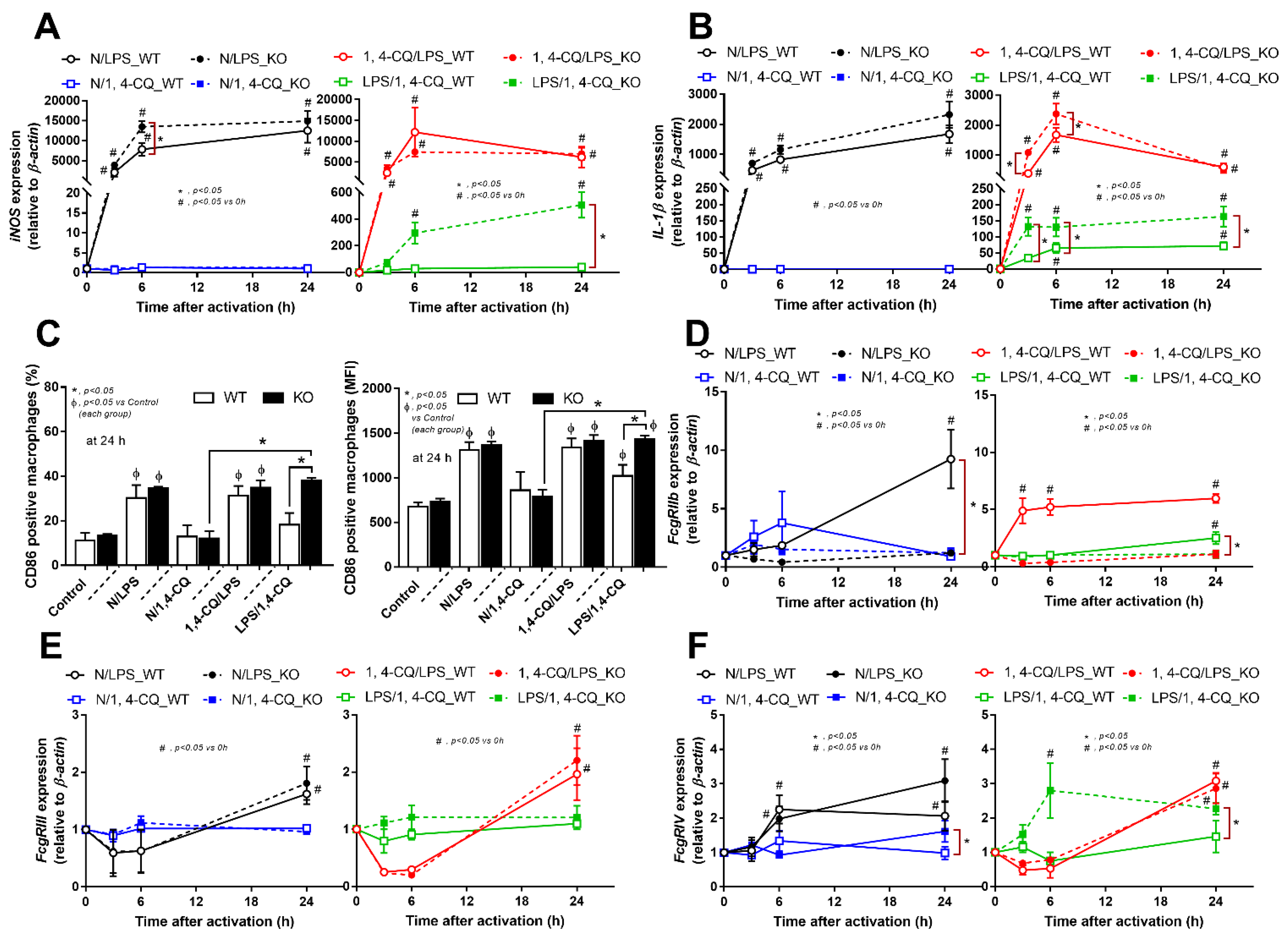
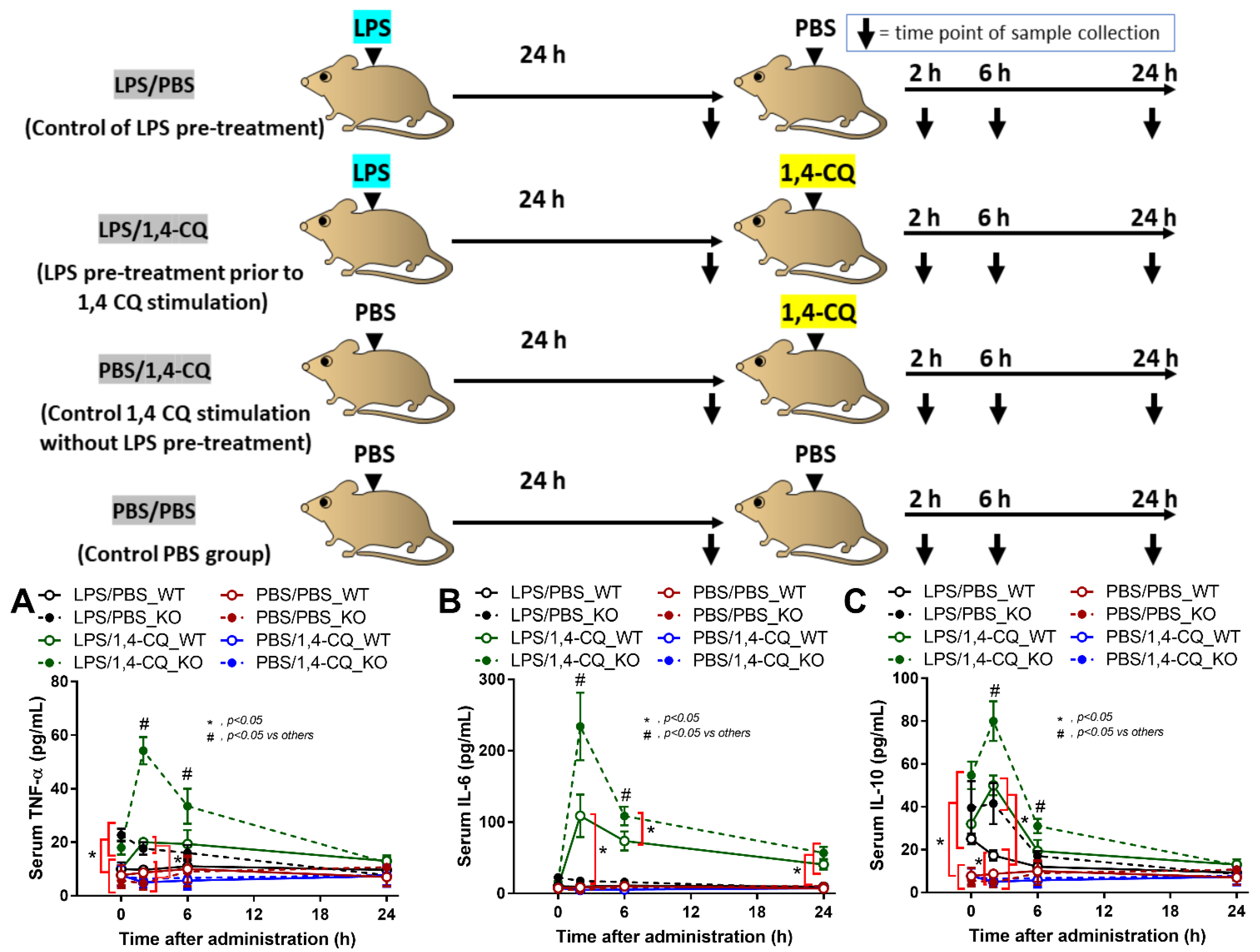
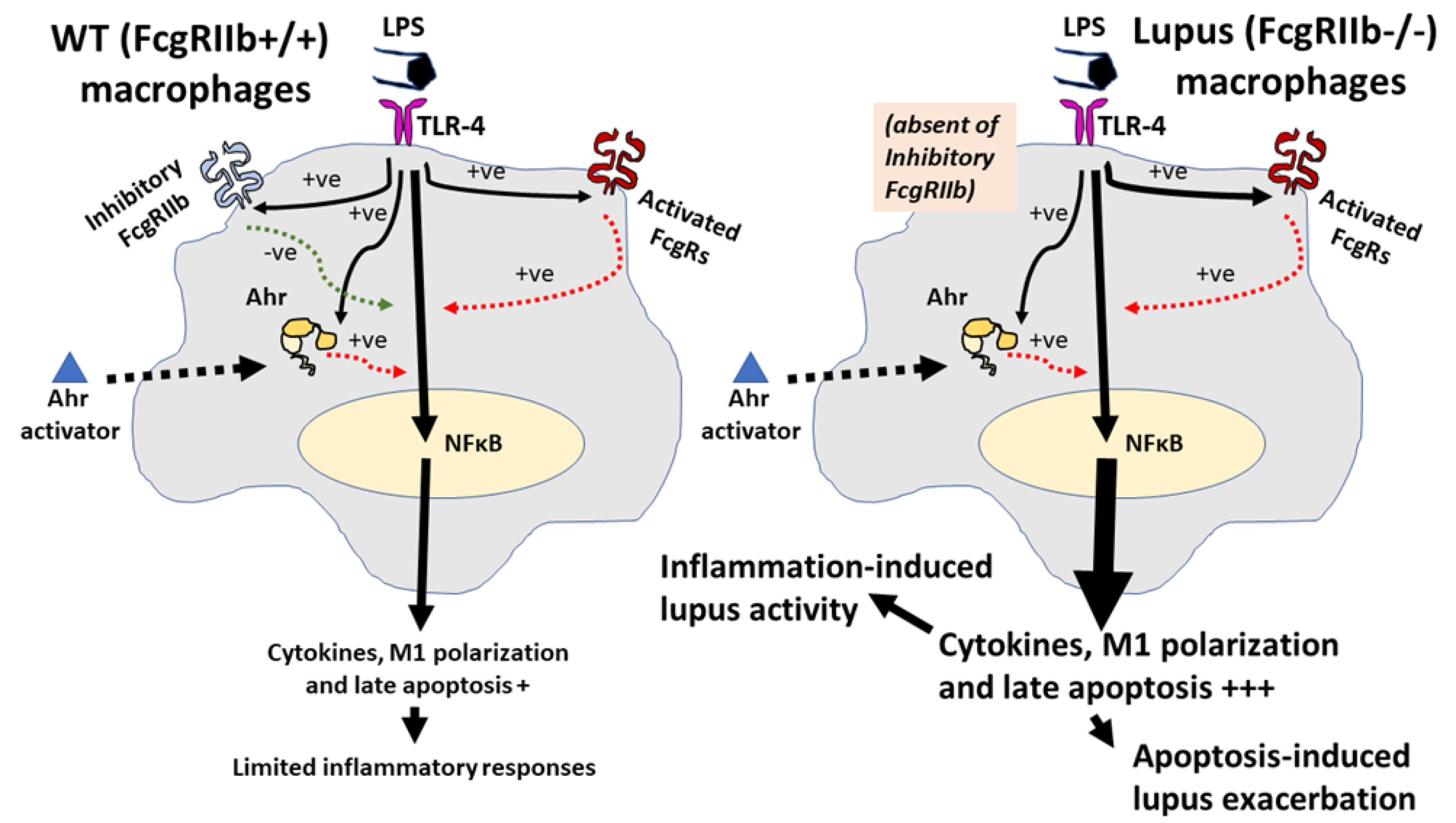
2.2. Pre-Treatment with LPS before Ahr Activation in FcgRIIb−/− Macrophages Induced More Severe Inflammation Than WT Cells
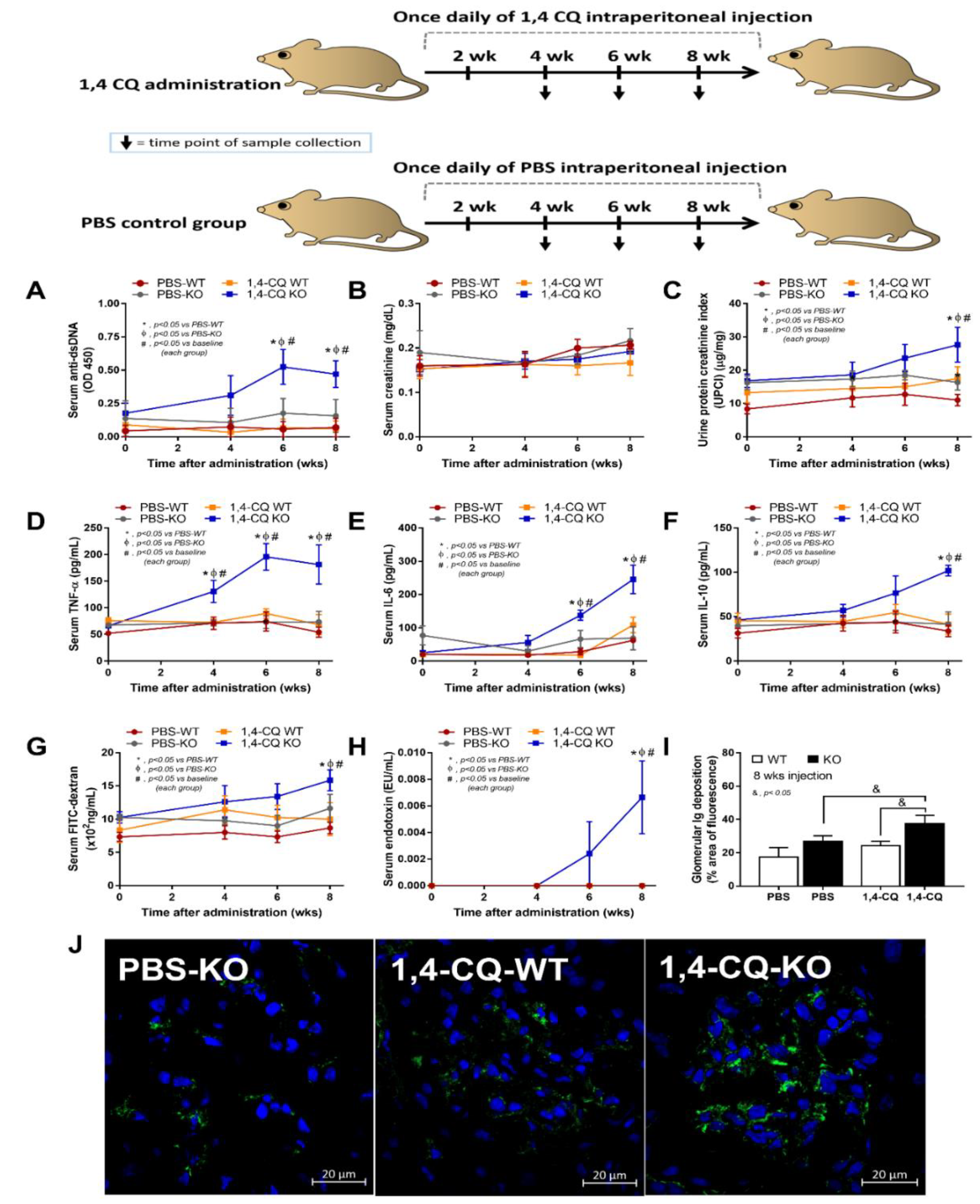
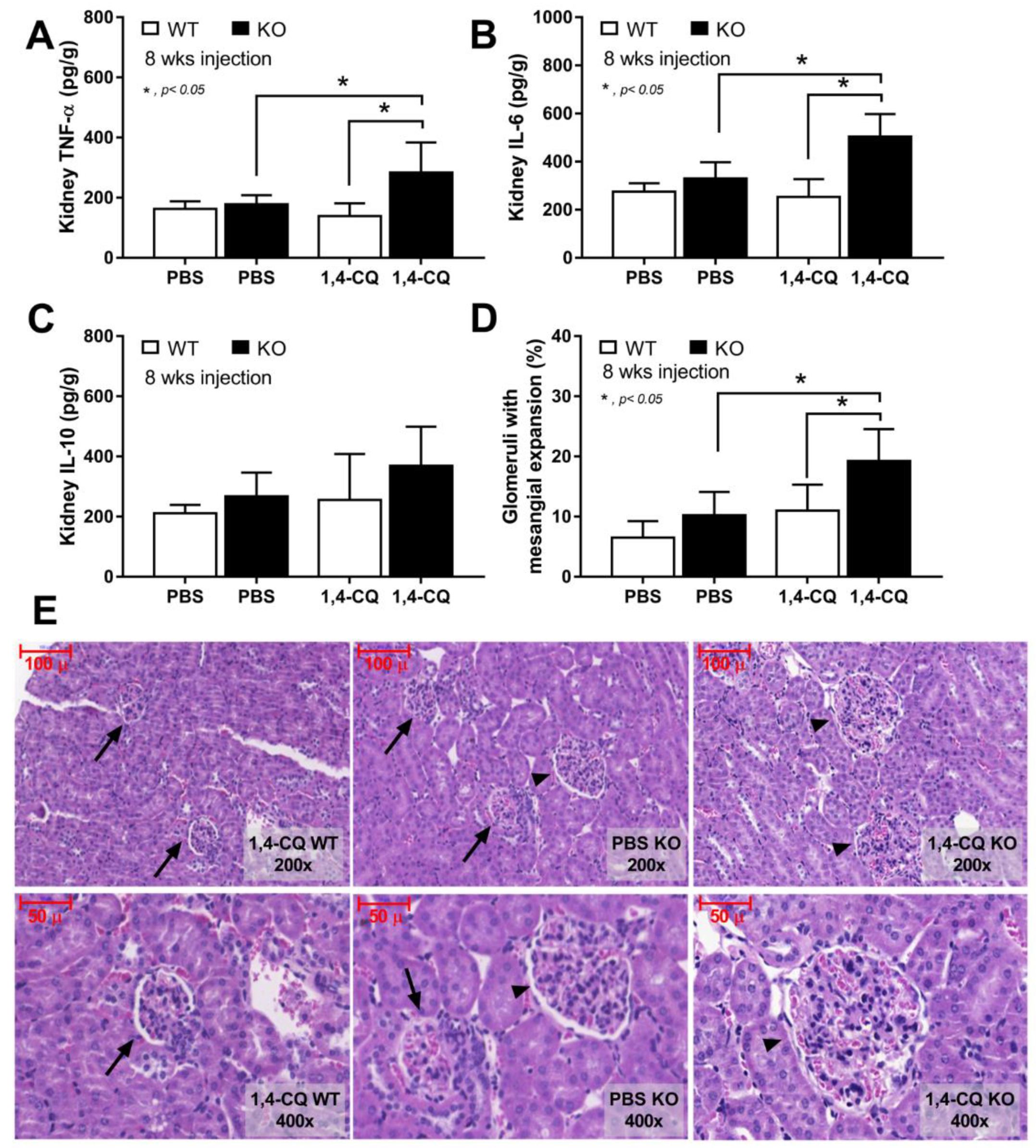
2.3. Prominent Inflammatory Activation and the Exacerbation of Lupus-Like Condition after the Activation of Aryl Hydrocarbon Receptor in FcgRIIb−/− Mice
3. Discussion
3.1. The Enhanced Macrophage Responses against an Aryl Hydrocarbon Receptor Activator by a Pre-Conditioning Immune Activation, an Impact of Inflammation from the Environmental Toxins
3.2. Prominent Inflammatory Responses in FcgRIIb−/− Macrophages over the Wild-Type Cells, an Inhibitory Effect of FcgRIIb
3.3. The Enhanced Activity of Lupus-Like Condition through the Aryl Hydrocarbon Receptor Activation, a Possible Impact of the Environmental Toxins in Lupus
4. Materials and Methods
4.1. The In Vitro Macrophage Experiments
4.2. Animal Model and Sample Collection
4.3. Gut Permeability Determination
4.4. Histology and Immunofluorescent Imaging
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, D.; Nishimura, N.; Kuo, V.; Fiehn, O.; Shahbaz, S.; Van Winkle, L.; Matsumura, F.; Vogel, C.F. Activation of aryl hydrocarbon receptor induces vascular inflammation and promotes atherosclerosis in apolipoprotein E−/− mice. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1260–1267. [Google Scholar] [CrossRef]
- Lawal, A.O. Air particulate matter induced oxidative stress and inflammation in cardiovascular disease and atherosclerosis: The role of Nrf2 and AhR-mediated pathways. Toxicol. Lett. 2017, 270, 88–95. [Google Scholar] [CrossRef]
- Rekefu, S.; Talifu, D.; Gao, B.; Turap, Y.; Maihemuti, M.; Wang, X.; Abulizi, A. Polycyclic Aromatic Hydrocarbons in PM2.5 and PM2.5–10 in Urumqi, China: Temporal Variations, Health Risk, and Sources. Atmosphere 2018, 9, 412. [Google Scholar] [CrossRef]
- Wakamatsu, T.; Yamamoto, S.; Ito, T.; Sato, Y.; Matsuo, K.; Takahashi, Y.; Kaneko, Y.; Goto, S.; Kazama, J.J.; Gejyo, F.; et al. Indoxyl Sulfate Promotes Macrophage IL-1β Production by Activating Aryl Hydrocarbon Receptor/NF-κ/MAPK Cascades, but the NLRP3 inflammasome Was Not Activated. Toxins 2018, 10, 124. [Google Scholar] [CrossRef]
- Hsu, Y.H.; Chuang, H.C.; Lee, Y.H.; Lin, Y.F.; Chen, Y.J.; Hsiao, T.C.; Wu, M.Y.; Chiu, H.W. Traffic-related particulate matter exposure induces nephrotoxicity in vitro and in vivo. Free Radic. Biol. Med. 2019, 135, 235–244. [Google Scholar] [CrossRef]
- Zhou, T.; Hu, Y.; Wang, Y.; Sun, C.; Zhong, Y.; Liao, J.; Wang, G. Fine particulate matter (PM(2.5)) aggravates apoptosis of cigarette-inflamed bronchial epithelium in vivo and vitro. Environ. Pollut. 2019, 248, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Rothhammer, V.; Quintana, F.J. The aryl hydrocarbon receptor: An environmental sensor integrating immune responses in health and disease. Nat. Rev. Immunol. 2019, 19, 184–197. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Omaye, S.T. Air pollutants, oxidative stress and human health. Mutat. Res. 2009, 674, 45–54. [Google Scholar] [CrossRef]
- Chowdhury, P.H.; Okano, H.; Honda, A.; Kudou, H.; Kitamura, G.; Ito, S.; Ueda, K.; Takano, H. Aqueous and organic extract of PM(2.5) collected in different seasons and cities of Japan differently affect respiratory and immune systems. Environ. Pollut. 2018, 235, 223–234. [Google Scholar] [CrossRef]
- Thaneeya Chetiyanukornkul, A.T.; Hayakawa, K. Atmospheric Polycyclic Aromatic Hydrocarbons and Nitropolycyclic Aromatic Hydrocarbons in Thailand; Springer: Berlin/Heidelberg, Germany, 2018. [Google Scholar]
- Alshaarawy, O.; Zhu, M.; Ducatman, A.; Conway, B.; Andrew, M.E. Polycyclic aromatic hydrocarbon biomarkers and serum markers of inflammation. A positive association that is more evident in men. Environ. Res. 2013, 126, 98–104. [Google Scholar] [CrossRef]
- Jia, Y.Y.; Wang, Q.; Liu, T. Toxicity Research of PM(2.5) Compositions In Vitro. Int. J. Environ. Res. Public Health 2017, 14, 232. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Li, Z.; Liao, Z.; Gao, S.; Hua, L.; Ye, X.; Wang, Y.; Jiang, S.; Wang, N.; Zhou, D.; et al. PM(2.5) induced pulmonary fibrosis in vivo and in vitro. Ecotoxicol. Environ. Saf. 2019, 171, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Kawajiri, K.; Fujii-Kuriyama, Y. The aryl hydrocarbon receptor: A multifunctional chemical sensor for host defense and homeostatic maintenance. Exp. Anim. 2017, 66, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Xue, P.; Fu, J.; Zhou, Y. The Aryl Hydrocarbon Receptor and Tumor Immunity. Front. Immunol. 2018, 9, 286. [Google Scholar] [CrossRef] [PubMed]
- Takano, H.; Inoue, K.I. Environmental pollution and allergies. J. Toxicol. Pathol. 2017, 30, 193–199. [Google Scholar] [CrossRef]
- Memari, B.; Bouttier, M.; Dimitrov, V.; Ouellette, M.; Behr, M.A.; Fritz, J.H.; White, J.H. Engagement of the Aryl Hydrocarbon Receptor in Mycobacterium tuberculosis-Infected Macrophages Has Pleiotropic Effects on Innate Immune Signaling. J. Immunol. 2015, 195, 4479–4491. [Google Scholar] [CrossRef]
- Zhang, Y.; Salam, M.T.; Berhane, K.; Eckel, S.P.; Rappaport, E.B.; Linn, W.S.; Habre, R.; Bastain, T.M.; Gilliland, F.D. Genetic and epigenetic susceptibility of airway inflammation to PM(2.5) in school children: New insights from quantile regression. Environ. Health 2017, 16, 88. [Google Scholar] [CrossRef]
- Karavitis, J.; Kovacs, E.J. Macrophage phagocytosis: Effects of environmental pollutants, alcohol, cigarette smoke, and other external factors. J. Leukoc. Biol. 2011, 90, 1065–1078. [Google Scholar] [CrossRef]
- Soukup, J.M.; Becker, S. Human alveolar macrophage responses to air pollution particulates are associated with insoluble components of coarse material, including particulate endotoxin. Toxicol. Appl. Pharmacol. 2001, 171, 20–26. [Google Scholar] [CrossRef]
- González, Y.; Doens, D.; Cruz, H.; Santamaría, R.; Gutiérrez, M.; Llanes, A.; Fernández, P.L. A Marine Diterpenoid Modulates the Proteasome Activity in Murine Macrophages Stimulated with LPS. Biomolecules 2018, 8, 109. [Google Scholar] [CrossRef]
- Rosales, C.; Uribe-Querol, E. Phagocytosis: A Fundamental Process in Immunity. Biomed. Res. Int. 2017, 2017, 9042851. [Google Scholar] [CrossRef] [PubMed]
- Flannagan, R.S.; Jaumouillé, V.; Grinstein, S. The cell biology of phagocytosis. Annu. Rev. Pathol. 2012, 7, 61–98. [Google Scholar] [CrossRef] [PubMed]
- Oishi, Y.; Manabe, I. Macrophages in inflammation, repair and regeneration. Int. Immunol. 2018, 30, 511–528. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, N.; Kobayashi, K. Macrophages in inflammation. Curr. Drug Targets Inflamm. Allergy 2005, 4, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, S.Z.; Plevy, S.E. The role of the macrophage in sentinel responses in intestinal immunity. Curr. Opin. Gastroenterol. 2010, 26, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Franken, L.; Schiwon, M.; Kurts, C. Macrophages: Sentinels and regulators of the immune system. Cell Microbiol. 2016, 18, 475–487. [Google Scholar] [CrossRef]
- Gordon, S.; Plüddemann, A. Tissue macrophages: Heterogeneity and functions. BMC Biol. 2017, 15, 53. [Google Scholar] [CrossRef]
- Bolland, S.; Ravetch, J.V. Spontaneous autoimmune disease in Fc(gamma)RIIB-deficient mice results from strain-specific epistasis. Immunity 2000, 13, 277–285. [Google Scholar] [CrossRef]
- Chu, Z.T.; Tsuchiya, N.; Kyogoku, C.; Ohashi, J.; Qian, Y.P.; Xu, S.B.; Mao, C.Z.; Chu, J.Y.; Tokunaga, K. Association of Fcgamma receptor IIb polymorphism with susceptibility to systemic lupus erythematosus in Chinese: A common susceptibility gene in the Asian populations. Tissue Antigens 2004, 63, 21–27. [Google Scholar] [CrossRef]
- Clatworthy, M.R.; Willcocks, L.; Urban, B.; Langhorne, J.; Williams, T.N.; Peshu, N.; Watkins, N.A.; Floto, R.A.; Smith, K.G. Systemic lupus erythematosus-associated defects in the inhibitory receptor FcgammaRIIb reduce susceptibility to malaria. Proc. Natl. Acad. Sci. USA 2007, 104, 7169–7174. [Google Scholar] [CrossRef]
- Crispín, J.C.; Hedrich, C.M.; Tsokos, G.C. Gene-function studies in systemic lupus erythematosus. Nat. Rev. Rheumatol. 2013, 9, 476–484. [Google Scholar] [CrossRef] [PubMed]
- Surawut, S.; Ondee, T.; Taratummarat, S.; Palaga, T.; Pisitkun, P.; Chindamporn, A.; Leelahavanichkul, A. The role of macrophages in the susceptibility of Fc gamma receptor IIb deficient mice to Cryptococcus neoformans. Sci. Rep. 2017, 7, 40006. [Google Scholar] [CrossRef] [PubMed]
- Issara-Amphorn, J.; Surawut, S.; Worasilchai, N.; Thim-Uam, A.; Finkelman, M.; Chindamporn, A.; Palaga, T.; Hirankarn, N.; Pisitkun, P.; Leelahavanichkul, A. The Synergy of Endotoxin and (1→3)-β-D-Glucan, from Gut Translocation, Worsens Sepsis Severity in a Lupus Model of Fc Gamma Receptor IIb-Deficient Mice. J. Innate Immun. 2018, 10, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Surawut, S.; Makjaroen, J.; Thim-Uam, A.; Wongphoom, J.; Palaga, T.; Pisitkun, P.; Chindamporn, A.; Leelahavanichkul, A. Increased susceptibility against Cryptococcus neoformans of lupus mouse models (pristane-induction and FcGRIIb deficiency) is associated with activated macrophage, regardless of genetic background. J. Microbiol. 2019, 57, 45–53. [Google Scholar] [CrossRef]
- Anania, J.C.; Chenoweth, A.M.; Wines, B.D.; Hogarth, P.M. The Human FcγRII (CD32) Family of Leukocyte FcR in Health and Disease. Front. Immunol. 2019, 10, 464. [Google Scholar] [CrossRef]
- Maglione, P.J.; Xu, J.; Casadevall, A.; Chan, J. Fc gamma receptors regulate immune activation and susceptibility during Mycobacterium tuberculosis infection. J. Immunol. 2008, 180, 3329–3338. [Google Scholar] [CrossRef]
- Clatworthy, M.R.; Smith, K.G. FcgammaRIIb balances efficient pathogen clearance and the cytokine-mediated consequences of sepsis. J. Exp. Med. 2004, 199, 717–723. [Google Scholar] [CrossRef]
- Willcocks, L.C.; Carr, E.J.; Niederer, H.A.; Rayner, T.F.; Williams, T.N.; Yang, W.; Scott, J.A.; Urban, B.C.; Peshu, N.; Vyse, T.J.; et al. A defunctioning polymorphism in FCGR2B is associated with protection against malaria but susceptibility to systemic lupus erythematosus. Proc. Natl. Acad. Sci. USA 2010, 107, 7881–7885. [Google Scholar] [CrossRef]
- Kuechle, M.K.; Elkon, K.B. Shining light on lupus and UV. Arthritis Res. Ther. 2007, 9, 101. [Google Scholar] [CrossRef]
- Parks, C.G.; Cooper, G.S. Occupational exposures and risk of systemic lupus erythematosus. Autoimmunity 2005, 38, 497–506. [Google Scholar] [CrossRef]
- Parks, C.G.; de Souza Espindola Santos, A.; Barbhaiya, M.; Costenbader, K.H. Understanding the role of environmental factors in the development of systemic lupus erythematosus. Best Pract. Res. Clin. Rheumatol. 2017, 31, 306–320. [Google Scholar] [CrossRef]
- Ondee, T.; Surawut, S.; Taratummarat, S.; Hirankarn, N.; Palaga, T.; Pisitkun, P.; Pisitkun, T.; Leelahavanichkul, A. Fc Gamma Receptor IIB Deficient Mice: A Lupus Model with Increased Endotoxin Tolerance-Related Sepsis Susceptibility. Shock 2017, 47, 743–752. [Google Scholar] [CrossRef]
- Ondee, T.; Gillen, J.; Visitchanakun, P.; Somparn, P.; Issara-Amphorn, J.; Dang Phi, C.; Chancharoenthana, W.; Gurusamy, D.; Nita-Lazar, A.; Leelahavanichkul, A. Lipocalin-2 (Lcn-2) Attenuates Polymicrobial Sepsis with LPS Preconditioning (LPS Tolerance) in FcGRIIb Deficient Lupus Mice. Cells 2019, 8, 1064. [Google Scholar] [CrossRef]
- Ondee, T.; Jaroonwitchawan, T.; Pisitkun, T.; Gillen, J.; Nita-Lazar, A.; Leelahavanichkul, A.; Somparn, P. Decreased Protein Kinase C-β Type II Associated with the Prominent Endotoxin Exhaustion in the Macrophage of FcGRIIb−/− Lupus Prone Mice is Revealed by Phosphoproteomic Analysis. Int. J. Mol. Sci. 2019, 20, 1354. [Google Scholar] [CrossRef]
- Shi, L.; Zhang, Z.; Yu, A.M.; Wang, W.; Wei, Z.; Akhter, E.; Maurer, K.; Costa Reis, P.; Song, L.; Petri, M.; et al. The SLE transcriptome exhibits evidence of chronic endotoxin exposure and has widespread dysregulation of non-coding and coding RNAs. PLoS ONE 2014, 9, e93846. [Google Scholar] [CrossRef] [PubMed]
- Bhunyakarnjanarat, T.; Udompornpitak, K.; Saisorn, W.; Chantraprapawat, B.; Visitchanakun, P.; Dang, C.P.; Issara-Amphorn, J.; Leelahavanichkul, A. Prominent Indomethacin-Induced Enteropathy in Fcgriib Defi-cient lupus Mice: An Impact of Macrophage Responses and Immune Deposition in Gut. Int. J. Mol. Sci. 2021, 22, 1377. [Google Scholar] [CrossRef] [PubMed]
- Gulati, G.; Brunner, H.I. Environmental triggers in systemic lupus erythematosus. Semin. Arthritis Rheum. 2018, 47, 710–717. [Google Scholar] [CrossRef] [PubMed]
- Deng, G.M.; Tsokos, G.C. Cholera toxin B accelerates disease progression in lupus-prone mice by promoting lipid raft aggregation. J. Immunol. 2008, 181, 4019–4026. [Google Scholar] [CrossRef]
- Podolska, M.J.; Biermann, M.H.; Maueröder, C.; Hahn, J.; Herrmann, M. Inflammatory etiopathogenesis of systemic lupus erythematosus: An update. J. Inflamm. Res. 2015, 8, 161–171. [Google Scholar] [CrossRef]
- Thim-Uam, A.; Surawut, S.; Issara-Amphorn, J.; Jaroonwitchawan, T.; Hiengrach, P.; Chatthanathon, P.; Wilantho, A.; Somboonna, N.; Palaga, T.; Pisitkun, P.; et al. Leaky-gut enhanced lupus progression in the Fc gamma receptor-IIb deficient and pristane-induced mouse models of lupus. Sci. Rep. 2020, 10, 777. [Google Scholar] [CrossRef]
- Issara-Amphorn, J.; Chancharoenthana, W.; Visitchanakun, P.; Leelahavanichkul, A. Syk Inhibitor Attenuates Polymicrobial Sepsis in FcgRIIb-Deficient Lupus Mouse Model, the Impact of Lupus Characteristics in Sepsis. J. Innate Immun. 2020, 12, 461–479. [Google Scholar] [CrossRef]
- Knapp, A.C.; Huang, J.; Starling, G.; Kiener, P.A. Inhibitors of HMG-CoA reductase sensitize human smooth muscle cells to Fas-ligand and cytokine-induced cell death. Atherosclerosis 2000, 152, 217–227. [Google Scholar] [CrossRef]
- Bretz, J.D.; Mezosi, E.; Giordano, T.J.; Gauger, P.G.; Thompson, N.W.; Baker, J.R., Jr. Inflammatory cytokine regulation of TRAIL-mediated apoptosis in thyroid epithelial cells. Cell Death Differ. 2002, 9, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, J.; Kabashima, K.; Kawanishi, M.; Toda, K.; Miyachi, Y. Cytotoxicity of IFN-gamma and TNF-alpha for vascular endothelial cell is mediated by nitric oxide. Biochem. Biophys. Res. Commun. 2002, 291, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Almaani, S.; Meara, A.; Rovin, B.H. Update on Lupus Nephritis. Clin. J. Am. Soc. Nephrol. 2017, 12, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Amornphimoltham, P.; Yuen, P.S.T.; Star, R.A.; Leelahavanichkul, A. Gut Leakage of Fungal-Derived Inflammatory Mediators: Part of a Gut-Liver-Kidney Axis in Bacterial Sepsis. Dig. Dis. Sci. 2019, 64, 2416–2428. [Google Scholar] [CrossRef] [PubMed]
- Panpetch, W.; Somboonna, N.; Palasuk, M.; Hiengrach, P.; Finkelman, M.; Tumwasorn, S.; Leelahavanichkul, A. Oral Candida administration in a Clostridium difficile mouse model worsens disease severity but is attenuated by Bifidobacterium. PLoS ONE 2019, 14, e0210798. [Google Scholar] [CrossRef]
- Panpetch, W.; Chancharoenthana, W.; Bootdee, K.; Nilgate, S.; Finkelman, M.; Tumwasorn, S.; Leelahavanichkul, A. Lactobacillus rhamnosus L34 Attenuates Gut Translocation-Induced Bacterial Sepsis in Murine Models of Leaky Gut. Infect. Immun. 2018, 86. [Google Scholar] [CrossRef] [PubMed]
- Panpetch, W.; Kullapanich, C.; Dang, C.P.; Visitchanakun, P.; Saisorn, W.; Wongphoom, J.; Wannigama, D.L.; Thim-Uam, A.; Patarakul, K.; Somboonna, N.; et al. Candida Administration Worsens Uremia-Induced Gut Leakage in Bilateral Nephrectomy Mice, an Impact of Gut Fungi and Organismal Molecules in Uremia. mSystems 2021, 6. [Google Scholar] [CrossRef]
- Panpetch, W.; Sawaswong, V.; Chanchaem, P.; Ondee, T.; Dang, C.P.; Payungporn, S.; Tumwasorn, S.; Leelahavanichkul, A. Corrigendum: Candida Administration Worsens Cecal Ligation and Puncture-Induced Sepsis in Obese Mice Through Gut Dysbiosis Enhanced Systemic Inflammation, Impact of Pathogen-Associated Molecules From Gut Translocation and Saturated Fatty Acid. Front. Immunol. 2020, 11, 613095. [Google Scholar] [CrossRef]
- André, P.; Laugerette, F.; Féart, C. Metabolic Endotoxemia: A Potential Underlying Mechanism of the Relationship between Dietary Fat Intake and Risk for Cognitive Impairments in Humans? Nutrients 2019, 11, 1887. [Google Scholar] [CrossRef]
- Yeh, Y.J.; Law, L.Y.; Lim, C.L. Gastrointestinal response and endotoxemia during intense exercise in hot and cool environments. Eur. J. Appl. Physiol. 2013, 113, 1575–1583. [Google Scholar] [CrossRef] [PubMed]
- Pendyala, S.; Walker, J.M.; Holt, P.R. A high-fat diet is associated with endotoxemia that originates from the gut. Gastroenterology 2012, 142, 1100–1101.e1102. [Google Scholar] [CrossRef]
- Leelahavanichkul, A.; Worasilchai, N.; Wannalerdsakun, S.; Jutivorakool, K.; Somparn, P.; Issara-Amphorn, J.; Tachaboon, S.; Srisawat, N.; Finkelman, M.; Chindamporn, A. Gastrointestinal Leakage Detected by Serum (1→3)-β-D-Glucan in Mouse Models and a Pilot Study in Patients with Sepsis. Shock 2016, 46, 506–518. [Google Scholar] [CrossRef] [PubMed]
- Rittirsch, D.; Flierl, M.A.; Day, D.E.; Nadeau, B.A.; Zetoune, F.S.; Sarma, J.V.; Werner, C.M.; Wanner, G.A.; Simmen, H.P.; Huber-Lang, M.S.; et al. Cross-talk between TLR4 and FcgammaReceptorIII (CD16) pathways. PLoS Pathog. 2009, 5, e1000464. [Google Scholar] [CrossRef] [PubMed]
- Stewart, R.; Hammond, S.A.; Oberst, M.; Wilkinson, R.W. The role of Fc gamma receptors in the activity of immunomodulatory antibodies for cancer. J. Immunother. Cancer 2014, 2, 29. [Google Scholar] [CrossRef]
- Domínguez-Acosta, O.; Vega, L.; Estrada-Muñiz, E.; Rodríguez, M.S.; Gonzalez, F.J.; Elizondo, G. Activation of aryl hydrocarbon receptor regulates the LPS/IFNγ-induced inflammatory response by inducing ubiquitin-proteosomal and lysosomal degradation of RelA/p65. Biochem. Pharmacol. 2018, 155, 141–149. [Google Scholar] [CrossRef]
- Kimura, A.; Naka, T.; Nakahama, T.; Chinen, I.; Masuda, K.; Nohara, K.; Fujii-Kuriyama, Y.; Kishimoto, T. Aryl hydrocarbon receptor in combination with Stat1 regulates LPS-induced inflammatory responses. J. Exp. Med. 2009, 206, 2027–2035. [Google Scholar] [CrossRef] [PubMed]
- Masuda, K.; Kimura, A.; Hanieh, H.; Nguyen, N.T.; Nakahama, T.; Chinen, I.; Otoyo, Y.; Murotani, T.; Yamatodani, A.; Kishimoto, T. Aryl hydrocarbon receptor negatively regulates LPS-induced IL-6 production through suppression of histamine production in macrophages. Int. Immunol. 2011, 23, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.Q.; Mei, X.D.; Feng, D. Air pollution and chronic airway diseases: What should people know and do? J. Thorac. Dis. 2016, 8, E31–E40. [Google Scholar] [CrossRef] [PubMed]
- Kurt, O.K.; Zhang, J.; Pinkerton, K.E. Pulmonary health effects of air pollution. Curr. Opin. Pulm. Med. 2016, 22, 138–143. [Google Scholar] [CrossRef]
- Gao, N.; Xu, W.; Ji, J.; Yang, Y.; Wang, S.T.; Wang, J.; Chen, X.; Meng, S.; Tian, X.; Xu, K.F. Lung function and systemic inflammation associated with short-term air pollution exposure in chronic obstructive pulmonary disease patients in Beijing, China. Environ. Health 2020, 19, 12. [Google Scholar] [CrossRef]
- Dennehy, K.M.; Ferwerda, G.; Faro-Trindade, I.; Pyz, E.; Willment, J.A.; Taylor, P.R.; Kerrigan, A.; Tsoni, S.V.; Gordon, S.; Meyer-Wentrup, F.; et al. Syk kinase is required for collaborative cytokine production induced through Dectin-1 and Toll-like receptors. Eur. J. Immunol. 2008, 38, 500–506. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Ravetch, J.V. Fcgamma receptors: Old friends and new family members. Immunity 2006, 24, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Koenderman, L. Inside-Out Control of Fc-Receptors. Front. Immunol. 2019, 10, 544. [Google Scholar] [CrossRef]
- Issara-Amphorn, J.; Somboonna, N.; Pisitkun, P.; Hirankarn, N.; Leelahavanichkul, A. Syk inhibitor attenuates inflammation in lupus mice from FcgRIIb deficiency but not in pristane induction: The influence of lupus pathogenesis on the therapeutic effect. Lupus 2020, 29, 1248–1262. [Google Scholar] [CrossRef]
- Tsokos, G.C.; Lo, M.S.; Costa Reis, P.; Sullivan, K.E. New insights into the immunopathogenesis of systemic lupus erythematosus. Nat. Rev. Rheumatol. 2016, 12, 716–730. [Google Scholar] [CrossRef]
- Kado, S.; Chang, W.L.W.; Chi, A.N.; Wolny, M.; Shepherd, D.M.; Vogel, C.F.A. Aryl hydrocarbon receptor signaling modifies Toll-like receptor-regulated responses in human dendritic cells. Arch. Toxicol. 2017, 91, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Marinković, N.; Pasalić, D.; Potocki, S. Polymorphisms of genes involved in polycyclic aromatic hydrocarbons’ biotransformation and atherosclerosis. Biochem. Med. 2013, 23, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Pernomian, L.; da Silva, C. Current basis for discovery and development of aryl hydrocarbon receptor antagonists for experimental and therapeutic use in atherosclerosis. Eur. J. Pharmacol. 2015, 764, 118–123. [Google Scholar] [CrossRef]
- Cho, C.C.; Hsieh, W.Y.; Tsai, C.H.; Chen, C.Y.; Chang, H.F.; Lin, C.S. In Vitro and In Vivo Experimental Studies of PM(2.5) on Disease Progression. Int. J. Environ. Res. Public Health 2018, 15, 1380. [Google Scholar] [CrossRef]
- Jin, X.T.; Chen, M.L.; Li, R.J.; An, Q.; Song, L.; Zhao, Y.; Xiao, H.; Cheng, L.; Li, Z.Y. Progression and inflammation of human myeloid leukemia induced by ambient PM2.5 exposure. Arch. Toxicol. 2016, 90, 1929–1938. [Google Scholar] [CrossRef]
- Salim, S.Y.; Kaplan, G.G.; Madsen, K.L. Air pollution effects on the gut microbiota: A link between exposure and inflammatory disease. Gut Microbes 2014, 5, 215–219. [Google Scholar] [CrossRef]
- Wang, W.; Zhou, J.; Chen, M.; Huang, X.; Xie, X.; Li, W.; Cao, Q.; Kan, H.; Xu, Y.; Ying, Z. Exposure to concentrated ambient PM(2.5) alters the composition of gut microbiota in a murine model. Part Fibre Toxicol. 2018, 15, 17. [Google Scholar] [CrossRef] [PubMed]
- Eppensteiner, J.; Kwun, J.; Scheuermann, U.; Barbas, A.; Limkakeng, A.T.; Kuchibhatla, M.; Elster, E.A.; Kirk, A.D.; Lee, J. Damage- and pathogen-associated molecular patterns play differential roles in late mortality after critical illness. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Neavin, D.R.; Liu, D.; Ray, B.; Weinshilboum, R.M. The Role of the Aryl Hydrocarbon Receptor (AHR) in Immune and Inflammatory Diseases. Int. J. Mol. Sci. 2018, 19, 3851. [Google Scholar] [CrossRef] [PubMed]
- Shu, T.; Song, X.; Cui, X.; Ge, W.; Gao, R.; Wang, J. Fc Gamma Receptor IIb Expressed in Hepatocytes Promotes Lipid Accumulation and Gluconeogenesis. Int. J. Mol. Sci. 2018, 19, 2932. [Google Scholar] [CrossRef]
- Roghanian, A.; Stopforth, R.J.; Dahal, L.N.; Cragg, M.S. New revelations from an old receptor: Immunoregulatory functions of the inhibitory Fc gamma receptor, FcγRIIB (CD32B). J. Leukoc. Biol. 2018, 103, 1077–1088. [Google Scholar] [CrossRef] [PubMed]
- Tsao, T.M.; Tsai, M.J.; Hwang, J.S.; Cheng, W.F.; Wu, C.F.; Chou, C.K.; Su, T.C. Health effects of a forest environment on natural killer cells in humans: An observational pilot study. Oncotarget 2018, 9, 16501–16511. [Google Scholar] [CrossRef]
- Kreitinger, J.M.; Beamer, C.A.; Shepherd, D.M. Environmental Immunology: Lessons Learned from Exposure to a Select Panel of Immunotoxicants. J. Immunol. 2016, 196, 3217–3225. [Google Scholar] [CrossRef]
- Larigot, L.; Juricek, L.; Dairou, J.; Coumoul, X. AhR signaling pathways and regulatory functions. Biochim. Open 2018, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.; Aslamkhan, A.G.; Pearson, K.; Tanis, K.Q.; Podtelezhnikov, A.; Frank, E.; Pacchione, S.; Pippert, T.; Glaab, W.E.; Sistare, F.D. AhR Activation in Pharmaceutical Development: Applying Liver Gene Expression Biomarker Thresholds to Identify Doses Associated with Tumorigenic Risks in Rats. Toxicol. Sci. 2019, 171, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Mihara, M.; Tan, I.; Chuzhin, Y.; Reddy, B.; Budhai, L.; Holzer, A.; Gu, Y.; Davidson, A. CTLA4Ig inhibits T cell-dependent B-cell maturation in murine systemic lupus erythematosus. J. Clin. Investig. 2000, 106, 91–101. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Forward Primer | Reverse Primer |
|---|---|---|
| Arginase-1 (Arg-1) | 5′-CTTGGCTTGCTTCGGAACTC-3′ | 5′-GGAGAAGGCGTTTGCTTAGTT-3′ |
| Aryl hydrocarbon receptor (Ahr) | 5′-GACCACTTAGAGCACCACTA-3′ | 5′-AGAACTTCAATCAGACATACACAA-3′ |
| Fc gamma receptor I (FcgRI) | 5′-CACAAATGCCCTTAGACCAC-3′ | 5′-ACCCTAGAGTTCCAGGGATG-3′ |
| Fc gamma receptor IIb (FcgRIIb) | 5′-TTCTCAAGCATCCCGAAGCC-3′ | 5′-TTCCCAATGCCAAGGGAGAC-3′ |
| Fc gamma receptor III (FcgRIII) | 5′-AGGGCCTCCATCTGGACTG-3′ | 5′-GTGGTTCTGGTAATCATGCTCTG-3′ |
| Fc gamma receptor IV (FcgRIV) | 5′-AACGGCAAAGGCAAGAAGTA-3′ | 5′-CCGCACAGAGAAATACAGCA-3′ |
| Inducible nitric oxide synthase (iNOS) | 5′-ACCCACATCTGGCAGAATGAG-3′ | 5′-AGCCATGACCTTTCGCATTAG-3′ |
| Interleukin-1β (IL-1β) | 5′-GAAATGCCACCTTTTGACAGTG-3′ | 5′-TGGATGCTCTCATCAGGACAG-3′ |
| Interleukin-6 (IL-6) | 5′-TACCACTTCACAAGTCGGAGGC-3′ | 5′-CTGCAAGTGCATCATCGTTGTTC-3′ |
| Interleukin-10 (IL-10) | 5′-GCTCTTACTGACTGGCATGAG-3′ | 5′-CGCAGCTCTAGGAGCATGTG-3′ |
| Nuclear factor-κB (NF-κB RelA) | 5′-CTTCCTCAGCCATGGTACCTCT-3′ | 5′-CAAGTCTTCATCAGCATCAAACTG-3′ |
| Resistin-like molecule-α (FIZZ-1) | 5′-GCCAGGTCCTGGAACCTTTC-3′ | 5′-GGAGCAGGGAGATGCAGATGA-3′ |
| Toll like receptor 4 (TLR-4) | 5′-GGCAGCAGGTGGAATTGTAT-3′ | 5′-AGGCCCCAGAGTTTTGTTCT-3′ |
| Transforming Growth Factor-β (TGF-β) | 5′-CAGAGCTGCGCTTGCAGAG-3′ | 5′-GTCAGCAGCCGGTTACCAAG-3′ |
| Tumor necrosis factor α (TNF-α) | 5′-CCTCACACTCAGATCATCTTCTC-3′ | 5′-AGATCCATGCCGTTGGCCAG-3′ |
| β-actin | 5′-CGGTTCCGATGCCCTGAGGCTCTT-3′ | 5′-CGTCACACTTCATGATGGAATTGA-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Udompornpitak, K.; Bhunyakarnjanarat, T.; Charoensappakit, A.; Dang, C.P.; Saisorn, W.; Leelahavanichkul, A. Lipopolysaccharide-Enhanced Responses against Aryl Hydrocarbon Receptor in FcgRIIb-Deficient Macrophages, a Profound Impact of an Environmental Toxin on a Lupus-Like Mouse Model. Int. J. Mol. Sci. 2021, 22, 4199. https://doi.org/10.3390/ijms22084199
Udompornpitak K, Bhunyakarnjanarat T, Charoensappakit A, Dang CP, Saisorn W, Leelahavanichkul A. Lipopolysaccharide-Enhanced Responses against Aryl Hydrocarbon Receptor in FcgRIIb-Deficient Macrophages, a Profound Impact of an Environmental Toxin on a Lupus-Like Mouse Model. International Journal of Molecular Sciences. 2021; 22(8):4199. https://doi.org/10.3390/ijms22084199
Chicago/Turabian StyleUdompornpitak, Kanyarat, Thansita Bhunyakarnjanarat, Awirut Charoensappakit, Cong Phi Dang, Wilasinee Saisorn, and Asada Leelahavanichkul. 2021. "Lipopolysaccharide-Enhanced Responses against Aryl Hydrocarbon Receptor in FcgRIIb-Deficient Macrophages, a Profound Impact of an Environmental Toxin on a Lupus-Like Mouse Model" International Journal of Molecular Sciences 22, no. 8: 4199. https://doi.org/10.3390/ijms22084199
APA StyleUdompornpitak, K., Bhunyakarnjanarat, T., Charoensappakit, A., Dang, C. P., Saisorn, W., & Leelahavanichkul, A. (2021). Lipopolysaccharide-Enhanced Responses against Aryl Hydrocarbon Receptor in FcgRIIb-Deficient Macrophages, a Profound Impact of an Environmental Toxin on a Lupus-Like Mouse Model. International Journal of Molecular Sciences, 22(8), 4199. https://doi.org/10.3390/ijms22084199