
Reprogramming: Emerging Strategies to Rejuvenate Aging Cells and Tissues



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Understanding the Aging Process
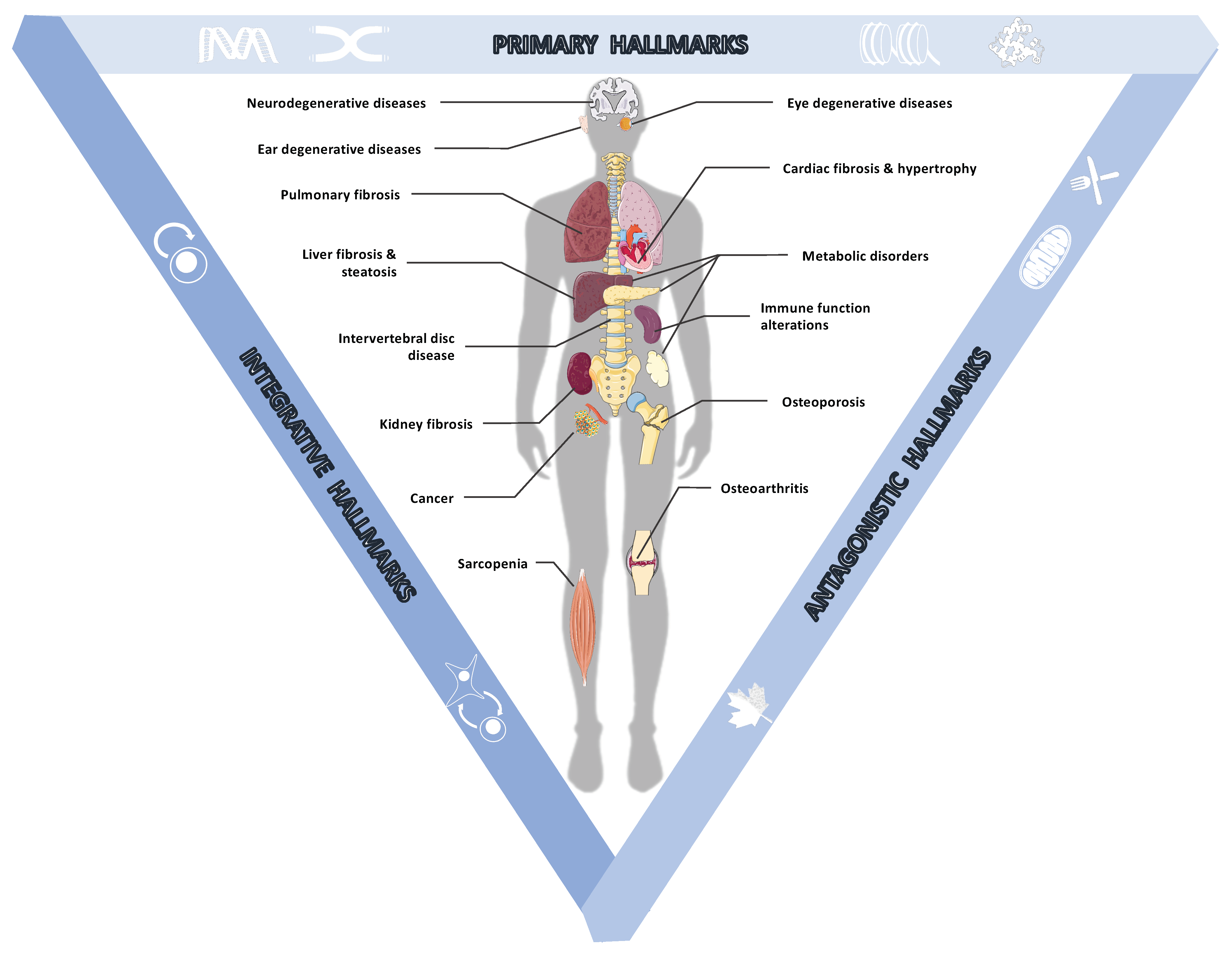
2.1. Age-Associated Pathologies
2.2. Cellular Damage at the Heart of Aging
2.2.1. The Primary Hallmarks of Aging Are the Triggering Events Whose Harmful Consequences Progressively Accumulate over Time
2.2.2. The Antagonistic Hallmarks Are Damage Response Mechanisms That Become Overwhelmed
2.2.3. The Integrative Hallmarks Are Tissue Homeostasis Failures
3. The Promise of Pluripotent Stem Cells
3.1. Human Embryonic Stem Cells
3.2. Cell Reprogramming
3.3. Human Pluripotent Cells as an Experimental Modelling Tool
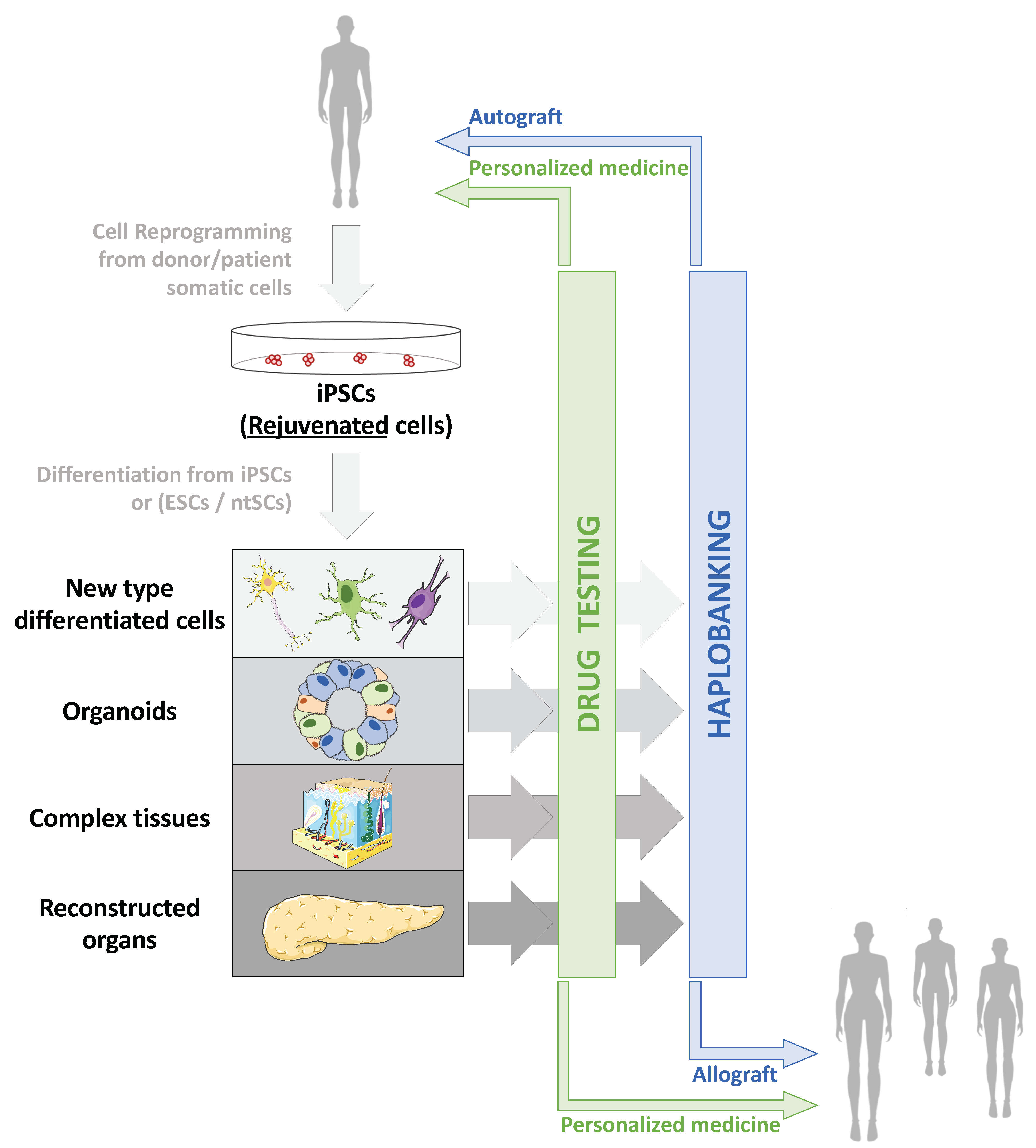
3.3.1. Organoids and Complex Tissues
3.3.2. Modelling Age-Related Pathologies In Vitro
3.3.3. New Models for the Screening of Therapeutic Molecules
4. New Strategies in Regenerative Medicine to Rejuvenate Cells and Tissues
4.1. Clinical Applications of Human Pluripotent Stem Cells
4.1.1. Production of hPSCs for Clinical Use
4.1.2. Cell and Tissue Replacement Therapies
4.1.3. Organ Production in In Vivo Models
4.2. Organismal Rejuvenation through Cellular Reprogramming
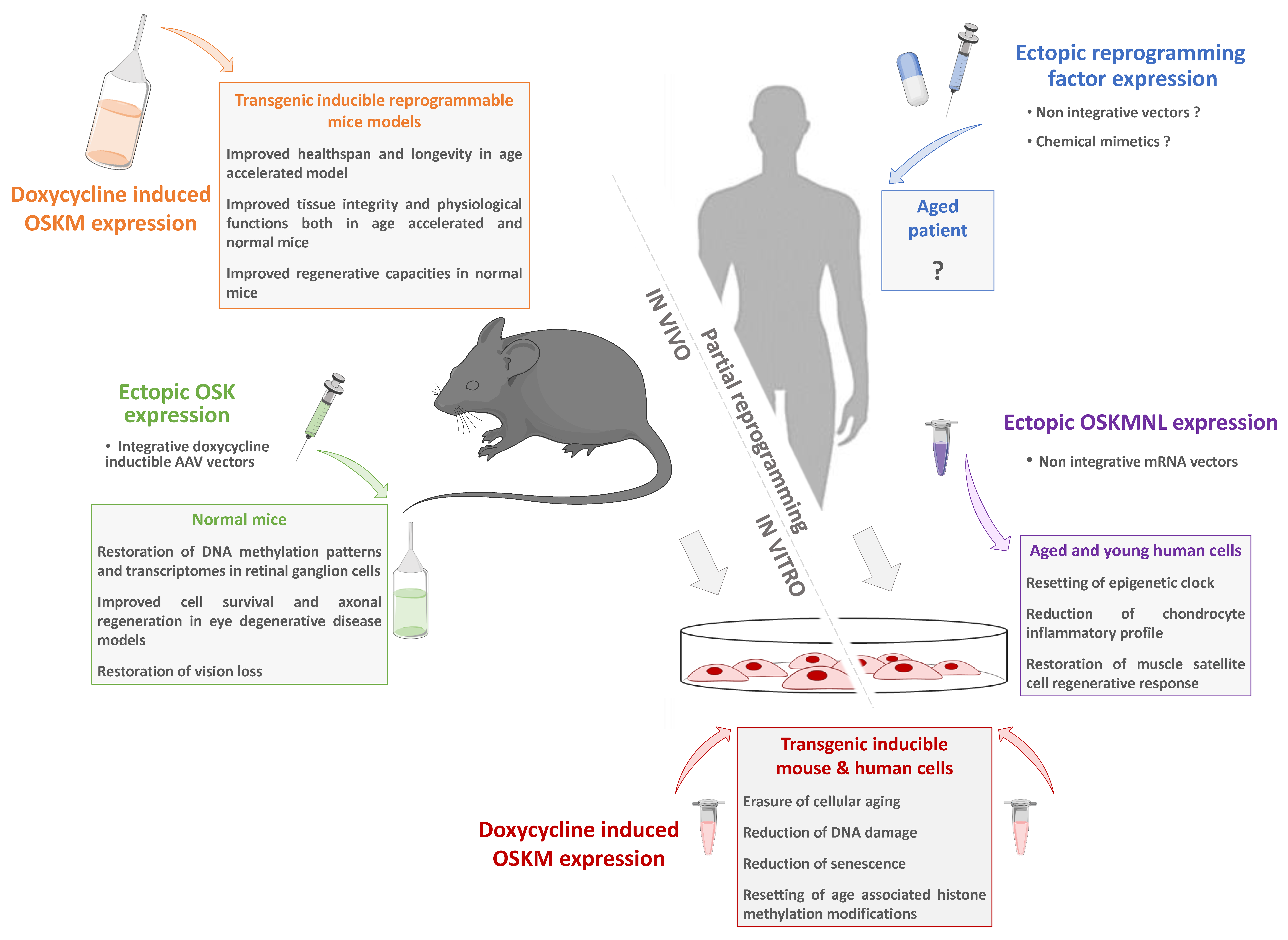
4.2.1. Aging and Senescence, Two Obstacles to Reprogramming
4.2.2. Cellular Reprogramming to Erase Cell Aging
4.2.3. Complete Cellular Reprogramming Causes Teratomas
4.2.4. Partial Cellular Reprogramming Rejuvenates Cells In Vitro and In Vivo
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef]
- Cai, Y.; Zhou, H.; Zhu, Y.; Sun, Q.; Ji, Y.; Xue, A.; Wang, Y.; Chen, W.; Yu, X.; Wang, L.; et al. Elimination of senescent cells by β-galactosidase-targeted prodrug attenuates inflammation and restores physical function in aged mice. Cell Res. 2020, 30, 574–589. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Seals, D.R.; Justice, J.N.; LaRocca, T.J. Physiological geroscience: Targeting function to increase healthspan and achieve optimal longevity. J. Physiol. 2016, 594, 2001–2024. [Google Scholar] [CrossRef]
- Eurostat, Measuring Progress towards a More Sustainable Europe. Sustainable Development Indicators for the European Union. Data 1990–2005; Office for Official Publications of the European Communities: Luxembourg, 2005. [Google Scholar]
- Pison, G. Pourquoi l’espérance de vie augmente-t-elle moins vite en France? Popul. Sociétés 2019, 564, 1. [Google Scholar] [CrossRef]
- DeSantis, C.E.; Miller, K.D.; Dale, W.; Mohile, S.G.; Cohen, H.J.; Leach, C.R.; Goding Sauer, A.; Jemal, A.; Siegel, R.L. Cancer statistics for adults aged 85 years and older. CA Cancer J. Clin. 2019, 69, 452–467. [Google Scholar] [CrossRef]
- Hazlehurst, J.M.; Woods, C.; Marjot, T.; Cobbold, J.F.; Tomlinson, J.W. Non-alcoholic fatty liver disease and diabetes. Metabolism 2016, 65, 1096–1108. [Google Scholar] [CrossRef]
- Birbrair, A.; Zhang, T.; Files, D.C.; Mannava, S.; Smith, T.; Wang, Z.M.; Messi, M.L.; Mintz, A.; Delbono, O. Type-1 pericytes accumulate after tissue injury and produce collagen in an organ-dependent manner. Stem Cell Res. Ther. 2014, 5, 122. [Google Scholar] [CrossRef]
- Espindola, M.S.; Habiel, D.M.; Narayanan, R.; Jones, I.; Coelho, A.L.; Murray, L.A.; Jiang, D.; Noble, P.W.; Hogaboam, C.M. Targeting of TAM Receptors Ameliorates Fibrotic Mechanisms in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2018, 197, 1443–1456. [Google Scholar] [CrossRef]
- Nastase, M.V.; Zeng-Brouwers, J.; Wygrecka, M.; Schaefer, L. Targeting renal fibrosis: Mechanisms and drug delivery systems. Adv. Drug. Deliv. Rev. 2018, 129, 295–307. [Google Scholar] [CrossRef]
- Lemmer, A.; VanWagner, L.B.; Ganger, D. Assessment of Advanced Liver Fibrosis and the Risk for Hepatic Decompensation in Patients With Congestive Hepatopathy. Hepatology 2018, 68, 1633–1641. [Google Scholar] [CrossRef]
- Li, L.; Zhao, Q.; Kong, W. Extracellular matrix remodeling and cardiac fibrosis. Matrix Biol. J. Int. Soc. Matrix Biol. 2018, 68–69, 490–506. [Google Scholar] [CrossRef]
- Palacio, L.; Goyer, M.L.; Maggiorani, D.; Espinosa, A.; Villeneuve, N.; Bourbonnais, S.; Moquin-Beaudry, G.; Le, O.; Demaria, M.; Davalos, A.R.; et al. Restored immune cell functions upon clearance of senescence in the irradiated splenic environment. Aging Cell 2019, 18, e12971. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef]
- Bowl, M.R.; Dawson, S.J. Age-Related Hearing Loss. Cold Spring Harb. Perspect Med. 2019, 9, a033217. [Google Scholar] [CrossRef]
- Pelletier, A.L.; Rojas-Roldan, L.; Coffin, J. Vision Loss in Older Adults. Am. Fam. Physician 2016, 94, 219–226. [Google Scholar]
- Hayflick, L. Biological Aging Is No Longer an Unsolved Problem. Ann. N. Y. Acad. Sci. 2007, 1100, 1–13. [Google Scholar] [CrossRef]
- Faggioli, F.; Wang, T.; Vijg, J.; Montagna, C. Chromosome-specific accumulation of aneuploidy in the aging mouse brain. Hum. Mol. Genet. 2012, 21, 5246–5253. [Google Scholar] [CrossRef]
- Forsberg, L.A.; Rasi, C.; Razzaghian, H.R.; Pakalapati, G.; Waite, L.; Thilbeault, K.S.; Ronowicz, A.; Wineinger, N.E.; Tiwari, H.K.; Boomsma, D.; et al. Age-related somatic structural changes in the nuclear genome of human blood cells. Am. J. Hum. Genet. 2012, 90, 217–228. [Google Scholar] [CrossRef]
- Park, C.B.; Larsson, N.-G. Mitochondrial DNA mutations in disease and aging. J. Cell Biol. 2011, 193, 809–818. [Google Scholar] [CrossRef]
- Rossi, D.J.; Bryder, D.; Seita, J.; Nussenzweig, A.; Hoeijmakers, J.; Weissman, I.L. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature 2007, 447, 725–729. [Google Scholar] [CrossRef]
- Martínez, P.; Blasco, M.A. Telomere-driven diseases and telomere-targeting therapies. J. Cell Biol. 2017, 216, 875–887. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, L.; Wang, Z.; Liu, J.-P. Roles of Telomere Biology in Cell Senescence, Replicative and Chronological Ageing. Cells 2019, 8, 54. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Liu, X.; Ding, X.; Wang, F.; Geng, X. Telomere and its role in the aging pathways: Telomere shortening, cell senescence and mitochondria dysfunction. Biogerontology 2019, 20, 1–16. [Google Scholar] [CrossRef]
- Field, A.E.; Robertson, N.A.; Wang, T.; Havas, A.; Ideker, T.; Adams, P.D. DNA Methylation Clocks in Aging: Categories, Causes, and Consequences. Mol. Cell 2018, 71, 882–895. [Google Scholar] [CrossRef]
- Jones, M.J.; Goodman, S.J.; Kobor, M.S. DNA methylation and healthy human aging. Aging Cell 2015, 14, 924–932. [Google Scholar] [CrossRef]
- Sati, S.; Bonev, B.; Szabo, Q.; Jost, D.; Bensadoun, P.; Serra, F.; Loubiere, V.; Papadopoulos, G.L.; Rivera-Mulia, J.-C.; Fritsch, L.; et al. 4D Genome Rewiring during Oncogene-Induced and Replicative Senescence. Mol. Cell 2020, 78, 522–538.e9. [Google Scholar] [CrossRef]
- Labbadia, J.; Morimoto, R.I. The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 2015, 84, 435–464. [Google Scholar] [CrossRef]
- Laskowska, E.; Kuczyńska-Wiśnik, D.; Lipińska, B. Proteomic analysis of protein homeostasis and aggregation. J. Proteom. 2019, 198, 98–112. [Google Scholar] [CrossRef] [PubMed]
- Klaips, C.L.; Jayaraj, G.G.; Hartl, F.U. Pathways of cellular proteostasis in aging and disease. J. Cell Biol. 2018, 217, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Leidal, A.M.; Levine, B.; Debnath, J. Autophagy and the cell biology of age-related disease. Nat. Cell Biol. 2018, 20, 1338–1348. [Google Scholar] [CrossRef] [PubMed]
- Tomaru, U.; Takahashi, S.; Ishizu, A.; Miyatake, Y.; Gohda, A.; Suzuki, S.; Ono, A.; Ohara, J.; Baba, T.; Murata, S.; et al. Decreased proteasomal activity causes age-related phenotypes and promotes the development of metabolic abnormalities. Am. J. Pathol. 2012, 180, 963–972. [Google Scholar] [CrossRef]
- Abdellatif, M.; Sedej, S.; Carmona-Gutierrez, D.; Madeo, F.; Kroemer, G. Autophagy in Cardiovascular Aging. Circ. Res. 2018, 123, 803–824. [Google Scholar] [CrossRef]
- Borghesan, M.; Hoogaars, W.M.H.; Varela-Eirin, M.; Talma, N.; Demaria, M. A Senescence-Centric View of Aging: Implications for Longevity and Disease. Trends Cell Biol. 2020, 10, 777–791. [Google Scholar] [CrossRef]
- Kujoth, G.C.; Bradshaw, P.C.; Haroon, S.; Prolla, T.A. The role of mitochondrial DNA mutations in mammalian aging. PLoS Genet. 2007, 3, e24. [Google Scholar] [CrossRef]
- Trifunovic, A.; Wredenberg, A.; Falkenberg, M.; Spelbrink, J.N.; Rovio, A.T.; Bruder, C.E.; Bohlooly, Y.M.; Gidlof, S.; Oldfors, A.; Wibom, R.; et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 2004, 429, 417–423. [Google Scholar] [CrossRef]
- Vermulst, M.; Wanagat, J.; Kujoth, G.C.; Bielas, J.H.; Rabinovitch, P.S.; Prolla, T.A.; Loeb, L.A. DNA deletions and clonal mutations drive premature aging in mitochondrial mutator mice. Nat. Genet. 2008, 40, 392–394. [Google Scholar] [CrossRef]
- López-Otín, C.; Galluzzi, L.; Freije, J.M.P.; Madeo, F.; Kroemer, G. Metabolic Control of Longevity. Cell 2016, 166, 802–821. [Google Scholar] [CrossRef]
- Chen, D.; Kerr, C. The Epigenetics of Stem Cell Aging Comes of Age. Trends Cell Biol. 2019, 29, 563–568. [Google Scholar] [CrossRef]
- Ren, R.; Ocampo, A.; Liu, G.-H.; Izpisua Belmonte, J.C. Regulation of Stem Cell Aging by Metabolism and Epigenetics. Cell Metab. 2017, 26, 460–474. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, E.L.; Dixit, V.D. Drivers of age-related inflammation and strategies for healthspan extension. Immunol. Rev. 2015, 265, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. Ser. A Biol. Sci. Med Sci. 2014, 69 (Suppl. 1), S4–S9. [Google Scholar] [CrossRef]
- Aw, D.; Silva, A.B.; Palmer, D.B. Immunosenescence: Emerging challenges for an ageing population. Immunology 2007, 120, 435–446. [Google Scholar] [CrossRef]
- Crooke, S.N.; Ovsyannikova, I.G.; Poland, G.A.; Kennedy, R.B. Immunosenescence and human vaccine immune responses. Immun. Ageing I A 2019, 16, 25. [Google Scholar] [CrossRef]
- Fedintsev, A.; Moskalev, A. Stochastic non-enzymatic modification of long-lived macromolecules—A missing hallmark of aging. Ageing Res. Rev. 2020, 62, 101097. [Google Scholar] [CrossRef]
- Yuan, Y.; Li, J.; He, Z.; Fan, X.; Mao, X.; Yang, M.; Yang, D. tRNA-derived fragments as New Hallmarks of Aging and Age-related Diseases. Aging Dis. 2021. [Google Scholar] [CrossRef]
- Knupp, D.; Miura, P. CircRNA accumulation: A new hallmark of aging? Mech. Ageing Dev. 2018, 173, 71–79. [Google Scholar] [CrossRef]
- Nagpal, R.; Mainali, R.; Ahmadi, S.; Wang, S.; Singh, R.; Kavanagh, K.; Kitzman, D.W.; Kushugulova, A.; Marotta, F.; Yadav, H. Gut microbiome and aging: Physiological and mechanistic insights. Nutr. Healthy Aging 2018, 4, 267–285. [Google Scholar] [CrossRef]
- Conboy, M.J.; Conboy, I.M.; Rando, T.A. Heterochronic parabiosis: Historical perspective and methodological considerations for studies of aging and longevity. Aging Cell 2013, 12, 525–530. [Google Scholar] [CrossRef]
- Villeda, S.A.; Luo, J.; Mosher, K.I.; Zou, B.; Britschgi, M.; Bieri, G.; Stan, T.M.; Fainberg, N.; Ding, Z.; Eggel, A.; et al. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature 2011, 477, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Carlson, M.E.; Hsu, M.; Conboy, I.M. Imbalance between pSmad3 and Notch induces CDK inhibitors in old muscle stem cells. Nature 2008, 454, 528–532. [Google Scholar] [CrossRef] [PubMed]
- Sinha, M.; Jang, Y.C.; Oh, J.; Khong, D.; Wu, E.Y.; Manohar, R.; Miller, C.; Regalado, S.G.; Loffredo, F.S.; Pancoast, J.R.; et al. Restoring systemic GDF11 levels reverses age-related dysfunction in mouse skeletal muscle. Science 2014, 344, 649–652. [Google Scholar] [CrossRef] [PubMed]
- Elabd, C.; Cousin, W.; Upadhyayula, P.; Chen, R.Y.; Chooljian, M.S.; Li, J.; Kung, S.; Jiang, K.P.; Conboy, I.M. Oxytocin is an age-specific circulating hormone that is necessary for muscle maintenance and regeneration. Nat. Commun. 2014, 5, 1–11. [Google Scholar] [CrossRef]
- Egerman, M.A.; Cadena, S.M.; Gilbert, J.A.; Meyer, A.; Nelson, H.N.; Swalley, S.E.; Mallozzi, C.; Jacobi, C.; Jennings, L.L.; Clay, I.; et al. GDF11 Increases with Age and Inhibits Skeletal Muscle Regeneration. Cell Metab. 2015, 22, 164–174. [Google Scholar] [CrossRef]
- Castellano, J.M.; Mosher, K.I.; Abbey, R.J.; McBride, A.A.; James, M.L.; Berdnik, D.; Shen, J.C.; Zou, B.; Xie, X.S.; Tingle, M.; et al. Human umbilical cord plasma proteins revitalize hippocampal function in aged mice. Nat. Cell Biol. 2017, 544, 488–492. [Google Scholar] [CrossRef]
- Mao, A.S.; Mooney, D.J. Regenerative Medicine: Current Therapies and Future Directions. Proc. Natl. Acad. Sci. USA 2015, 112, 14452–14459. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.J.; Kaufman, M.H. Establishment in culture of pluripotential cells from mouse embryos. Nat. Cell Biol. 1981, 292, 154–156. [Google Scholar] [CrossRef]
- Martin, G.R. Isolation of a Pluripotent Cell Line from Early Mouse Embryos Cultured in Medium Conditioned by Teratocarcinoma Stem Cells. Proc. Natl. Acad. Sci. USA 1981, 78, 7634–7638. [Google Scholar] [CrossRef]
- Thomson, J.A.; Kalishman, J.; Golos, T.G.; Durning, M.; Harris, C.P.; Becker, R.A.; Hearn, J.P. Isolation of a Primate Embryonic Stem Cell Line. Proc. Natl. Acad. Sci. USA 1995, 92, 7844–7848. [Google Scholar] [CrossRef]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic Stem Cell Lines Derived from Human Blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [PubMed]
- Eguizabal, C.; Aran, B.; Lopes, S.M.C.D.S.; Geens, M.; Heindryckx, B.; Panula, S.; Popovic, M.; Vassena, R.; Veiga, A. Two decades of embryonic stem cells: A historical overview. Hum. Rep. Open 2019, 2019, hoy024. [Google Scholar] [CrossRef]
- Colman, A. Profile of John Gurdon and Shinya Yamanaka, 2012 Nobel Laureates in Medicine or Physiology. Proc. Natl. Acad. Sci. USA 2013, 110, 5740–5741. [Google Scholar] [CrossRef]
- Gurdon, J.B.; Elsdale, T.R.; Fischberg, M. Sexually Mature Individuals of Xenopus laevis from the Transplantation of Single Somatic Nuclei. Nat. Cell Biol. 1958, 182, 64–65. [Google Scholar] [CrossRef] [PubMed]
- Gurdon, J. Adult frogs derived from the nuclei of single somatic cells. Dev. Biol. 1962, 4, 256–273. [Google Scholar] [CrossRef]
- Halley-Stott, R.P.; Pasque, V.; Gurdon, J.B. Nuclear reprogramming. Development 2013, 140, 2468–2471. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. A decade of transcription factor-mediated reprogramming to pluripotency. Nat. Rev. Mol. Cell Biol. 2016, 17, 183–193. [Google Scholar] [CrossRef]
- Yamanaka, S. Induced Pluripotent Stem Cells: Past, Present, and Future. Cell Stem Cell 2012, 10, 678–684. [Google Scholar] [CrossRef]
- Yamanaka, S.; Blau, H.M. Nuclear reprogramming to a pluripotent state by three approaches. Nat. Cell Biol. 2010, 465, 704–712. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Avilion, A.A.; Nicolis, S.K.; Pevny, L.H.; Perez, L.; Vivian, N.; Lovell-Badge, R. Multipotent cell lineages in early mouse development depend on SOX2 function. Genes Dev. 2003, 17, 126–140. [Google Scholar] [CrossRef] [PubMed]
- Nichols, J.; Zevnik, B.; Anastassiadis, K.; Niwa, H.; Klewe-Nebenius, D.; Chambers, I.; Schöler, H.; Smith, A. Formation of Pluripotent Stem Cells in the Mammalian Embryo Depends on the POU Transcription Factor. Cell 1998, 95, 379–391. [Google Scholar] [CrossRef]
- Niwa, H.; Miyazaki, J.-I.; Smith, A.G. Quantitative expression of Oct-3/4 defines differentiation, dedifferentiation or self-renewal of ES cells. Nat. Genet. 2000, 24, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Cartwright, P.; McLean, C.; Sheppard, A.; Rivett, D.; Jones, K.; Dalton, S. LIF/STAT3 controls ES cell self-renewal and pluripotency by a Myc-dependent mechanism. Development 2005, 132, 885–896. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; McClintick, J.; Zhong, L.; Edenberg, H.J.; Yoder, M.C.; Chan, R.J. Murine embryonic stem cell differentiation is promoted by SOCS-3 and inhibited by the zinc finger transcription factor. Blood 2005, 105, 635–637. [Google Scholar] [CrossRef]
- Rauch, C.; Feifel, E.; Kern, G.; Murphy, C.; Meier, F.; Parson, W.; Beilmann, M.; Jennings, P.; Gstraunthaler, G.; Wilmes, A. Differentiation of human iPSCs into functional podocytes. PLoS ONE 2018, 13, e0203869. [Google Scholar] [CrossRef]
- Song, B.; Smink, A.M.; Jones, C.V.; Callaghan, J.M.; Firth, S.D.; Bernard, C.A.; Laslett, A.L.; Kerr, P.G.; Ricardo, S.D. The Directed Differentiation of Human iPS Cells into Kidney Podocytes. PLoS ONE 2012, 7, e46453. [Google Scholar] [CrossRef]
- Fernandopulle, M.S.; Prestil, R.; Grunseich, C.; Wang, C.; Gan, L.; Ward, M.E. Transcription Factor-Mediated Differentiation of Human iPSCs into Neurons. Curr. Protoc. Cell Biol. 2018, 79, e51. [Google Scholar] [CrossRef] [PubMed]
- Yoder, M.C. Differentiation of pluripotent stem cells into endothelial cells. Curr. Opin. Hematol. 2015, 22, 252–257. [Google Scholar] [CrossRef]
- Acimovic, I.; Vilotic, A.; Pesl, M.; Lacampagne, A.; Dvorak, P.; Rotrekl, V.; Meli, A.C. Human Pluripotent Stem Cell-Derived Cardiomyocytes as Research and Therapeutic Tools. BioMed Res. Int. 2014, 2014, 1–14. [Google Scholar] [CrossRef]
- Deng, Y.I.; Verron, E.; Rohanizadeh, R. Molecular Mechanisms of Anti-metastatic Activity of Curcumin. Anticancer Res. 2016, 36, 5639–5648. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Xiao, L.; Zhan, X.; Zhou, H. Pdxl and its role in activating Ngn3 and Pax6 to induce differentiation of iPSCs into islet β cells. Genet. Mol. Res. 2015, 14, 8892–8900. [Google Scholar] [CrossRef]
- Mianné, J.; Ahmed, E.; Bourguignon, C.; Fieldes, M.; Vachier, I.; Bourdin, A.; Assou, S.; De Vos, J. Induced Pluripotent Stem Cells for Primary Ciliary Dyskinesia Modeling and Personalized Medicine. Am. J. Respir. Cell Mol. Biol. 2018, 59, 672–683. [Google Scholar] [CrossRef] [PubMed]
- Akkerman, N.; Defize, L.H. Dawn of the organoid era. BioEssays 2017, 39, 1600244. [Google Scholar] [CrossRef]
- Huch, M.; Knoblich, J.A.; Lutolf, M.P.; Martinez-Arias, A. The hope and the hype of organoid research. Development 2017, 144, 938–941. [Google Scholar] [CrossRef]
- Rossi, G.; Manfrin, A.; Lutolf, M.P. Progress and potential in organoid research. Nat. Rev. Genet. 2018, 19, 671–687. [Google Scholar] [CrossRef]
- Brazovskaja, A.; Treutlein, B.; Camp, J.G. High-throughput single-cell transcriptomics on organoids. Curr. Opin. Biotechnol. 2019, 55, 167–171. [Google Scholar] [CrossRef]
- Di Lullo, E.; Kriegstein, A.R. The use of brain organoids to investigate neural development and disease. Nat. Rev. Neurosci. 2017, 18, 573–584. [Google Scholar] [CrossRef]
- Heide, M.; Huttner, W.B.; Mora-Bermúdez, F. Brain organoids as models to study human neocortex development and evolution. Curr. Opin. Cell Biol. 2018, 55, 8–16. [Google Scholar] [CrossRef]
- Seto, Y.; Eiraku, M. Human brain development and its in vitro recapitulation. Neurosci. Res. 2019, 138, 33–42. [Google Scholar] [CrossRef]
- Serra, D.; Mayr, U.; Boni, A.; Lukonin, I.; Rempfler, M.; Meylan, L.C.; Stadler, M.B.; Strnad, P.; Papasaikas, P.; Vischi, D.; et al. Self-organization and symmetry breaking in intestinal organoid development. Nat. Cell Biol. 2019, 569, 66–72. [Google Scholar] [CrossRef]
- Yin, Y.-B.; De Jonge, H.R.; Wu, X.; Yin, Y.-L. Mini-gut: A promising model for drug development. Drug Discov. Today 2019, 24, 1784–1794. [Google Scholar] [CrossRef]
- Nishinakamura, R. Human kidney organoids: Progress and remaining challenges. Nat. Rev. Nephrol. 2019, 15, 613–624. [Google Scholar] [CrossRef]
- Hulot, J.-S. Modeling Cardiac Arrhythmias with Organoids. J. Am. Coll. Cardiol. 2019, 73, 2325–2327. [Google Scholar] [CrossRef]
- Nugraha, B.; Buono, M.F.; Von Boehmer, L.; Hoerstrup, S.P.; Emmert, M.Y. Human Cardiac Organoids for Disease Modeling. Clin. Pharmacol. Ther. 2019, 105, 79–85. [Google Scholar] [CrossRef]
- Pesl, M.; Acimovic, I.; Pribyl, J.; Hezova, R.; Vilotic, A.; Fauconnier, J.; Vrbsky, J.; Kruzliak, P.; Skladal, P.; Kara, T.; et al. Forced aggregation and defined factors allow highly uniform-sized embryoid bodies and functional cardiomyocytes from human embryonic and induced pluripotent stem cells. Heart Vessel. 2013, 29, 834–846. [Google Scholar] [CrossRef]
- Brooks, M.J.; Chen, H.Y.; Kelley, R.A.; Mondal, A.K.; Nagashima, K.; De Val, N.; Li, T.; Chaitankar, V.; Swaroop, A. Improved Retinal Organoid Differentiation by Modulating Signaling Pathways Revealed by Comparative Transcriptome Analyses with Development In Vivo. Stem Cell Rep. 2019, 13, 891–905. [Google Scholar] [CrossRef]
- Lee, J.-S.; Kim, B.S.; Seo, D.; Park, J.H.; Cho, D.-W. Three-Dimensional Cell Printing of Large-Volume Tissues: Application to Ear Regeneration. Tissue Eng. Part C Methods 2017, 23, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.-K.; Pang, Y.; Zhou, Z.-Z.; Yao, R.; Sun, W. An integrated cell printing system for the construction of heterogeneous tissue models. Acta Biomater. 2019, 95, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Chou, C.-K.; Xia, X.; Hung, M.-C.; Qin, L. Block-Cell-Printing for Live Single-Cell Printing. Proc. Natl. Acad. Sci. USA 2014, 111, 2948–2953. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced Pluripotent Stem Cell Lines Derived from Human Somatic Cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- Acimovic, I.; Refaat, M.M.; Moreau, A.; Salykin, A.; Reiken, S.; Sleiman, Y.; Souidi, M.; Přibyl, J.; Kajava, A.V.; Richard, S.; et al. Post-Translational Modifications and Diastolic Calcium Leak Associated to the Novel RyR2-D3638A Mutation Lead to CPVT in Patient-Specific hiPSC-Derived Cardiomyocytes. J. Clin. Med. 2018, 7, 423. [Google Scholar] [CrossRef] [PubMed]
- Jelinkova, S.; Vilotic, A.; Pribyl, J.; Aimond, F.; Salykin, A.; Acimovic, I.; Pesl, M.; Caluori, G.; Klimovic, S.; Urban, T.; et al. DMD Pluripotent Stem Cell Derived Cardiac Cells Recapitulate in vitro Human Cardiac Pathophysiology. Front. Bioeng. Biotechnol. 2020, 8, 535. [Google Scholar] [CrossRef] [PubMed]
- Park, I.-H.; Arora, N.; Huo, H.; Maherali, N.; Ahfeldt, T.; Shimamura, A.; Lensch, M.W.; Cowan, C.; Hochedlinger, K.; Daley, G.Q. Disease-Specific Induced Pluripotent Stem Cells. Cell 2008, 134, 877–886. [Google Scholar] [CrossRef] [PubMed]
- Gatinois, V.; Desprat, R.; Becker, F.; Pichard, L.; Bernex, F.; Corsini, C.; Pellestor, F.; Lemaitre, J.-M. Reprogramming of Human Peripheral Blood Mononuclear Cell (PBMC) from a patient suffering of a Werner syndrome resulting in iPSC line (REGUi003-A) maintaining a short telomere length. Stem Cell Res. 2019, 39, 101515. [Google Scholar] [CrossRef] [PubMed]
- Gatinois, V.; Desprat, R.; Becker, F.; Pichard, L.; Bernex, F.; Isidor, B.; Pellestor, F.; Lemaitre, J.-M. iPSC line derived from a Bloom syndrome patient retains an increased disease-specific sister-chromatid exchange activity. Stem Cell Res. 2020, 43, 101696. [Google Scholar] [CrossRef]
- Zhang, J.; Lian, Q.; Zhu, G.; Zhou, F.; Sui, L.; Tan, C.; Mutalif, R.A.; Navasankari, R.; Zhang, Y.; Tse, H.-F.; et al. A Human iPSC Model of Hutchinson Gilford Progeria Reveals Vascular Smooth Muscle and Mesenchymal Stem Cell Defects. Cell Stem Cell 2011, 8, 31–45. [Google Scholar] [CrossRef]
- Liu, G.-H.; Barkho, B.Z.; Ruiz, S.Z.; Diep, D.; Qu, J.; Yang, S.-L.; Panopoulos, A.D.; Suzuki, K.; Kurian, L.; Walsh, C.T.; et al. Recapitulation of premature ageing with iPSCs from Hutchinson–Gilford progeria syndrome. Nat. Cell Biol. 2011, 472, 221–225. [Google Scholar] [CrossRef]
- Batista, L.F.Z.; Pech, M.F.; Zhong, F.L.; Nguyen, H.N.; Xie, K.T.; Zaug, A.J.; Crary, S.M.; Choi, J.; Sebastiano, V.; Cherry, A.M.; et al. Telomere shortening and loss of self-renewal in dyskeratosis congenita induced pluripotent stem cells. Nat. Cell Biol. 2011, 474, 399–402. [Google Scholar] [CrossRef]
- Agarwal, S.; Loh, Y.-H.; McLoughlin, E.M.; Huang, J.; Park, I.-H.; Miller, J.D.; Huo, H.; Okuka, M.; Dos Reis, R.M.; Loewer, S.; et al. Telomere elongation in induced pluripotent stem cells from dyskeratosis congenita patients. Nat. Cell Biol. 2010, 464, 292–296. [Google Scholar] [CrossRef]
- Cheung, H.-H.; Liu, X.; Canterel-Thouennon, L.; Li, L.; Edmonson, C.; Rennert, O.M. Telomerase Protects Werner Syndrome Lineage-Specific Stem Cells from Premature Aging. Stem Cell Rep. 2014, 2, 534–546. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Li, J.; Suzuki, K.; Qu, J.; Wang, P.; Zhou, J.; Liu, X.; Ren, R.; Xu, X.; Ocampo, A.; et al. A Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging. Science 2015, 348, 1160–1163. [Google Scholar] [CrossRef]
- Jung, C.B.; Moretti, A.; Schnitzler, M.M.Y.; Iop, L.; Storch, U.; Bellin, M.; Dorn, T.; Ruppenthal, S.; Pfeiffer, S.; Goedel, A.; et al. Dantrolene rescues arrhythmogenic RYR2 defect in a patient-specific stem cell model of catecholaminergic polymorphic ventricular tachycardia. EMBO Mol. Med. 2012, 4, 180–191. [Google Scholar] [CrossRef]
- Sasaki, K.; Makiyama, T.; Yoshida, Y.; Wuriyanghai, Y.; Kamakura, T.; Nishiuchi, S.; Hayano, M.; Harita, T.; Yamamoto, Y.; Kohjitani, H.; et al. Patient-Specific Human Induced Pluripotent Stem Cell Model Assessed with Electrical Pacing Validates S107 as a Potential Therapeutic Agent for Catecholaminergic Polymorphic Ventricular Tachycardia. PLoS ONE 2016, 11, e0164795. [Google Scholar] [CrossRef]
- Del Álamo, J.C.; Lemons, D.; Serrano, R.; Savchenko, A.; Cerignoli, F.; Bodmer, R.; Mercola, M. High throughput physiological screening of iPSC-derived cardiomyocytes for drug development. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2016, 1863, 1717–1727. [Google Scholar] [CrossRef]
- Grimm, F.A.; Iwata, Y.; Sirenko, O.; Bittner, M.; Rusyn, I. High-Content Assay Multiplexing for Toxicity Screening in Induced Pluripotent Stem Cell-Derived Cardiomyocytes and Hepatocytes. Assay Drug Dev. Technol. 2015, 13, 529–546. [Google Scholar] [CrossRef]
- Pesl, M.; Pribyl, J.; Caluori, G.; Cmiel, V.; Acimovic, I.; Jelinkova, S.; Dvorak, P.; Starek, Z.; Skladal, P.; Rotrekl, V. Phenotypic assays for analyses of pluripotent stem cell-derived cardiomyocytes. J. Mol. Recognit. 2017, 30, e2602. [Google Scholar] [CrossRef]
- Sharma, A.; McKeithan, W.L.; Serrano, R.; Kitani, T.; Burridge, P.W.; Del Álamo, J.C.; Mercola, M.; Wu, J.C. Use of human induced pluripotent stem cell–derived cardiomyocytes to assess drug cardiotoxicity. Nat. Protoc. 2018, 13, 3018–3041. [Google Scholar] [CrossRef]
- Sharma, A.; Burridge, P.W.; McKeithan, W.L.; Serrano, R.; Shukla, P.; Sayed, N.; Churko, J.M.; Kitani, T.; Wu, H.; Holmström, A.; et al. High-throughput screening of tyrosine kinase inhibitor cardiotoxicity with human induced pluripotent stem cells. Sci. Transl. Med. 2017, 9, eaaf2584. [Google Scholar] [CrossRef]
- Paik, D.T.; Chandy, M.; Wu, J.C. Patient and Disease–Specific Induced Pluripotent Stem Cells for Discovery of Personalized Cardiovascular Drugs and Therapeutics. Pharmacol. Rev. 2020, 72, 320–342. [Google Scholar] [CrossRef]
- Perkhofer, L.; Frappart, P.-O.; Müller, M.; Kleger, A. Importance of organoids for personalized medicine. Pers. Med. 2018, 15, 461–465. [Google Scholar] [CrossRef]
- Takahashi, T. Organoids for Drug Discovery and Personalized Medicine. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 447–462. [Google Scholar] [CrossRef]
- Yokouchi, Y.; Suzuki, S.; Ohtsuki, N.; Yamamoto, K.; Noguchi, S.; Soejima, Y.; Goto, M.; Ishioka, K.; Nakamura, I.; Suzuki, S.; et al. Rapid repair of human disease-specific single-nucleotide variants by One-SHOT genome editing. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef]
- Beghini, D.G.; Horita, S.I.; Cascabulho, C.M.; Alves, L.A.; Henriques-Pons, A. Induced Pluripotent Stem Cells: Hope in the Treatment of Diseases, including Muscular Dystrophies. Int. J. Mol. Sci. 2020, 21, 5467. [Google Scholar] [CrossRef] [PubMed]
- Li, H.L.; Fujimoto, N.; Sasakawa, N.; Shirai, S.; Ohkame, T.; Sakuma, T.; Tanaka, M.; Amano, N.; Watanabe, A.; Sakurai, H.; et al. Precise Correction of the Dystrophin Gene in Duchenne Muscular Dystrophy Patient Induced Pluripotent Stem Cells by TALEN and CRISPR-Cas. Stem Cell Rep. 2015, 4, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Long, C.; Li, H.; Tiburcy, M.; Rodriguez-Caycedo, C.; Kyrychenko, V.; Zhou, H.; Zhang, Y.; Min, Y.-L.; Shelton, J.M.; Mammen, P.P.A.; et al. Correction of diverse muscular dystrophy mutations in human engineered heart muscle by single-site genome editing. Sci. Adv. 2018, 4, eaap9004. [Google Scholar] [CrossRef] [PubMed]
- Lapasset, L.; Milhavet, O.; Prieur, A.; Besnard, E.; Babled, A.; Aït-Hamou, N.; Leschik, J.; Pellestor, F.; Ramirez, J.-M.; De Vos, J.; et al. Rejuvenating senescent and centenarian human cells by reprogramming through the pluripotent state. Genes Dev. 2011, 25, 2248–2253. [Google Scholar] [CrossRef]
- Okita, K.; Ichisaka, T.; Yamanaka, S. Generation of germline-competent induced pluripotent stem cells. Nat. Cell Biol. 2007, 448, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Rebuzzini, P.; Zuccotti, M.; Redi, C.A.; Garagna, S. Achilles’ heel of pluripotent stem cells: Genetic, genomic and epigenetic variations during prolonged culture. Cell. Mol. Life Sci. 2016, 73, 2453–2466. [Google Scholar] [CrossRef] [PubMed]
- Tosca, L.; Féraud, O.; Magniez, A.; Bas, C.; Griscelli, F.; Bennaceur-Griscelli, A.; Tachdjian, G. Genomic instability of human embryonic stem cell lines using different passaging culture methods. Mol. Cytogenet. 2015, 8, 1–11. [Google Scholar] [CrossRef]
- Wang, J.; Hao, J.; Bai, D.; Gu, Q.; Han, W.; Wang, L.; Tan, Y.; Li, X.; Xue, K.; Han, P.; et al. Generation of clinical-grade human induced pluripotent stem cells in Xeno-free conditions. Stem Cell Res. Ther. 2015, 6, 223. [Google Scholar] [CrossRef] [PubMed]
- Mallon, B.S.; Park, K.-Y.; Chen, K.G.; Hamilton, R.S.; McKay, R.D.G. Toward xeno-free culture of human embryonic stem cells. Int. J. Biochem. Cell Biol. 2006, 38, 1063–1075. [Google Scholar] [CrossRef] [PubMed]
- Gagliano, O.; Luni, C.; Qin, W.; Bertin, E.; Torchio, E.; Galvanin, S.; Urciuolo, A.; Elvassore, N. Microfluidic reprogramming to pluripotency of human somatic cells. Nat. Protoc. 2019, 14, 722–737. [Google Scholar] [CrossRef] [PubMed]
- Luni, C.; Giulitti, S.; Serena, E.; Ferrari, L.; Zambon, A.; Gagliano, O.; Giobbe, G.G.; Michielin, F.; Knöbel, S.; Bosio, A.; et al. High-efficiency cellular reprogramming with microfluidics. Nat. Chem. Biol. 2016, 13, 446–452. [Google Scholar] [CrossRef]
- Barry, J.; Hyllner, J.; Stacey, G.; Taylor, C.J.; Turner, M. Setting Up a Haplobank: Issues and Solutions. Curr. Stem Cell Rep. 2015, 1, 110–117. [Google Scholar] [CrossRef]
- Gourraud, P.-A.; Gilson, L.; Girard, M.; Peschanski, M. The Role of Human Leukocyte Antigen Matching in the Development of Multiethnic “Haplobank” of Induced Pluripotent Stem Cell Lines. Stem Cells 2012, 30, 180–186. [Google Scholar] [CrossRef]
- Lee, S.; Huh, J.Y.; Turner, D.M.; Lee, S.; Robinson, J.; Stein, J.E.; Shim, S.H.; Hong, C.P.; Kang, M.S.; Nakagawa, M.; et al. Repurposing the Cord Blood Bank for Haplobanking of HLA-Homozygous iPSCs and Their Usefulness to Multiple Populations. Stem Cells 2018, 36, 1552–1566. [Google Scholar] [CrossRef]
- Deuse, T.; Hu, X.; Gravina, A.; Wang, D.; Tediashvili, G.; De, C.; Thayer, W.O.; Wahl, A.; Garcia, J.V.; Reichenspurner, H.; et al. Hypoimmunogenic derivatives of induced pluripotent stem cells evade immune rejection in fully immunocompetent allogeneic recipients. Nat. Biotechnol. 2019, 37, 252–258. [Google Scholar] [CrossRef]
- Jones, B.S.; Lamb, L.S.; Egoldman, F.; Stasi, A.E. Improving the safety of cell therapy products by suicide gene transfer. Front. Pharmacol. 2014, 5, 254. [Google Scholar] [CrossRef]
- Liang, Q.; Monetti, C.; Shutova, M.V.; Neely, E.J.; Hacibekiroglu, S.; Yang, H.; Kim, C.; Zhang, P.; Li, C.; Nagy, K.; et al. Linking a cell-division gene and a suicide gene to define and improve cell therapy safety. Nat. Cell Biol. 2018, 563, 701–704. [Google Scholar] [CrossRef]
- Yamanaka, S. Pluripotent Stem Cell-Based Cell Therapy—Promise and Challenges. Cell Stem Cell 2020, 27, 523–531. [Google Scholar] [CrossRef]
- Iwanami, A.; Kaneko, S.; Nakamura, M.; Kanemura, Y.; Mori, H.; Kobayashi, S.; Yamasaki, M.; Momoshima, S.; Ishii, H.; Ando, K.; et al. Transplantation of human neural stem cells for spinal cord injury in primates. J. Neurosci. Res. 2005, 80, 182–190. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Okada, Y.; Itakura, G.; Iwai, H.; Nishimura, S.; Yasuda, A.; Nori, S.; Hikishima, K.; Konomi, T.; Fujiyoshi, K.; et al. Pre-Evaluated Safe Human iPSC-Derived Neural Stem Cells Promote Functional Recovery after Spinal Cord Injury in Common Marmoset without Tumorigenicity. PLoS ONE 2012, 7, e52787. [Google Scholar] [CrossRef]
- Nori, S.; Okada, Y.; Yasuda, A.; Tsuji, O.; Takahashi, Y.; Kobayashi, Y.; Fujiyoshi, K.; Koike, M.; Uchiyama, Y.; Ikeda, E.; et al. Grafted human-induced pluripotent stem-cell-derived neurospheres promote motor functional recovery after spinal cord injury in mice. Proc. Natl. Acad. Sci. USA 2011, 108, 16825–16830. [Google Scholar] [CrossRef]
- Chen, W.; Jongkamonwiwat, N.; Abbas, L.; Eshtan, S.J.; Johnson, S.L.; Kuhn, S.; Milo, M.; Thurlow, J.K.; Andrews, P.W.; Marcotti, W.; et al. Restoration of auditory evoked responses by human ES-cell-derived otic progenitors. Nat. Cell Biol. 2012, 490, 278–282. [Google Scholar] [CrossRef]
- Duncan, T.; Valenzuela, M. Alzheimer’s disease, dementia, and stem cell therapy. Stem Cell Res. Ther. 2017, 8, 1–9. [Google Scholar] [CrossRef]
- González, C.; Bonilla, S.; Flores, A.I.; Cano, E.; Liste, I. An Update on Human Stem Cell-Based Therapy in Parkinson’;s Disease. Curr. Stem Cell Res. Ther. 2016, 11, 561–568. [Google Scholar] [CrossRef]
- Sonntag, K.-C.; Song, B.; Lee, N.; Jung, J.H.; Cha, Y.; Leblanc, P.; Neff, C.; Kong, S.W.; Carter, B.S.; Schweitzer, J.; et al. Pluripotent stem cell-based therapy for Parkinson’s disease: Current status and future prospects. Prog. Neurobiol. 2018, 168, 1–20. [Google Scholar] [CrossRef]
- Hallett, P.J.; Deleidi, M.; Astradsson, A.; Smith, G.A.; Cooper, O.; Osborn, T.M.; Sundberg, M.; Moore, M.A.; Perez-Torres, E.; Brownell, A.-L.; et al. Successful Function of Autologous iPSC-Derived Dopamine Neurons following Transplantation in a Non-Human Primate Model of Parkinson’s Disease. Cell Stem Cell 2015, 16, 269–274. [Google Scholar] [CrossRef]
- Ben M’Barek, K.; Monville, C. Cell Therapy for Retinal Dystrophies: From Cell Suspension Formulation to Complex Retinal Tissue Bioengineering. Stem Cells Int. 2019, 2019, 1–14. [Google Scholar] [CrossRef]
- Morizur, L.; Herardot, E.; Monville, C.; Ben M’Barek, K.; Elise, H.; Christelle, M.; Karim, B.M. Human pluripotent stem cells: A toolbox to understand and treat retinal degeneration. Mol. Cell. Neurosci. 2020, 107, 103523. [Google Scholar] [CrossRef]
- Mandai, M.; Watanabe, A.; Kurimoto, Y.; Hirami, Y.; Morinaga, C.; Daimon, T.; Fujihara, M.; Akimaru, H.; Sakai, N.; Shibata, Y.; et al. Autologous Induced Stem-Cell–Derived Retinal Cells for Macular Degeneration. N. Engl. J. Med. 2017, 376, 1038–1046. [Google Scholar] [CrossRef]
- Suchy, F.; Nakauchi, H. Interspecies chimeras. Curr. Opin. Genet. Dev. 2018, 52, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Suchy, F.; Yamaguchi, T.; Nakauchi, H. iPSC-Derived Organs In Vivo: Challenges and Promise. Cell Stem Cell 2018, 22, 21–24. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Yamaguchi, T.; Hamanaka, S.; Kato-Itoh, M.; Yamazaki, Y.; Ibata, M.; Sato, H.; Lee, Y.-S.; Usui, J.-I.; Knisely, A.; et al. Generation of Rat Pancreas in Mouse by Interspecific Blastocyst Injection of Pluripotent Stem Cells. Cell 2010, 142, 787–799. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Sato, H.; Kato-Itoh, M.; Goto, T.; Hara, H.; Sanbo, M.; Mizuno, N.; Kobayashi, T.; Yanagida, A.; Umino, A.; et al. Interspecies organogenesis generates autologous functional islets. Nat. Cell Biol. 2017, 542, 191–196. [Google Scholar] [CrossRef]
- Usui, J.-I.; Kobayashi, T.; Yamaguchi, T.; Knisely, A.; Nishinakamura, R.; Nakauchi, H. Generation of Kidney from Pluripotent Stem Cells via Blastocyst Complementation. Am. J. Pathol. 2012, 180, 2417–2426. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Platero-Luengo, A.; Sakurai, M.; Sugawara, A.; Gil, M.A.; Yamauchi, T.; Suzuki, K.; Bogliotti, Y.S.; Cuello, C.; Valencia, M.M.; et al. Interspecies Chimerism with Mammalian Pluripotent Stem Cells. Cell 2017, 168, 473–486.e15. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, T.; Suchy, F.P.; Bhadury, J.; Igarashi, K.J.; Charlesworth, C.T.; Nakauchi, H. Generation of Functional Organs Using a Cell-Competitive Niche in Intra- and Inter-species Rodent Chimeras. Cell Stem Cell 2021, 28, 141–149.e3. [Google Scholar] [CrossRef] [PubMed]
- Brons, I.G.M.; Smithers, L.E.; Trotter, M.W.B.; Rugg-Gunn, P.; Sun, B.; Lopes, S.M.C.D.S.; Howlett, S.K.; Clarkson, A.; Ahrlund-Richter, L.; Pedersen, R.A.; et al. Derivation of pluripotent epiblast stem cells from mammalian embryos. Nat. Cell Biol. 2007, 448, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Tesar, P.J.; Chenoweth, J.G.; Brook, F.A.; Davies, T.J.; Evans, E.P.; Mack, D.L.; Gardner, R.L.; McKay, R.D.G. New cell lines from mouse epiblast share defining features with human embryonic stem cells. Nat. Cell Biol. 2007, 448, 196–199. [Google Scholar] [CrossRef] [PubMed]
- Angeles, A.D.L.; Ferrari, F.; Xi, R.; Fujiwara, Y.; Benvenisty, N.; Deng, H.; Hochedlinger, K.; Jaenisch, R.; Lee, S.; Leitch, H.G.; et al. Hallmarks of pluripotency. Nat. Cell Biol. 2015, 525, 469–478. [Google Scholar] [CrossRef]
- Hu, Z.; Li, H.; Jiang, H.; Ren, Y.; Yu, X.; Qiu, J.; Stablewski, A.B.; Zhang, B.; Buck, M.J.; Feng, J. Transient inhibition of mTOR in human pluripotent stem cells enables robust formation of mouse-human chimeric embryos. Sci. Adv. 2020, 6, eaaz0298. [Google Scholar] [CrossRef]
- Kilens, S.; Meistermann, D.; Moreno, D.; Chariau, C.; Gaignerie, A.; Reignier, A.; Lelièvre, Y.; Casanova, M.; Vallot, C.; Nedellec, S.; et al. Parallel derivation of isogenic human primed and naive induced pluripotent stem cells. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Theunissen, T.W.; Friedli, M.; He, Y.; Planet, E.; O’Neil, R.C.; Markoulaki, S.; Pontis, J.; Wang, H.; Iouranova, A.; Imbeault, M.; et al. Molecular Criteria for Defining the Naive Human Pluripotent State. Cell Stem Cell 2016, 19, 502–515. [Google Scholar] [CrossRef] [PubMed]
- Banito, A.; Rashid, S.T.; Acosta, J.C.; Li, S.; Pereira, C.F.; Geti, I.; Pinho, S.; Silva, J.C.; Azuara, V.; Walsh, M.; et al. Senescence impairs successful reprogramming to pluripotent stem cells. Genes Dev. 2009, 23, 2134–2139. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Collado, M.; Villasante, A.; Strati, K.; Ortega, S.; Cañamero, M.; Blasco, M.A.; Serrano, M. The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nat. Cell Biol. 2009, 460, 1136–1139. [Google Scholar] [CrossRef]
- Marión, R.M.; Strati, K.; Li, H.; Murga, M.; Blanco, R.; Ortega, S.; Fernandez-Capetillo, O.; Serrano, M.; Blasco, M.A. A p53-mediated DNA damage response limits reprogramming to ensure iPS cell genomic integrity. Nat. Cell Biol. 2009, 460, 1149–1153. [Google Scholar] [CrossRef]
- Hayashi, Y.; Hsiao, E.C.; Sami, S.; Lancero, M.; Schlieve, C.R.; Nguyen, T.; Yano, K.; Nagahashi, A.; Ikeya, M.; Matsumoto, Y.; et al. BMP-SMAD-ID promotes reprogramming to pluripotency by inhibiting p16/INK4A-dependent senescence. Proc. Natl. Acad. Sci. USA 2016, 113, 13057–13062. [Google Scholar] [CrossRef]
- Hong, H.; Takahashi, K.; Ichisaka, T.; Aoi, T.; Kanagawa, O.; Nakagawa, M.; Okita, K.; Yamanaka, S. Suppression of induced pluripotent stem cell generation by the p53–p21 pathway. Nat. Cell Biol. 2009, 460, 1132–1135. [Google Scholar] [CrossRef] [PubMed]
- Utikal, J.; Polo, J.M.; Stadtfeld, M.; Maherali, N.; Kulalert, W.; Walsh, R.M.; Khalil, A.; Rheinwald, J.G.; Hochedlinger, K. Immortalization eliminates a roadblock during cellular reprogramming into iPS cells. Nat. Cell Biol. 2009, 460, 1145–1148. [Google Scholar] [CrossRef]
- Kawamura, T.; Suzuki, J.; Wang, Y.V.; Menendez, S.; Morera, L.B.; Raya, A.; Wahl, G.M.; Belmonte, J.C.I. Linking the p53 tumour suppressor pathway to somatic cell reprogramming. Nat. Cell Biol. 2009, 460, 1140–1144. [Google Scholar] [CrossRef]
- Mahmoudi, S.; Mancini, E.; Xu, L.; Moore, A.; Jahanbani, F.; Hebestreit, K.; Srinivasan, R.; Li, X.; Devarajan, K.; Prélot, L.; et al. Heterogeneity in old fibroblasts is linked to variability in reprogramming and wound healing. Nat. Cell Biol. 2019, 574, 553–558. [Google Scholar] [CrossRef]
- Marion, R.M.; Strati, K.; Li, H.; Tejera, A.; Schoeftner, S.; Ortega, S.; Serrano, M.; Blasco, M.A. Telomeres Acquire Embryonic Stem Cell Characteristics in Induced Pluripotent Stem Cells. Cell Stem Cell 2009, 4, 141–154. [Google Scholar] [CrossRef]
- Suhr, S.T.; Chang, E.A.; Tjong, J.; Alcasid, N.; Perkins, G.A.; Goissis, M.D.; Ellisman, M.H.; Perez, G.I.; Cibelli, J.B. Mitochondrial Rejuvenation After Induced Pluripotency. PLoS ONE 2010, 5, e14095. [Google Scholar] [CrossRef]
- Abad, M.; Mosteiro, L.; Pantoja, C.; Cañamero, M.; Rayon, T.; Ors, I.; Graña, O.; Megías, D.; Domínguez, O.; Martínez, D.; et al. Reprogramming in vivo produces teratomas and iPS cells with totipotency features. Nat. Cell Biol. 2013, 502, 340–345. [Google Scholar] [CrossRef]
- Ohnishi, K.; Semi, K.; Yamamoto, T.; Shimizu, M.; Tanaka, A.; Mitsunaga, K.; Okita, K.; Osafune, K.; Arioka, Y.; Maeda, T.; et al. Premature Termination of Reprogramming In Vivo Leads to Cancer Development through Altered Epigenetic Regulation. Cell 2014, 156, 663–677. [Google Scholar] [CrossRef] [PubMed]
- Mosteiro, L.; Pantoja, C.; Alcazar, N.; Marión, R.M.; Chondronasiou, D.; Rovira, M.; Fernandez-Marcos, P.J.; Muñoz-Martin, M.; Blanco-Aparicio, C.; Pastor, J.; et al. Tissue damage and senescence provide critical signals for cellular reprogramming in vivo. Science 2016, 354, aaf4445. [Google Scholar] [CrossRef]
- Chiche, A.; Le Roux, I.; von Joest, M.; Sakai, H.; Aguín, S.B.; Cazin, C.; Salam, R.; Fiette, L.; Alegria, O.; Flamant, P.; et al. Injury-Induced Senescence Enables In Vivo Reprogramming in Skeletal Muscle. Cell Stem Cell 2017, 20, 407–414.e4. [Google Scholar] [CrossRef] [PubMed]
- Ocampo, A.; Reddy, P.; Martinez-Redondo, P.; Platero-Luengo, A.; Hatanaka, F.; Hishida, T.; Li, M.; Lam, D.; Kurita, M.; Beyret, E.; et al. In Vivo Amelioration of Age-Associated Hallmarks by Partial Reprogramming. Cell 2016, 167, 1719–1733.e12. [Google Scholar] [CrossRef]
- Carey, B.W.; Markoulaki, S.; Beard, C.; Hanna, J.; Jaenisch, R. Single-gene transgenic mouse strains for reprogramming adult somatic cells. Nat. Methods 2009, 7, 56–59. [Google Scholar] [CrossRef]
- Osorio, F.G.; Navarro, C.L.; Cadiñanos, J.; López-Mejía, I.C.; Quirós, P.M.; Bartoli, C.; Rivera, J.; Tazi, J.; Guzmán, G.; Varela, I.; et al. Splicing-Directed Therapy in a New Mouse Model of Human Accelerated Aging. Sci. Transl. Med. 2011, 3, 106ra107. [Google Scholar] [CrossRef]
- Doeser, M.C.; Schöler, H.R.; Wu, G. Reduction of Fibrosis and Scar Formation by Partial Reprogramming In Vivo. Stem Cells 2018, 36, 1216–1225. [Google Scholar] [CrossRef]
- Rodríguez-Matellán, A.; Alcazar, N.; Hernández, F.; Serrano, M.; Ávila, J. In Vivo Reprogramming Ameliorates Aging Features in Dentate Gyrus Cells and Improves Memory in Mice. Stem Cell Rep. 2020, 15, 1056–1066. [Google Scholar] [CrossRef]
- Sarkar, T.J.; Quarta, M.; Mukherjee, S.; Colville, A.; Paine, P.; Doan, L.; Tran, C.M.; Chu, C.R.; Horvath, S.; Qi, L.S.; et al. Transient non-integrative expression of nuclear reprogramming factors promotes multifaceted amelioration of aging in human cells. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Senís, E.; Mosteiro, L.; Wilkening, S.; Wiedtke, E.; Nowrouzi, A.; Afzal, S.; Fronza, R.; Landerer, H.; Abad, M.; Niopek, D.; et al. AAV vector-mediated in vivo reprogramming into pluripotency. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Brommer, B.; Tian, X.; Krishnan, A.; Meer, M.; Wang, C.; Vera, D.L.; Zeng, Q.; Yu, D.; Bonkowski, M.S.; et al. Reprogramming to recover youthful epigenetic information and restore vision. Nat. Cell Biol. 2020, 588, 124–129. [Google Scholar] [CrossRef]
- Olova, N.; Simpson, D.J.; Marioni, R.E.; Chandra, T. Partial reprogramming induces a steady decline in epigenetic age before loss of somatic identity. Aging Cell 2018, 18, e12877. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alle, Q.; Le Borgne, E.; Milhavet, O.; Lemaitre, J.-M. Reprogramming: Emerging Strategies to Rejuvenate Aging Cells and Tissues. Int. J. Mol. Sci. 2021, 22, 3990. https://doi.org/10.3390/ijms22083990
Alle Q, Le Borgne E, Milhavet O, Lemaitre J-M. Reprogramming: Emerging Strategies to Rejuvenate Aging Cells and Tissues. International Journal of Molecular Sciences. 2021; 22(8):3990. https://doi.org/10.3390/ijms22083990
Chicago/Turabian StyleAlle, Quentin, Enora Le Borgne, Ollivier Milhavet, and Jean-Marc Lemaitre. 2021. "Reprogramming: Emerging Strategies to Rejuvenate Aging Cells and Tissues" International Journal of Molecular Sciences 22, no. 8: 3990. https://doi.org/10.3390/ijms22083990
APA StyleAlle, Q., Le Borgne, E., Milhavet, O., & Lemaitre, J.-M. (2021). Reprogramming: Emerging Strategies to Rejuvenate Aging Cells and Tissues. International Journal of Molecular Sciences, 22(8), 3990. https://doi.org/10.3390/ijms22083990