Abstract
Skin disorders showing abnormal pigmentation are often difficult to manage because of their uncertain etiology or pathogenesis. Abnormal pigmentation is a common symptom accompanying aging skin. The association between skin aging and skin pigmentation abnormalities can be attributed to certain inherited disorders characterized by premature aging and abnormal pigmentation in the skin and some therapeutic modalities effective for both. Several molecular mechanisms, including oxidative stress, mitochondrial DNA mutations, DNA damage, telomere shortening, hormonal changes, and autophagy impairment, have been identified as involved in skin aging. Although each of these skin aging-related mechanisms are interconnected, this review examined the role of each mechanism in skin hyperpigmentation or hypopigmentation to propose the possible association between skin aging and pigmentation abnormalities.
1. Introduction
Melanin pigmentation plays a critical role in protecting the skin from the harmful effects of ultraviolet (UV) radiation. A variety of genetic and environmental factors are involved in the regulation of pigmentation. Excessive or deficient amounts of melanin cause disorders showing abnormal skin pigmentation. Although wrinkling and laxity are well-identified symptoms [1], abnormal pigmentation is also a common symptom in aging skin [2,3,4,5]. Pigmentation abnormalities associated with skin aging have recently drawn attention, however, not many reports have described the changes in melanin pigmentation in aged human skin, and the role of skin aging in pigmentation abnormalities remains to be clarified.
Human skin undergoes chronological aging and environmental aging. Chronological aging is dependent upon the passage of time. Ultraviolet (UV) irradiation is the primary factor in environmental aging, which is also called photoaging, although other factors such as tobacco smoking and air pollution are involved in environmental aging [6,7]. With age, senescent cells accumulate in human skin, which can compromise skin function and integrity. Chronological aging and photoaging share certain molecular mechanisms (Figure 1). Skin aging is influenced by several factors including oxidative stress, mitochondrial DNA mutations, DNA damage, telomere shortening, and hormonal changes [8,9,10]. Autophagy impairment is also involved in aging and the senescence of skin cells [11,12]. After a brief description of skin pigmentation and skin conditions showing abnormal pigmentation accompanied by aging, this review covers the association between each of the mechanisms implicated in skin aging and skin hyperpigmentation or hypopigmentation to provide a basis for their possible relationship.
Figure 1.
Schematic view of causative factors involved in skin aging. Skin aging is influenced by several factors including oxidative stress, mitochondrial DNA mutations, DNA damage, telomere shortening, hormonal changes, and autophagy impairment.
2. Melanin Pigmentation and Its Abnormalities Accompanied by Skin Aging
Skin pigmentation is largely determined by melanin synthesis in melanocytes, melanosome transfer to keratinocytes, and melanosome degradation. Melanin synthesis is restricted to melanosomes, which contain tyrosinase, the key regulatory enzyme of melanogenesis, and tyrosine-related proteins (TRPs). Tyrosinase is involved in the early steps of melanin synthesis to dopaquinone, which are common to two types of melanin, brown-black eumelanin and yellow-red pheomelanin. Eumelanin is photoprotective by scattering and absorbing UV light. In contrast, pheomelanin is photo-unstable and phototoxic, promoting UV-induced damage including photoaging [13]. Eumelanin and pheomelanin can be produced at different proportions within the same cell. A paucity of eumelanin production makes the skin vulnerable to UV-induced damage [14]. The biogenesis of melanin is controlled by complex regulatory mechanisms. The tyrosinase and TRPs genes have binding sites for microphthalmia-associated transcription factor (MITF), which plays a fundamental role in the transcriptional regulation of melanogenesis via the cAMP, protein kinase C, and nitrogen oxide intracellular signal transduction pathways [15].
Abnormalities in melanin pigmentation are divided into two types, hypermelanosis and hypomelanosis, based on the amount of melanin in the skin. Reduced melanin synthesis with the downregulation of TRP-1, TRP-2, and MITF in senescent melanocytes [16], and decreased melanosome degradation by declining autophagy activity in aging [17] indicate the association of pigmentation abnormalities with skin aging. Their association has also been suggested in inherited and acquired disorders. Inherited disorders showing accelerated aging and skin hyperpigmentation or hypopigmentation provide strong evidence that skin aging can be associated with abnormalities in melanin pigmentation. The favorable therapeutic effects of chemicals and lasers such as polyphenol compounds and fractional photothermolysis, respectively, on both skin aging and pigmentation abnormalities [18,19] also provide reliable evidence for their association. Among the acquired skin disorders of hypermelanosis, melasma is a common skin condition. Although a complex interaction of causative factors is likely to be involved in melasma development [20], a certain proportion of melasma cases are associated with skin aging, either UV-related or UV-unrelated [21,22]. Seborrheic keratosis, which is a benign skin tumor presenting as sharply demarcated light brown-to-black papules containing a greater amount of melanin, develops in elderly individuals, mainly in sun-exposed areas. Senile lentigo is another example of a common benign skin tumor with hyperpigmentation in aged people associated with chronic UV irradiation. As for hypomelanosis, vitiligo is a representative skin condition associated with premature senescence of the skin cells [22]. The role of melanocyte senescence has also been suggested in idiopathic guttate hypomelanosis [23].
3. Association of Mechanisms Implicated in Skin Aging with Skin Pigmentation Abnormalities
3.1. Role of Oxidative Stress in Skin Pigmentation Abnormalities
Oxidative stress is considered one of the most important mechanisms involved in skin aging [24,25,26,27,28]. Whenever the cellular accumulation of reactive oxygen species (ROS), a family of oxygen-based free radicals including hydrogen peroxide (H2O2), overcomes the cellular antioxidant capacity, oxidative stress develops. ROS are generated by exogenous stimuli such as UV irradiation [29] and endogenous stimuli including cellular metabolism [30]. The skin is a high turnover, metabolically active organ, requiring energy. Because ROS are produced as the by-products of energy generation by mitochondrial respiration, mitochondria can play a critical role in oxidative stress [28,31]. Conversely, ROS can cause mitochondrial DNA mutations [32], leading to mitochondrial dysfunction. This interconnection provides a reasonable basis for considering mitochondrial DNA mutations in conjunction with oxidative stress. The skin is rich in potential biological targets for oxidative damage such as lipids, proteins, and DNA. Oxidative damage leads to cellular senescence, and thus, skin aging [33,34], therefore, the use of antioxidants can induce anti-aging effects [35,36,37].
3.1.1. Oxidative Stress and Skin Hypermelanosis
The regulatory role of H2O2 in melanocyte tyrosinase and the amelioration of hyperpigmentation by antioxidants support the effect of oxidative damage in skin hyperpigmentation [38,39,40,41]. The role of oxidative stress in skin pigmentation abnormalities can be predicted by the clinical findings of abnormal skin pigmentation in certain congenital skin diseases caused by mutations in DNA affecting mitochondrial function (Table 1). Mutation of the cytochrome c oxidase subunit 7B (COX7B) induces oxidative phosphorylation defects and mitochondrial respiration deficiency [42]. Patients with COX7B mutations show eye symptoms and linear skin defects with hyperpigmented streaks [28,43]. Mutations in excision repair cross-complementing group 6 (ERCC6) cause defective mitophagy and the accumulation of damaged mitochondria [44]. The ERCC6 gene encoding a DNA excision repair protein is responsible for Cockayne syndrome group B and can also cause UV-sensitive syndrome, characterized by photosensitivity, hyperpigmentation, freckling, and dryness in sun-exposed areas [45]. A mutation in Fanconi anemia complementation group A (FANCA) can reduce the electron transfer between respiratory complex I-III and ROS detoxification enzymes [46,47] and impair mitophagy [48]. Patients with FANCA mutation show morphological abnormalities of the skin, including hyperpigmentation [28]. Mutations in the human Harvey rat sarcoma viral oncogene homolog (HRAS) can cause mitochondrial dysfunction and defective oxidative phosphorylation [49,50]. HRAS mutations and skin hyperpigmentation have been anecdotally associated with the presence of nevi and phacomatosis pigmentokeratotica in patients with the mutation [51,52,53]. Holocytochrome c synthase (HCCS) is an enzyme located in the inner mitochondrial membrane, which adds heme to apocytochrome c and c1. Mutations in HCCS mutation reduce oxidative phosphorylation efficiency [54] and cause microphthalmia with linear skin defects syndrome exhibiting skin pigmentation [55]. Protoporphyrinogen oxidase (PPOX), the penultimate enzyme in heme biosynthesis, is a nucleus-encoded flavoprotein associated with the outer surface of the inner mitochondrial membrane. PPOX mutations cause mitochondrial abnormalities [56,57,58] and variegate porphyria, which has cutaneous manifestations characterized by photosensitivity, skin fragility, hypertrichosis, and hyperpigmentation [59]. Mutations in BRAF, NRAS, KIT, GNAQ, CDK4, PTK2B, ERBB4, GNA11, MEK, MITF, AKT3, MMP, CXCR4, EPHA3, FAS, PIK3CA, MET, CTNNB1, NEK10, and PDGFRA have been reported to induce mitochondrial dysfunction, mitochondrial respiratory complex deficiency, and decrease mitochondrial oxidative phosphorylation [60,61], which are associated with skin pigmentation [28]. Mitochondrial DNA polymerase gamma (POLG1) is responsible for mitochondrial DNA replication and repair. POLG1 mutations result in the depletion of mitochondrial DNA and can cause Alpers-Huttenlocher syndrome, which is characterized by the classic triad of seizures, cognitive degeneration, and hepatopathy. Most patients with this syndrome develop other system involvement [62], such as hyperpigmentation in the antecubital and popliteal fossae and dorsa of the feet [63].
Acquired abnormal skin pigmentation can be related to oxidative stress (Table 2). Sunlight stimulates melanin production for protection against the harmful effects from UV radiation such as from tanning. The substantial role of oxidative stress in UV-induced melanogenesis and the alleviation of UV-induced melanogenesis by antioxidant therapy [64,65,66] indicates an association between oxidative stress and UV-induced hyperpigmentation. UV irradiation can generate the peroxidation products of sebum, squalene monohydroperoxides. The hyperpigmentation of reconstructed human skin equivalents by exposure to squalene monohydroperoxides and the amelioration of squalene monohydroperoxide-induced hyperpigmentation by co-treatment with 12-hydroxystearic acid support a pivotal role of antioxidant mechanisms in UV-induced hyperpigmentation [65]. Although the exact pathogenesis of melasma is uncertain, oxidative stress has been implicated based on increased serum malondialdehyde but reduced serum catalase levels in the patient groups [67,68] and the efficacy of antioxidant drugs identified in a clinical trial [69,70]. The role of oxidative stress has also been identified in seborrheic keratosis. Guanine deaminase, shown to be upregulated in seborrheic keratosis, stimulated UV-induced keratinocyte senescence via ROS generation [71].
3.1.2. Oxidative Stress and Skin Hypomelanosis
Oxidative stress caused by mitochondrial redox imbalance or antioxidant enzyme deficiency is also associated with skin hypopigmentation (Table 1). The loss of ATPase copper-transporting alpha (ATP7A) activity, which causes Menkes disease, results in the accumulation of copper in cellular organelles, including mitochondria, thereby disrupting the mitochondrial redox balance [72]. Tyrosinase is a copper-dependent enzyme involved in melanogenesis. Because ATP7A protein is required for tyrosinase activity, the loss of ATP7A activity by gene mutations impairs melanogenesis [73]. Mutations in SURF1 cytochrome C oxidase assembly factor (SURF1) are associated with Leigh syndrome, a rare progressive neurodegenerative disorder caused by mitochondrial cytopathy. Skin and hair abnormalities, including hypopigmentation are often accompanying symptoms [74]. The deletion of mitochondrial DNA induces Kearns-Sayre syndrome, a mitochondrial multisystem disorder characterized by the clinical symptoms of ophthalmoplegia and pigmentary retinopathy. Hypopigmented patches like Hypomelanosis of Ito have been reported as a possible symptom [75]. Piebaldism, characterized by depigmented patches presenting at birth, is caused by KIT gene mutations. An association between H2O2-mediated stress and piebaldism has been identified, although its mechanism in oxidative stress differs from that in vitiligo [76].
Concerning the role of oxidative stress in acquired hypomelanosis, most data are related to vitiligo (Table 2). Oxidative stress caused by ROS production and low levels of antioxidants are considered the pivotal mechanisms in vitiligo development [77,78], although the therapeutic effects of antioxidants have been controversial [79]. Elevations in superoxide dismutase and malondialdehyde [80], reductions in catalase or catalase gene polymorphism [81,82,83], increases in advanced oxidation protein products [84], nuclear factor erythroid 2-like 2 (Nrf2) gene polymorphism [85], and reduced Nrf2 activation [86,87] are associated with vitiligo susceptibility. Increased mitochondrial DNA copy numbers [88], mitochondrial dysfunction by sirtuin3 (SIRT3) deficiency [89], and increased calcium influx by the upregulation of transient receptor potential cation channel subfamily M member 2 (TRPM2), a calcium channel sensitive to oxidative stress [90], contribute to oxidative stress-induced melanocyte damage. Autophagy impairment also plays a role in oxidative stress by disrupting the antioxidant defense system [91,92]. ROS accumulation damages melanocytes by the generation of autoantigens, which leads to the CD8+ T cell-mediated destruction of melanocytes.

Table 1.
Abnormal skin pigmentation in congenital disorders related to oxidative stress.
Table 2.
Acquired abnormal skin pigmentation related to oxidative stress.
3.2. Role of DNA Damage in Skin Pigmentation Abnormalities
DNA repair mechanisms such as the repair of nucleotide excision and double-strand breaks should be activated to maintain homeostasis whenever DNA damage occurs. The downregulation of DNA repair and the accumulation of DNA damage drives the aging process [93,94]. Impaired DNA replication and repair by mutations in a group of premature aging syndromes known as progeroid syndromes provide strong evidence for the role of DNA damage in aging [95]. UV radiation is a notorious inducer of DNA damage in all skin types despite its inverse relationship with constitutive skin pigmentation [96].
3.2.1. DNA Damage and Skin Hypermelanosis
The association between DNA damage and hyperpigmentation, particularly aging-related effects, has been identified in certain DNA repair syndromes caused by pathogenic variants encoding proteins in DNA replication and cellular responses to DNA damage (Table 3). Mutations in RecQ protein-like 4 (RECQL4) belonging to the RECQ DNA helicase family, which participate in many aspects of DNA metabolism [97], cause Rothmund-Thomson syndrome [98]. The syndrome shows clinical features of accelerated aging, such as atrophic skin and pigment changes [97]. Mutations in one of the eight genes involved in nucleotide excision repair and DNA polymerase cause Xeroderma pigmentosum [99]. Xeroderma pigmentosum is characterized by the extreme sensitivity to sunlight, resulting in sunburn, unusually increased numbers of lentigines in sun-exposed areas, areas containing both hyperpigmentation and hypopigmentation in the absence of rigorous sunlight protection, accelerated photoaging, and increased skin cancer risk over time [98]. Mutations in several genes involved in DNA repair, particularly interstrand crosslink repair and telomere maintenance, cause Fanconi anemia. A poikilodermatic change with hypopigmentation, hyperpigmentation, and telangiectasia appears in the skin of patients with Fanconi anemia [100,101]. Inactivating mutations of the tumor suppressor serine-threonine kinase 11/liver kinase B1 (STK11/LKB1), which can play a role in the regulation of the UV-induced DNA damage response in mice skin [102], underlie Peutz-Jeghers syndrome. Hyperpigmentation of mucous membranes and the skin is one of the characteristic symptoms of Peutz-Jeghers syndrome.
Acquired abnormal skin pigmentation can be related to DNA damage (Table 4). Skin keratinocytes with DNA damage induced by UV exposure secrete α–melanocyte-stimulating hormone (α-MSH) [103]. α-MSH interacts with melanocortin 1 receptor (MC1R), which enhances nucleotide excision repair in melanocytes [104]. The role of α-MSH in melanogenesis suggests an association between DNA damage and melanogenesis. Defects in DNA repair have not been reported in melasma, seborrheic keratosis, and senile lentigo, although microsatellite instability indicating DNA repair defects was detected in seborrheic keratosis developed in a patient with hereditary nonpolyposis colorectal cancer [105].
Table 3.
Abnormal skin pigmentation in congenital disorders related to DNA damage.
Table 4.
Acquired abnormal skin pigmentation related to DNA damage.
3.2.2. DNA Damage and Skin Hypomelanosis
Certain inherited disorders caused by DNA damage can exhibit aging-related skin hypopigmentation alone or both hypopigmentation and hyperpigmentation (Table 3). As described above, xeroderma pigmentosum patients have hypopigmented areas in addition to hyperpigmented skin [98]. Fanconi anemia shows poikilodermatic changes consisting of hypopigmentation, hyperpigmentation, atrophy, and telangiectasia as the characteristic skin symptoms [100,101]. Poikilodermatic changes also appear in the skin of Rothmund-Thomson syndrome patients [98]. A chromosomal deletion on 15q11.2-q13, which induces shortening of leukocyte telomere length, a marker of biological age, causes Prader-Willi syndrome [106]; as one of the premature aging syndromes, Prader-Willi syndrome patients can exhibit hypopigmentation [107]. Oculocutaneous albinism and Hermansky-Pudlak syndrome are hereditary diseases with DNA mutations [108,109]. Both diseases exhibit oculocutaneous albinism caused by the increased degradation of tyrosinase through the ubiquitin-proteasome system [110]. Photoaging is accelerated in albino hairless mice by exposure to solar simulation [111].
DNA damage and defects in DNA repair have not been identified in vitiligo, although an association between apurinic/apyrimidinic endonuclease 1 (APE1) polymorphism with leukotrichia suggests the role of this polymorphism in vitiligo [112] (Table 4).
3.3. Role of Telomere Shortening in Skin Pigmentation Abnormalities
Telomeres are dynamic nucleoprotein-DNA structures composed of long tandem TTAGGG repeat, guanine-rich sequences located at the end of chromosomes to cap and protect the linear chromosome ends. Although the length of telomeres differs between chromosome arms, telomeres shorten by cell division, and the initial length of telomeres correlates with the cellular replicative capacity. Accelerated aging in short telomere syndromes caused by inheritable gene mutations resulting in decreased telomere lengths [113] indicates that telomere shortening can be one of the primary hallmarks of aging [114].
3.3.1. Telomere Shortening and Skin Hypermelanosis
The genetically inherited diseases with much shorter telomeres compared to age-matched controls are referred to as telomeropathies. Primary telomeropathies are defined as disorders caused by mutations in the telomere maintenance machinery. Secondary telomeropathies are caused by mutations in genes encoding proteins involved in telomere DNA repair, leading to telomere aberrations and loss [115]. However, overlap exists between telomeropathies, particularly secondary telomeropathies, and DNA mutations.
The role of telomeres in skin pigmentation has been identified in telomeropathies (Table 5). The first disorder identified as a primary telomeropathy was dyskeratosis congenita, which is caused by mutations in the DKC1 gene encoding the dyskerin protein, or one of the core telomerase genes encoding a protein involved in telomere maintenance [116], resulting in telomere shortening, and thereby, premature aging [117,118]. Reticular hyperpigmentation is a symptom of the dyskeratosis congenita diagnostic triad along with oral leukoplakia and nail dystrophy. Xeroderma pigmentosum and Fanconi anemia are included in secondary telomeropathies, indicating an overlap between telomere shortening and DNA repair syndromes. Among the eight complementation groups in xeroderma pigmentosum, mutations in complementation group C (XPC), can cause the instability of telomere upon UV exposure [117]. Fanconi anemia is caused by genetic mutations involved in DNA repair for telomere maintenance. As described before, patients with these disorders manifest abnormal skin pigmentation [100,101]. A point mutation in the LMNA gene, which produces abnormal lamin A protein associated with rapid telomere erosion, can cause Hutchinson-Gilford progeria syndrome [119,120]. As one of the most severe segmental progeroid syndromes, patients with Hutchinson-Gilford syndrome demonstrate premature aging with distinct dermatologic features including hyper- and hypopigmentation over areas of sclerodermoid changes [95,121].
Table 5.
Abnormal skin pigmentation in congenital disorders related to telomere shortening.
Although telomere attrition contributes to certain age-related disorders, the association of melasma, seborrheic keratosis, or senile lentigo with telomere shortening has not been reported.
3.3.2. Telomere Shortening and Skin Hypomelanosis
Telomere shortening can be associated with skin hypomelanosis, although hypopigmentation usually coexists with hyperpigmentation as shown in patients with xeroderma pigmentosum and Hutchinson-Gilford progeria syndrome [95,98] (Table 5).
In contrast, in vitiligo, telomerase activity is lower in lesional skin compared to non-lesional skin and healthy control skin, which was proposed as a reason for the low incidence of sun damage and cancer in the lesional skin of vitiligo patients [122].
3.4. Role of Hormones in Skin Pigmentation Abnormalities
Cutaneous neuroendocrine systems can regulate skin function in response to environmental stresses, such as UV radiation and pollutants. Melatonin belongs to the family of neurohormones [123]. The pineal gland is the main organ of melatonin synthesis and secretion. However, human skin can produce and rapidly metabolize melatonin to maintain homeostasis against environmental stresses [124]. Melatonin is involved in the regulation of various physiological activities, including circadian rhythm, immune responses, oxidative process, apoptosis, and mitochondrial homeostasis. Antioxidant actions of melatonin protect skin from UVB-induced harm [125,126], indicating the critical role of melatonin in photoaging.
Receptors for sex steroids, particularly estrogen receptors, are present in human skin, including epidermal keratinocytes and dermal fibroblasts. Many findings, such as the gradual decline of these receptors with chronological and environmental aging [127,128], skin aging accelerated by the loss of estradiol production after menopause, and UV-induced collagen degradation reversed by estradiol replacement [128,129,130], indicate the role of estrogens in protecting skin from aging.
3.4.1. Hormones and Skin Hypermelanosis
Oxidative stress is the main mechanism involved in skin aging and pigmentation. The antioxidant actions of melatonin [125,126] are implicated in the association between melatonin and skin pigmentation. Although significantly lower serum levels of melatonin have been seen in melasma patients compared to controls [68], relevant reports are rare.
Sex hormones, particularly estrogens, play a role, which is stimulatory but not inhibitory, in pigmentation in cultured skin cells [131] and in melasma [20,132]. These results suggest that estrogens enhance skin pigmentation and delay rather than accelerate skin aging.
3.4.2. Hormones and Skin Hypomelanosis
The association of melatonin with vitiligo has been described [133]. A connection between reduced testosterone due to secondary hypogonadism and vitiligo has also been suggested [134].
Above all, vitiligo patients frequently suffer from serious psychological problems such as depression and anxiety. The levels of dehydroepiandrosterone sulfate, which has antioxidant properties, were lower but the ratios of cortisol to dehydroepiandrosterone sulfate were higher in vitiligo patients compared to healthy controls [135]. The results suggest the role of dehydroepiandrosterone sulfate in vitiligo development and provide reliable evidence for the association of vitiligo with stress-related hormones.
3.5. Role of Autophagy in Skin Pigmentation Abnormalities
Autophagy is an essential cellular process for homeostasis accomplished by degrading and recycling damaged cellular components. Autophagic activity declines with aging [136]. However, defects in autophagy can also cause skin aging with an accumulation of damaged cellular components [137]. The association between reduced autophagy and skin aging has mostly been investigated in photoaging. The phenotype of skin aging induced by UVA irradiation in human fibroblasts and mice with Cockayne syndrome type B deficiency is accompanied by reduced autophagic activity and is rescued by the improvement in autophagic function [138]. Repeated UVA irradiation results in the accumulation of autophagosomes and lysosomes, suggesting that photoaging can reduce autophagic activity at the degradation stage [139,140]. Oxidative stress-induced cellular senescence in murine melanocytes and keratinocytes associated with autophagy-related 7 (Atg7) deficiency [12,141] and the protective role of caffeine in oxidative stress-induced senescence of human epidermal keratinocytes through autophagy induction [142] suggest that autophagy defects can accelerate skin aging by increasing oxidative stress.
3.5.1. Autophagy and Skin Hypermelanosis
The melanosome is a lysosome-related organelle. Autophagy-related regulators are involved in the formation, maturation, and degradation of melanosomes in melanocytes [143]. In vitro and in vivo results have demonstrated the association between autophagy and skin pigmentation abnormalities (Table 6).
Table 6.
Abnormal skin pigmentation in disorders related to autophagy impairment.
The diversity of skin color was affected by epidermal heat shock 70 kDa protein 1A (Hsp70-1A), which modulates autophagic melanosome degradation in keratinocytes [144]. Accelerated keratinocyte senescence by arginase-2 upregulation impairs autophagy and reduces melanosome degradation, resulting in hyperpigmentation in melasma [145]. Association between premature skin aging with autophagy reduction was also detected in the hyperpigmented lesions of patients with senile lentigo, whereas autophagy activation ameliorated pigmentation in ex vivo lesional skin [17]. Chemical agents such as tranexamic acid, β–mangostin, 3-O-glyceryl-2-O-hexyl ascorbate, and Melasolv induced skin lightening through autophagy activation [146,147,148,149]. Light-emitting diodes can also activate autophagy, resulting in skin lightening [150].
3.5.2. Autophagy and Skin Hypomelanosis
Recessive mutations in EPG5 encoding the ectopic P-granules autophagy protein 5 homolog, a key regulator of autophagy, cause Vici syndrome. As one of the most typical examples of congenital autophagy disorders, patients with the syndrome exhibit characteristic features that include oculocutaneous hypopigmentation [151]. Although the mechanism of hypopigmentation remains to be clarified, defective autophagosome-lysosome fusion in EPG5-depleted melanocytes [152] may disrupt the antioxidant defense system. Autophagic dysregulation by activation of the mTOR signaling pathway in melanocytes has been identified as a mechanism of reduced pigmentation in hypopigmented macules in patients with tuberous sclerosis complex (TSC) gene mutations [153,154]. The loss of glycoprotein nonmetastatic melanoma protein B (GPNMB) causes amyloidosis cutis dyschromica, which is characterized by generalized hyperpigmentation mottled with hypopigmented macules [155]. GPNMB is a melanocytic cell marker involved in multiple functions, including melanosome formation and autophagy [156]. GPNMB knockdown reduced the number of melanosomes in melanocytes [157].
GPNMB has also been downregulated in vitiligo keratinocytes, causing the loss of melanocytes [158]. The finding indicates the regulatory role of GPNMB in skin hypomelanosis by reducing melanocyte survival in addition to reducing melanogenesis.
4. Conclusions
Skin diseases with pigmentation abnormalities are difficult to treat primarily due to unknown causes or pathogenesis. The pathomechanisms in individual patients can be different even in the same disease. It is important to identify the cause to manage skin pigmentation abnormalities. To propose the possible role of skin aging in abnormal pigmentation, the association between the identified mechanisms involved in skin aging and skin pigmentation abnormalities in various inherited and acquired disorders were reviewed. The mechanisms implicated in skin aging include oxidative stress, which is the most pivotal cause of skin aging, DNA damage, telomere shortening, decreased melatonin, and autophagy impairment. Both skin aging and pigmentation abnormalities in various inherited disorders caused by DNA damage or telomere shortening indicate the relevant relationship between skin aging and pigmentation abnormalities. However, other mechanisms may not yet sufficiently support the relationship between skin aging and pigmentation abnormalities (Figure 2). Epigenetic changes, particularly DNA methylation [159,160] and microRNAs [161], are also proposed to be involved in skin aging without an identified role in skin pigmentation. More studies are needed to prove the reliable role of skin aging in various conditions showing abnormal skin pigmentation.
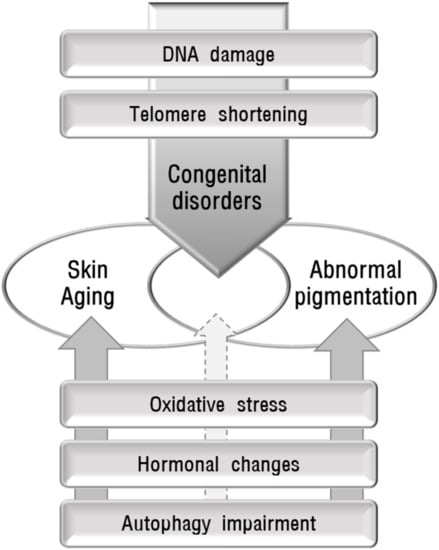
Figure 2.
Schematic view of the possible association between abnormal pigmentation and skin aging. Some causative mechanisms, such as DNA damage and telomere shortening, are involved in hereditary disorders that show accelerated aging and skin hyperpigmentation or hypopigmentation, providing reliable evidence for the association between skin aging and pigmentation abnormalities. However, oxidative stress, hormonal changes, and autophagy impairment, may not yet sufficiently support the relationship between skin aging and pigmentation abnormalities.
Funding
This research was supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number: HP20C0131). This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. NRF 2018R1A5A2023127).
Conflicts of Interest
The author states no conflict of interest.
Abbreviations
| α-MSH | α–melanocyte-stimulating hormone |
| APE1 | apurinic/apyrimidinic endonuclease 1 |
| Atg | autophagy-related |
| ATP7A | ATPase copper transporting alpha |
| CAT | catalase |
| COX7B | cytochrome c oxidase subunit 7B |
| DKC1 | dyskeratosis congenita 1 |
| EPG5 | ectopic P-granules autophagy protein 5 homolog |
| ERCC6 | excision repair cross-complementing group 6 |
| FANCA | Fanconi anemia complementation group A |
| GPNMB | glycoprotein nonmetastatic melanoma protein B |
| H2O2 | hydrogen peroxide |
| HCCS | holocytochrome c synthase |
| HRAS | Harvey rat sarcoma viral oncogene homolog |
| Hsp70-1A | heat shock 70kDa protein 1A |
| LMNA | lamin A |
| MC1R | melanocortin 1 receptor |
| MITF | microphthalmia-associated transcription factor |
| Nrf2 | nuclear factor erythroid 2-like 2 |
| POLG1 | polymerase gamma |
| PPOX | protoporphyrinogen oxidase |
| RECQL4 | RecQ protein-like 4 |
| ROS | reactive oxygen species |
| SIRT3 | sirtuin3 |
| STK11/LKB1 | serine-threonine kinase 11/liver kinase B1 |
| SURF1 | SURF1 cytochrome C oxidase assembly factor |
| TERT | telomerase reverse transcriptase |
| TRP | tyrosine-related protein |
| TRPM2 | transient receptor potential cation channel subfamily M member 2 |
| TSC | tuberous sclerosis complex |
| UV | ultraviolet |
| XPC | Xeroderma pigmentosum complementation group C |
References
- McCullough, J.L.; Kelly, K.M. Prevention and treatment of skin aging. In Aging Interventions and Therapies; World Scientific Publishing: Singapore, 2006; Volume 1067, pp. 323–331. [Google Scholar]
- Okazaki, M.; Yoshimura, K.; Uchida, G.; Harii, K. Correlation between age and the secretions of melanocyte-stimulating cytokines in cultured keratinocytes and fibroblasts. Br. J. Dermatol. 2005, 153 (Suppl. 2), 23–29. [Google Scholar] [CrossRef] [PubMed]
- Nakama, M.; Murakami, Y.; Tanaka, H.; Nakata, S. Decrease in nicotinamide adenine dinucleotide dehydrogenase is related to skin pigmentation. J. Cosmet. Dermatol. 2012, 11, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Duval, C.; Cohen, C.; Chagnoleau, C.; Flouret, V.; Bourreau, E.; Bernerd, F. Key regulatory role of dermal fibroblasts in pigmentation as demonstrated using a reconstructed skin model: Impact of photo-aging. PLoS ONE 2014, 9, e114182. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Kim, J.Y.; Oh, S.H. Advanced glycation end products (AGEs) promote melanogenesis through receptor for AGEs. Sci. Rep. 2016, 6, 27848. [Google Scholar] [CrossRef]
- Krutmann, J.; Bouloc, A.; Sore, G.; Bernard, B.A.; Passeron, T.J. The skin aging exposome. J. Dermatol. Sci. 2017, 85, 152–161. [Google Scholar] [CrossRef]
- Schikowski, T.; Hüls, A. Air Pollution and Skin Aging. Curr. Environ. Health Rep. 2020, 7, 58–64. [Google Scholar] [CrossRef]
- Fisher, G.J.; Kang, S.; Varani, J.; Bata-Csorgo, Z.; Wan, Y.; Datta, S.; Voorhees, J.J. Mechanisms of photoaging and chronological skin aging. Arch. Dermatol. 2002, 138, 1462–1470. [Google Scholar] [CrossRef]
- Tobin, D.J. Introduction to skin aging. J. Tissue Viability 2017, 26, 37–46. [Google Scholar] [CrossRef]
- Schuch, A.P.; Moreno, N.C.; Schuch, N.J.; Menck, C.F.M.; Garcia, C.C.M. Sunlight damage to cellular DNA: Focus on oxidatively generated lesions. Free Radic. Biol. Med. 2017, 107, 110–124. [Google Scholar] [CrossRef]
- Deruy, E.; Nassour, J.; Martin, N.; Vercamer, C.; Malaquin, N.; Bertout, J.; Chelli, F.; Pourtier, A.; Pluquet, O.; Abbadie, C. Level of macroautophagy drives senescent keratinocytes into cell death or neoplastic evasion. Cell Death Dis. 2014, 5, e1577. [Google Scholar] [CrossRef]
- Zhang, C.F.; Gruber, F.; Ni, C.; Mildner, M.; Koenig, U.; Karner, S.; Barresi, C.; Rossiter, H.; Narzt, M.S.; Nagelreiter, I.M.; et al. Suppression of autophagy dysregulates the antioxidant response and causes premature senescence of melanocytes. J. Investig. Dermatol. 2015, 135, 1348–1357. [Google Scholar] [CrossRef]
- Ito, S.; Wakamatsu, K.; Sarna, T. Photodegradation of Eumelanin and Pheomelanin and Its Pathophysiological Implications. Photochem. Photobiol. 2018, 94, 409–420. [Google Scholar] [CrossRef]
- Nasti, T.H.; Timares, L. MC1R, eumelanin and pheomelanin: Their role in determining the susceptibility to skin cancer. Photochem. Photobiol. 2015, 91, 188–200. [Google Scholar] [CrossRef]
- Slominski, A.; Tobin, D.J.; Shibahara, S.; Wortsman, J. Melanin pigmentation in mammalian skin and its hormonal regulation. Physiol. Rev. 2004, 84, 1155–1228. [Google Scholar] [CrossRef]
- Ko, H.; Kim, M.M. H2O2 promotes the aging process of melanogenesis through modulation of MITF and Nrf2. Mol. Biol. Rep. 2019, 46, 2461–2471. [Google Scholar] [CrossRef]
- Murase, D.; Kusaka-Kikushima, A.; Hachiya, A.; Fullenkamp, R.; Stepp, A.; Imai, A.; Ueno, M.; Kawabata, K.; Takahashi, Y.; Hase, T.; et al. Autophagy Declines with Premature Skin Aging resulting in Dynamic Alterations in Skin Pigmentation and Epidermal Differentiation. Int. J. Mol. Sci. 2020, 21, 5708. [Google Scholar] [CrossRef]
- Narurkar, V.A.; Alster, T.S.; Bernstein, E.F.; Lin, T.J.; Loncaric, A. Safety and Efficacy of a 1550 nm/1927 nm Dual Wavelength Laser for the Treatment of Photodamaged Skin. J. Drugs Dermatol. 2018, 17, 41–46. [Google Scholar]
- Boo, Y.C. Human Skin Lightening Efficacy of Resveratrol and Its Analogs: From in Vitro Studies to Cosmetic Applications. Antioxidants 2019, 8, 332. [Google Scholar] [CrossRef]
- Lee, A.Y. Recent progress in melasma pathogenesis. Pigment Cell Melanoma Res. 2015, 28, 648–660. [Google Scholar] [CrossRef]
- Kim, N.H.; Choi, S.H.; Lee, T.R.; Lee, C.H.; Lee, A.Y. Cadherin 11 involved in basement membrane damage and dermal changes in melasma. Acta Derm. Venereol. 2016, 96, 635–640. [Google Scholar] [CrossRef]
- Bellei, B.; Picardo, M. Premature cell senescence in human skin: Dual face in chronic acquired pigmentary disorders. Ageing Res. Rev. 2020, 57, 100981. [Google Scholar] [CrossRef] [PubMed]
- Rani, S.; Kumar, R.; Kumarasinghe, P.; Bhardwaj, S.; Srivastava, N.; Madaan, A.; Parsad, D. Melanocyte abnormalities and senescence in the pathogenesis of idiopathic guttate hypomelanosis. Int. J. Dermatol. 2018, 57, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Bogdan, A.I.; Baumann, L. Antioxidants used in skin care formulations. Skin Therapy Lett. 2008, 13, 5–9. [Google Scholar]
- Sasaki, M.; Kajiya, H.; Ozeki, S.; Okabe, K.; Ikebe, T. Reactive oxygen species promotes cellular senescence in normal human epidermal keratinocytes through epigenetic regulation of p16(INK4a.). Biochem. Biophys. Res. Commun. 2014, 452, 622–628. [Google Scholar] [CrossRef]
- Kammeyer, A.; Luiten, R.M. Oxidation events and skin aging. Ageing Res. Rev. 2015, 21, 16–29. [Google Scholar] [CrossRef]
- De Jager, T.L.; Cockrell, A.E.; Du Plessis, S.S. Ultraviolet Light Induced Generation of Reactive Oxygen Species. Adv. Exp. Med. Biol. 2017, 996, 15–23. [Google Scholar]
- Sreedhar, A.; Aguilera-Aguirre, L.; Singh, K.K. Mitochondria in skin health, aging, and disease. Cell Death Dis. 2020, 11, 444. [Google Scholar] [CrossRef]
- Zhang, X.; Rosenstein, B.S.; Wang, Y.; Lebwohl, M.; Wei, H. Identification of possible reactive oxygen species involved in ultraviolet radiation-induced oxidative DNA damage. Free Radic. Biol. Med. 1997, 23, 980–985. [Google Scholar] [CrossRef]
- Trouba, K.J.; Hamadeh, H.K.; Amin, R.P.; Germolec, D.R. Oxidative stress and its role in skin disease. Antioxid. Redox Signal. 2002, 4, 665–673. [Google Scholar] [CrossRef]
- Hudson, L.; Bowman, A.; Rashdan, E.; Birch-Machin, M.A. Mitochondrial damage and ageing using skin as a model organ. Maturitas 2016, 93, 34–40. [Google Scholar] [CrossRef]
- Shen, J.; Wan, J.; Huff, C.; Fang, S.; Lee, J.E.; Zhao, H. Mitochondrial DNA 4977-base pair common deletion in blood leukocytes and melanoma risk. Pigment Cell Melanoma Res. 2016, 29, 372–378. [Google Scholar] [CrossRef]
- Birch-Machin, M.A.; A Bowman, A. Oxidative stress and ageing. Br. J. Dermatol. 2016, 175 (Suppl. 2), 26–29. [Google Scholar] [CrossRef]
- Gu, Y.; Han, J.; Jiang, C.; Zhang, Y. Biomarkers, oxidative stress and autophagy in skin aging. Ageing Res. Rev. 2020, 59, 101036. [Google Scholar] [CrossRef]
- Masaki, H. Role of antioxidants in the skin: Anti-aging effects. J. Dermatol. Sci. 2010, 58, 85–90. [Google Scholar] [CrossRef]
- Lephart, E.D. Skin aging and oxidative stress: Equol’s anti-aging effects via biochemical and molecular mechanisms. Ageing Res. Rev. 2016, 31, 36–54. [Google Scholar] [CrossRef]
- Zouboulis, C.C.; Ganceviciene, R.; Liakou, A.I.; Theodoridis, A.; Elewa, R.; Makrantonaki, E. Aesthetic aspects of skin aging, prevention, and local treatment. Clin. Dermatol. 2019, 37, 365–372. [Google Scholar] [CrossRef]
- Schallreuter, K.U.; Kothari, S.; Chavan, B.; Spencer, J.D. Regulation of melanogenesis--controversies and new concepts. Exp. Dermatol. 2008, 17, 395–404. [Google Scholar] [CrossRef]
- Park, D.J.; Sekhon, S.S.; Yoon, J.; Kim, Y.H.; Min, J. Color reduction of melanin by lysosomal and peroxisomal enzymes isolated from mammalian cells. Mol. Cell Biochem. 2016, 413, 119–125. [Google Scholar] [CrossRef]
- Jiang, R.; Xu, X.H.; Wang, K.; Yang, X.Z.; Bi, Y.F.; Yan, Y.; Liu, J.Z.; Chen, X.N.; Wang, Z.Z.; Guo, X.L.; et al. Ethyl acetate extract from Panax ginseng C.A. Meyer and its main constituents inhibit α-melanocyte-stimulating hormone-induced melanogenesis by suppressing oxidative stress in B16 mouse melanoma cells. J. Ethnopharmacol. 2017, 208, 149–156. [Google Scholar] [CrossRef]
- Rodboon, T.; Okada, S.; Suwannalert, P. Germinated Riceberry Rice Enhanced Protocatechuic Acid and Vanillic Acid to Suppress Melanogenesis through Cellular Oxidant-Related Tyrosinase Activity in B16 Cells. Antioxidants 2020, 9, 247. [Google Scholar] [CrossRef]
- Gronow, S.; Noah, C.; Blumenthal, A.; Lindner, B.; Brade, H. Construction of a deep-rough mutant of Burkholderia cepacia ATCC 25416 and characterization of its chemical and biological properties. J. Biol. Chem. 2003, 278, 1647–1655. [Google Scholar] [CrossRef] [PubMed]
- Temple, I.K.; Hurst, J.A.; Hing, S.; Butler, L.; Baraitser, M. De novo deletion of Xp22.2-pter in a female with linear skin lesions of the face and neck, microphthalmia, and anterior chamber eye anomalies. J. Med. Genet. 1990, 27, 56–58. [Google Scholar] [CrossRef] [PubMed]
- Scheibye-Knudsen, M.; Ramamoorthy, M.; Sykora, P.; Maynard, S.; Lin, P.-C.; Minor, R.K.; Wilson, D.M., 3rd; Cooper, M.; Spencer, R.; de Cabo, R.; et al. Cockayne syndrome group B protein prevents the accumulation of damaged mitochondria by promoting mitochondrial autophagy. J. Exp. Med. 2012, 209, 855–869. [Google Scholar] [CrossRef] [PubMed]
- Sin, Y.; Makimura, F.; Saijo, M.; Obika, S. Generation of splice switching oligonucleotides targeting the Cockayne syndrome group B gene product in order to change the diseased cell state. Biochem. Biophys. Res. Commun. 2018, 500, 163–169. [Google Scholar] [CrossRef]
- Cappelli, E.; Ravera, S.; Vaccaro, D.; Cuccarolo, P.; Bartolucci, M.; Panfoli, I.; Dufour, C.; Degan, P. Mitochondrial respiratory complex I defects in Fanconi anemia. Trends Mol. Med. 2013, 19, 513–514. [Google Scholar] [CrossRef]
- Ravera, S.; Vaccaro, D.; Cuccarolo, P.; Columbaro, M.; Capanni, C.; Bartolucci, M.; Panfoli, I.; Morelli, A.; Dufour, C.; Cappelli, E.; et al. Mitochondrial respiratory chain Complex I defects in Fanconi anemia complementation group A. Biochimie 2013, 95, 1828–1837. [Google Scholar] [CrossRef]
- Sumpter, R., Jr.; Sirasanagandla, S.; Fernández, Á.F.; Wei, Y.; Dong, X.; Franco, L.; Zou, Z.; Marchal, C.; Lee, M.Y.; Clapp, D.W.; et al. Fanconi Anemia Proteins Function in Mitophagy and Immunity. Cell 2016, 165, 867–881. [Google Scholar] [CrossRef]
- Aeby, A.; Sznajer, Y.; Cavé, H.; Rebuffat, E.; Coster, R.V.; Rigal, O.; Bogaert, P.V. Cardiofaciocutaneous (CFC) syndrome associated with muscular coenzyme Q10 deficiency. J. Inherit. Metab. Dis. 2007, 30, 827. [Google Scholar] [CrossRef]
- Champion, K.J.; Bunag, C.; Estep, A.L.; Jones, J.R.; Bolt, C.H.; Rogers, R.C.; Rauen, K.A.; Everman, D.B. Germline mutation in BRAF codon 600 is compatible with human development: De novo p.V600G mutation identified in a patient with CFC syndrome. Clin. Genet. 2011, 79, 468–474. [Google Scholar] [CrossRef]
- Groesser, L.; Herschberger, E.; Sagrera, A.; Shwayder, T.; Flux, K.; Ehmann, L.; Wollenberg, A.; Torrelo, A.; Bagazgoitia, L.; Blanca Diaz-Ley, B.; et al. Phacomatosis pigmentokeratotica is caused by a postzygotic HRAS mutation in a multipotent progenitor cell. J. Investig. Dermatol. 2013, 133, 1998–2003. [Google Scholar] [CrossRef]
- Sarin, K.Y.; McNiff, J.M.; Kwok, S.; Kim, J.; Khavari, P.A. Activating HRAS mutation in nevus spilus. J. Investig. Dermatol. 2014, 134, 1766–1768. [Google Scholar] [CrossRef]
- Martin, R.J.; Arefi, M.; Splitt, M.; Redford, L.; Moss, C.; Rajan, N. Phacomatosis pigmentokeratotica and precocious puberty associated with HRAS mutation. Br. J. Dermatol. 2018, 178, 289–291. [Google Scholar] [CrossRef]
- Amary, M.F.; Damato, S.; Halai, D.; Eskandarpour, M.; Berisha, F.; Bonar, F.; McCarthy, S.; Fantin, V.R.; Straley, K.S.; Lobo, S.; et al. Ollier disease and Maffucci syndrome are caused by somatic mosaic mutations of IDH1 and IDH2. Nat. Genet. 2011, 43, 1262–1265. [Google Scholar] [CrossRef]
- Slavotinek, A.M. Eye development genes and known syndromes. Mol. Genet. Metab. 2011, 104, 448–456. [Google Scholar] [CrossRef]
- Lange, H.; Mühlenhoff, U.; Denzel, M.; Kispal, G.; Lill, R. The heme synthesis defect of mutants impaired in mitochondrial iron-sulfur protein biogenesis is caused by reversible inhibition of ferrochelatase. J. Biol. Chem. 2004, 279, 29101–29108. [Google Scholar] [CrossRef]
- Nilsson, R.; Schultz, I.J.; Pierce, E.L.; Soltis, K.A.; Naranuntarat, A.; Ward, D.M.; Baughman, J.M.; Paradkar, P.N.; Kingsley, P.D.; Culotta, V.C.; et al. Discovery of genes essential for heme biosynthesis through large-scale gene expression analysis. Cell Metab. 2009, 10, 119–130. [Google Scholar] [CrossRef]
- Bareth, B.; Dennerlein, S.; Mick, D.U.; Nikolov, M.; Urlaub, H.; Rehling, P. The heme a synthase Cox15 associates with cytochrome c oxidase assembly intermediates during Cox1 maturation. Mol. Cell Biol. 2013, 33, 4128–4137. [Google Scholar] [CrossRef]
- Dawe, R. An overview of the cutaneous porphyrias. F1000Reserach 2017, 6, 1906. [Google Scholar] [CrossRef]
- Deichmann, M.; Kahle, B.; Benner, A.; Thome, M.; Helmke, B.; Helmut Näher, H. Somatic mitochondrial mutations in melanoma resection specimens. Int. J. Oncol. 2004, 24, 137–141. [Google Scholar] [CrossRef]
- Dutton-Regester, K.; Irwin, D.; Hunt, P.; Aoude, L.G.; Tembe, V.; Pupo, G.M.; Lanagan, C.; Carter, C.D.; O’Connor, L.; O’Rourke, M.; et al. A high-throughput panel for identifying clinically relevant mutation profiles in melanoma. Mol. Cancer Ther. 2012, 11, 888–897. [Google Scholar] [CrossRef]
- Saneto, R.P. Alpers-Huttenlocher syndrome: The role of a multidisciplinary health care team. J. Multidiscip. Healthc. 2016, 9, 323–333. [Google Scholar] [CrossRef]
- Campuzano-García, A.E.; Rodríguez-Arámbula, A.; Torres-Alvarez, B.; Castanedo-Cázares, J.P. Hyperpigmentation and atrophy in folds as cutaneous manifestation in a case of mitochondrial myopathy. Dermatol. Online J. 2015, 21, 5. [Google Scholar]
- Onkoksoong, T.; Jeayeng, S.; Poungvarin, N.; Limsaengurai, S.; Thamsermsang, O.; Tripatara, P.; Akarasereenont, P.; Panich, U. Thai herbal antipyretic 22 formula (APF22) inhibits UVA-mediated melanogenesis through activation of Nrf2-regulated antioxidant defense. Phytother. Res. 2018, 32, 1546–1554. [Google Scholar] [CrossRef]
- Mi, T.; Dong, Y.; Santhanam, U.; Huang, N. Niacinamide and 12-hydroxystearic acid prevented benzo(a)pyrene and squalene peroxides induced hyperpigmentation in skin equivalent. Exp. Dermatol. 2019, 28, 742–746. [Google Scholar] [CrossRef]
- Nahhas, A.F.; Abdel-Malek, Z.A.; Kohli, I.; Braunberger, T.L.; Lim, H.W.; Hamzavi, I.H. The potential role of antioxidants in mitigating skin hyperpigmentation resulting from ultraviolet and visible light-induced oxidative stress. Photodermatol. Photoimmunol. Photomed. 2019, 35, 420–428. [Google Scholar] [CrossRef]
- Choubey, V.; Sarkar, R.; Garg, V.; Kaushik, S.; Ghunawat, S.; Sonthalia, S. Role of oxidative stress in melasma: A prospective study on serum and blood markers of oxidative stress in melasma patients. Int. J. Dermatol. 2017, 56, 939–943. [Google Scholar] [CrossRef]
- Sarkar, R.; Devadasan, S.; Choubey, V.; Goswami, B. Melatonin and oxidative stress in melisma—An unexplored territory; a prospective study. Int. J. Dermatol. 2020, 59, 572–575. [Google Scholar] [CrossRef]
- Nofal, A.; Ibrahim, A.M.; Nofal, E.; Gamal, N.; Osman, S. Topical silymarin versus hydroquinone in the treatment of melasma: A comparative study. J. Cosmet. Dermatol. 2019, 18, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Babbush, K.M.; Babbush, R.A.; Khachemoune, A. The Therapeutic Use of Antioxidants for Melasma. J. Drugs Dermatol. 2020, 19, 788–792. [Google Scholar] [CrossRef] [PubMed]
- Cheong, K.A.; Lee, A.Y. Guanine Deaminase Stimulates Ultraviolet-induced Keratinocyte Senescence in Seborrhoeic Keratosis via Guanine Metabolites. Acta. Derm. Venereol. 2020, 100, adv00109. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, A.; Yang, H.; Duffy, M.; Robinson, E.; Conrad-Antoville, A.; Lu, Y.-W.; Capps, T.; Braiterman, L.; Wolfgang, M.; Murphy, M.P.; et al. The Activity of Menkes Disease Protein ATP7A Is Essential for Redox Balance in Mitochondria. J. Biol. Chem. 2016, 291, 16644–16658. [Google Scholar] [CrossRef]
- Petris, M.J.; Strausak, D.; Mercer, J.F. The Menkes copper transporter is required for the activation of tyrosinase. Hum. Mol. Genet. 2000, 9, 2845–2851. [Google Scholar] [CrossRef]
- Sonam, K.; Khan, N.A.; Bindu, P.S.; Taly, A.B.; Gayathri, N.; Bharath, M.M.S.; Govindaraju, C.; Arvinda, H.R.; Nagappa, M.; Sinha, S.; et al. Clinical and magnetic resonance imaging findings in patients with Leigh syndrome and SURF1 mutations. Brain Dev. 2014, 36, 807–812. [Google Scholar] [CrossRef]
- Kakourou, T.; Garoufi, A.; Nicolaidou, P.; Dafni, E.; Tsamouri, M.; Papadimitriou, A.; Karpathios, T. Kearns Sayre syndrome initially presenting as hypomelanosis of Ito. Arch. Dis. Child. 1999, 81, 280–281. [Google Scholar]
- Vafaee, T.; Rokos, H.; Salem, M.M.; Schallreuter, K.U. In vivo and in vitro evidence for epidermal H2O2-mediated oxidative stress in piebaldism. Exp. Dermatol. 2010, 19, 883–887. [Google Scholar] [CrossRef]
- Xie, H.; Zhou, F.; Liu, L.; Zhu, G.; Li, Q.; Li, C.; Gao, T. Vitiligo: How do oxidative stress-induced autoantigens trigger autoimmunity? J. Dermatol. Sci. 2016, 81, 3–9. [Google Scholar] [CrossRef]
- Wang, Y.; Li, S.; Li, C. Perspectives of New Advances in the Pathogenesis of Vitiligo: From Oxidative Stress to Autoimmunity. Med. Sci. Monit. 2019, 25, 1017–1023. [Google Scholar] [CrossRef]
- Speeckaert, R.; Dugardin, J.; Lambert, J.; Lapeere, H.; Verhaeghe, E.; Speeckaert, M.M.; van Geel, N. Critical appraisal of the oxidative stress pathway in vitiligo: A systematic review and meta-analysis. J. Eur. Acad. Dermatol. Venereol. 2018, 32, 1089–1098. [Google Scholar] [CrossRef]
- Shi., M.-H.; Wu, Y.; Li, L.; Cai, Y.-F.; Liu, M.; Gao, X.-H.; Chen, H.-D. Meta-analysis of the association between vitiligo and the level of superoxide dismutase or malondialdehyde. Clin. Exp. Dermatol. 2017, 42, 21–29. [Google Scholar] [CrossRef]
- Mansuri, M.S.; Jadeja, S.D.; Singh, M.; Laddha, N.C.; Dwivedi, M.; Begum, R. The catalase gene promoter and 5′-untranslated region variants lead to altered gene expression and enzyme activity in vitiligo. Br. J. Dermatol. 2017, 177, 1590–1600. [Google Scholar] [CrossRef]
- Caputo, V.; Niceta, M.; Fiorella, S.; Vecchia, M.L.; Bastonini, E.; Bongiorno, M.R.; Pistone, G. Vitiligo susceptibility and catalase gene polymorphisms in Sicilian population. G. Ital. Dermatol. Venereol. 2018, 153, 619–623. [Google Scholar] [CrossRef] [PubMed]
- Ochoa-Ramírez, L.A.; Díaz-Camacho, S.P.; Becerra-Loaiza, D.S.; Verdugo-Nieto, L.; Muñoz-Estrada, V.F.; Servín-Vázquez, L.A.; Osuna-Ramírez, I.; Rodríguez-Millán, J.; Velarde-Félix, J.S. Catalase but not vitamin D receptor gene polymorphisms are associated with nonsegmental vitiligo in Northwestern Mexicans. Int. J. Dermatol. 2019, 58, 1264–1269. [Google Scholar] [CrossRef] [PubMed]
- Vaccaro, M.; Bagnato, G.; Cristani, M.; Borgia, F.; Spatari, G.; Tigano, V.; Saja, A.; Guarneri, F.; Cannavò, S.P.; Gangemi, S. Oxidation products are increased in patients affected by non-segmental generalized vitiligo. Arch. Dermatol. Res. 2017, 309, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Li, K.; Liu, L.; Wang, X.; Jian, Z.; Zhang, W.; Wang, G.; Li, C.; Gao, T. Genetic polymorphism of the Nrf2 promoter region is associated with vitiligo risk in Han Chinese populations. J. Cell Mol. Med. 2016, 20, 1840–1850. [Google Scholar] [CrossRef]
- Kim, H.; Park, C.S.; Lee, A.Y. Reduced Nrf2 activation in PI3K phosphorylation-impaired vitiliginous keratinocytes increases susceptibility to ROS-generating chemical-induced apoptosis. Environ. Toxicol. 2017, 32, 2481–2491. [Google Scholar] [CrossRef]
- Arowojolu, O.A.; Orlow, S.J.; Elbuluk, N.; Manga, P. The nuclear factor (erythroid-derived 2)-like 2 (NRF2) antioxidant response promotes melanocyte viability and reduces toxicity of the vitiligo-inducing phenol monobenzone. Exp. Dermatol. 2017, 26, 637–644. [Google Scholar] [CrossRef]
- Vaseghi, H.; Houshmand, M.; Jadali, Z. Increased levels of mitochondrial DNA copy number in patients with vitiligo. Clin. Exp. Dermatol. 2017, 42, 749–754. [Google Scholar] [CrossRef]
- Yi, X.; Guo, W.; Shi, Q.; Yang, Y.; Zhang, W.; Chen, X.; Kang, P.; Chen, J.; Cui, T.; Ma, J.; et al. SIRT3-Dependent Mitochondrial Dynamics Remodeling Contributes to Oxidative Stress-Induced Melanocyte Degeneration in Vitiligo. Theranostics 2019, 9, 1614–1633. [Google Scholar] [CrossRef]
- Kang, P.; Zhang, W.; Chen, X.; Yi, X.; Song, P.; Chang, Y.; Zhang, S.; Gao, T.; Li, C.; Li, S. TRPM2 mediates mitochondria-dependent apoptosis of melanocytes under oxidative stress. Free Radic. Biol. Med. 2018, 126, 259–268. [Google Scholar] [CrossRef]
- Qiao, Z.; Wang, X.; Xiang, L.; Zhang, C. Dysfunction of Autophagy: A Possible Mechanism Involved in the Pathogenesis of Vitiligo by Breaking the Redox Balance of Melanocytes. Oxid. Med. Cell Longev. 2016, 2016, 3401570. [Google Scholar] [CrossRef]
- He, Y.; Li, S.; Zhang, W.; Dai, W.; Cui, T.; Wang, G.; Gao, T.; Li, C. Dysregulated autophagy increased melanocyte sensitivity to H2O2-induced oxidative stress in vitiligo. Sci. Rep. 2017, 7, 42394. [Google Scholar] [CrossRef]
- Moriwaki, S.; Takahashi, Y. Photoaging and DNA repair. J. Dermatol. Sci. 2008, 50, 169–176. [Google Scholar] [CrossRef]
- Ashapkin, V.V.; Kutueva, L.I.; Kurchashova, S.Y.; Kireev, I.I. Are There Common Mechanisms Between the Hutchinson-Gilford Progeria Syndrome and Natural Aging? Front. Genet. 2019, 10, 455. [Google Scholar] [CrossRef]
- Foo, M.X.R.; Ong, P.F.; Dreesen, O. Premature aging syndromes: From patients to mechanism. J. Dermatol. Sci. 2019, 96, 58–65. [Google Scholar] [CrossRef]
- Cestari, T.; Buster, K. Photoprotection in specific populations: Children and people of color. J. Am. Acad. Dermatol. 2017, 76, S110–S121. [Google Scholar] [CrossRef]
- Lu, L.; Jin, W.; Wang, L.L. Aging in Rothmund-Thomson syndrome and related RECQL4 genetic disorders. Ageing Res. Rev. 2017, 33, 30–35. [Google Scholar] [CrossRef]
- Walsh, M.F.; Chang, V.Y.; Kohlmann, W.K.; Scott, H.S.; Cunniff, C.; Bourdeaut, F.; Molenaar, J.J.; Porter, C.C.; Sandlund, J.T.; Plon, S.E.; et al. Recommendations for Childhood Cancer Screening and Surveillance in DNA Repair Disorders. Clin. Cancer Res. 2017, 23, e23–e31. [Google Scholar] [CrossRef]
- Lehmann, A.R.; McGibbon, D.; Stefanini, M. Xeroderma pigmentosum. Orphanet. J. Rare Dis. 2011, 6, 70. [Google Scholar] [CrossRef]
- Che, R.; Zhang, J.; Nepal, M.; Han, B.; Fei, P. Multifaceted Fanconi Anemia Signaling. Trends Genet. 2018, 34, 171–183. [Google Scholar] [CrossRef]
- Ruggiero, J.L.; Dodds, M.; Freese, R.; Polcari, I.C.; Maguiness, S.; Hook, K.P.; Boull, C. Cutaneous Findings in Fanconi Anemia. J. Am. Acad. Dermatol. 2020. [Google Scholar] [CrossRef]
- Esteve-Puig, R.; Gil, R.; González-Sánchez, E.; Bech-Serra, J.J.; Grueso, J.; Hernández-Losa, J.; Moliné, T.; Canals, F.; Ferrer, B.; Cortés, J.; et al. A mouse model uncovers LKB1 as an UVB-induced DNA damage sensor mediating CDKN1A (p21WAF1/CIP1) degradation. PLoS Genet. 2014, 10, e1004721. [Google Scholar] [CrossRef]
- Nguyen, N.T.; Fisher, D.E. MITF and UV responses in skin: From pigmentation to addiction. Pigment Cell Melanoma Res. 2019, 32, 224–236. [Google Scholar] [CrossRef]
- Jarrett, S.G.; Horrell, E.M.W.; Boulanger, M.C.; D’Orazio, J.A. Defining the Contribution of MC1R Physiological Ligands to ATR Phosphorylation at Ser435, a Predictor of DNA Repair in Melanocytes. J. Investig. Dermatol. 2015, 135, 3086–3095. [Google Scholar] [CrossRef]
- Takata, M.; Shirasaki, F.; Nakatani, T.; Takehara, K. Hereditary non-polyposis colorectal cancer associated with disseminated superficial porokeratosis. Microsatellite instability in skin tumours. Br. J. Dermatol. 2000, 143, 851–855. [Google Scholar] [CrossRef]
- Donze, S.H.; Codd, V.; Damen, L.; Goedegebuure, W.J.; Denniff, M.; Samani, N.J.; van der Velden, J.A.E.M.; Hokken-Koelega, A.C.S. Evidence for Accelerated Biological Aging in Young Adults with Prader-Willi Syndrome. J. Clin. Endocrinol. Metab. 2020, 105, 2053–2059. [Google Scholar] [CrossRef]
- Ehrhart, F.; Janssen, K.J.M.; Coort, S.L.; Evelo, C.T.; Curfs, L.M.G. Prader-Willi syndrome and Angelman syndrome: Visualisation of the molecular pathways for two chromosomal disorders. World J. Biol. Psychiatry 2019, 20, 670–682. [Google Scholar] [CrossRef]
- Seward, S.L., Jr.; Gahl, W.A. Hermansky-Pudlak syndrome: Health care throughout life. Pediatrics 2013, 132, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Kamaraj, B.; Purohit, R. Mutational analysis of oculocutaneous albinism: A compact review. Biomed. Res. Int. 2014, 2014, 905472. [Google Scholar] [CrossRef]
- Ando, H.; Ichihashi, M.; Hearing, V.J. Role of the ubiquitin proteasome system in regulating skin pigmentation. Int. J. Mol. Sci. 2009, 10, 4428–4434. [Google Scholar] [CrossRef]
- Hossy, B.H.; da Costa Leitão, A.A.; Luz, F.B.; dos Santos, E.P.; Allodi, S.; de Pádula, M.; de Oliveira Miguel, N.C. Effects of a sunscreen formulation on albino hairless mice: A morphological approach. Arch. Dermatol. Res. 2013, 305, 535–544. [Google Scholar] [CrossRef]
- Aydin, A.F.; Aydıngöz, I.E.; Doğru-Abbasoğlu, S.; Vural, P.; Uysal, M. Association of Leukotrichia in Vitiligo and Asp148Glu Polymorphism of Apurinic/Apyrimidinic Endonuclease 1. Int. J. Trichol. 2017, 9, 171–176. [Google Scholar]
- Mangaonkar, A.A.; Patnaik, M.M. Short Telomere syndromes in clinical practice: Bridging bench and bedside. Mayo Clin. Proc. 2018, 93, 904–916. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.S.; Dreesen, O. Biomarkers of Cellular Senescence and Skin Aging. Front. Genet. 2018, 9, 247. [Google Scholar] [CrossRef]
- Opresko, P.L.; Shay, J.W. Telomere-associated aging disorders. Ageing Res. Rev. 2017, 33, 52–66. [Google Scholar] [CrossRef] [PubMed]
- AlSabbagh, M.M. Dyskeratosis congenita: A literature review. J. Dtsch. Dermatol. Ges. 2020, 18, 943–967. [Google Scholar] [CrossRef] [PubMed]
- Stout, G.J.; Blasco, M.A. Telomere length and telomerase activity impact the UV sensitivity syndrome xeroderma pigmentosum C. Cancer Res. 2013, 73, 1844–1854. [Google Scholar] [CrossRef]
- Abdollahi, M.; Gao, M.Y.M.; Munoz, D.G. Distinct pattern of neostriatal calcifications in dyskeratosis congenita: A case report and literature review. Clin. Neuropathol. 2018, 37, 277–282. [Google Scholar] [CrossRef]
- Chojnowski, A.; Ong, P.F.; Wong, E.S.M.; Lim, J.S.Y.; Mutalif, R.A.; Navasankari, R.; Dutta, B.; Yang, H.; Liow, Y.Y.; Sze, S.K.; et al. Progerin reduces LAP2α-telomere association in Hutchinson-Gilford progeria. eLife 2015, 4, e07759. [Google Scholar] [CrossRef]
- Li, Y.; Zhou, G.; Bruno, I.G.; Cooke, J.P. Telomerase mRNA Reverses Senescence in Progeria Cells. J. Am. Coll. Cardiol. 2017, 70, 804–805. [Google Scholar] [CrossRef]
- Rork, J.F.; Huang, J.T.; Gordon, L.B.; Kleinman, M.; Kieran, M.W.; Liang, M.G. Initial cutaneous manifestations of Hutchinson-Gilford progeria syndrome. Pediatr. Dermatol. 2014, 31, 196–202. [Google Scholar] [CrossRef]
- Moftah, N.H.; El-Barbary, E.; Rashed, L.; El-Sammad, N. Assessment of telomerase activity in nonsegmental vitiligo tissue: A pilot study. Clin. Exp. Dermatol. 2019, 44, 747–752. [Google Scholar] [CrossRef]
- Bocheva, G.; Slominski, R.M.; Slominski, A.T. Neuroendocrine Aspects of Skin Aging. Int. J. Mol. Sci. 2019, 20, 2798. [Google Scholar] [CrossRef]
- Rusanova, I.; Martínez-Ruiz, L.; Florido, J.; Rodríguez-Santana, C.; Guerra-Librero, A.; Acuña-Castroviejo, D.; Escames, G. Protective Effects of Melatonin on the Skin: Future Perspectives. Int. J. Mol. Sci. 2019, 20, 4948. [Google Scholar] [CrossRef]
- Scheuer, C. Melatonin for prevention of erythema and oxidative stress in response to ultraviolet radiation. Dan. Med. J. 2017, 64, B5358. [Google Scholar]
- Skobowiat, C.; Brożyna, A.A.; Janjetovic, Z.; Jeayeng, S.; Oak, A.S.W.; Kim, T.K.; Panich, U.; Reiter, R.J.; Slominski, A.T. Melatonin and its derivatives counteract the ultraviolet B radiation-induced damage in human and porcine skin ex vivo. J. Pineal Res. 2018, 65, e12501. [Google Scholar] [CrossRef]
- Seleit, I.; Bakry, O.A.; Repey, H.S.E.; Ali, R. Intrinsic versus Extrinsic Aging: A Histopathological, Morphometric and Immunohistochemical Study of Estrogen Receptor β and Androgen Receptor. Skin Pharmacol. Physiol. 2016, 29, 178–189. [Google Scholar] [CrossRef]
- Draelos, Z.D. A Double-Blind Randomized Pilot Study Evaluating the Safety and Efficacy of Topical MEP in the Facial Appearance Improvement of Estrogen Deficient Females. J. Drugs Dermatol. 2018, 17, 1186–1189. [Google Scholar]
- Röck, K.; Joosse, S.A.; Müller, J.; Heinisch, N.; Fuchs, N.; Meusch, M.; Zipper, P.; Reifenberger, J.; Pantel, K.; Fischer, J.W. Chronic UVB-irradiation actuates perpetuated dermal matrix remodeling in female mice: Protective role of estrogen. Sci. Rep. 2016, 6, 30482. [Google Scholar] [CrossRef]
- Cohen, J.L. Evaluation of Efficacy of a Skin Care Regimen Containing Methyl Estradiolpropanoate (MEP) for Treating Estrogen Deficient Skin. J. Drugs Dermatol. 2019, 18, 1226–1230. [Google Scholar]
- Cario, M. How hormones may modulate human skin pigmentation in melasma: An in vitro perspective. Exp. Dermatol. 2019, 28, 709–718. [Google Scholar] [CrossRef]
- Mobasher, P.; Foulad, D.P.; Raffi, J.; Zachary, C.; Fackler, N.; Zohuri, N.; Juhasz, M.; Mesinkovska, N.A. Catamenial Hyperpigmentation: A Review. J. Clin. Aesthet. Dermatol. 2020, 13, 18–21. [Google Scholar] [PubMed]
- Patel, S.; Rauf, A.; Khan, H.; Meher, B.R.; Hassan, S.S.U. A holistic review on the autoimmune disease vitiligo with emphasis on the causal factors. Biomed. Pharmacother. 2017, 92, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Paolino, G.; Bearzi, P.; Mercuri, S.R. Onset of vitiligo in a patient with acquired secondary hypogonadism under treatment with testosterone gel 2%: Inside the pathogenesis. An. Bras. Dermatol. 2020, 95, 661–662. [Google Scholar] [CrossRef] [PubMed]
- Gürpınar, A.; Günaydın, S.D.; Kılıç, C.; Karaduman, A. Association of serum cortisol and dehydroepiandrosterone sulfate (DHEAS) levels with psychological stress in patients with vitiligo. Turk. J. Med. Sci. 2019, 49, 832–837. [Google Scholar] [CrossRef] [PubMed]
- Kalfalah, F.; Janke, L.; Schiavi, A.; Tigges, J.; Ix, A.; Ventura, N.; Boege, F.; Reinke, H. Crosstalk of clock gene expression and autophagy in aging. Aging 2016, 8, 1876–1895. [Google Scholar] [CrossRef] [PubMed]
- Jeong, D.; Qomaladewi, N.P.; Lee, J.; Park, S.H.; Cho, J.Y. The Role of Autophagy in Skin Fibroblasts, Keratinocytes, Melanocytes, and Epidermal Stem Cells. J. Investig. Dermatol. 2020, 140, 1691–1697. [Google Scholar] [CrossRef] [PubMed]
- Majora, M.; Sondenheimer, K.; Knechten, M.; Uthe, I.; Esser, C.; Schiavi, A.; Ventura, N.; Krutmann, J. HDAC inhibition improves autophagic and lysosomal function to prevent loss of subcutaneous fat in a mouse model of Cockayne syndrome. Sci. Transl. Med. 2018, 10, eaam7510. [Google Scholar] [CrossRef]
- Huang, Y.; Li, Y.; Qu, Y.; Zheng, Y.; Ouyang, M.; Zhang, Y.; Lai, W.; Xu, Q. UVA-induced photoaging inhibits autophagic degradation by impairing lysosomal function in dermal fibroblasts. Biochem. Biophys. Res. Commun. 2019, 518, 611–618. [Google Scholar] [CrossRef]
- Endo, K.; Katsuyama, Y.; Taira, N.; Yoshioka, M.; Okano, Y.; Masaki, H. Impairment of the autophagy system in repetitively UVA-irradiated fibroblasts. Photodermatol. Photoimmunol. Photomed. 2020, 36, 111–117. [Google Scholar] [CrossRef]
- Song, X.; Narzt, M.S.; Nagelreiter, I.M.; Hohensinner, P.; Terlecki-Zaniewicz, L.; Tschachler, E.; Grillari, J.; Gruber, F. Autophagy deficient keratinocytes display increased DNA damage, senescence and aberrant lipid composition after oxidative stress in vitro and in vivo. Redox Biol. 2017, 11, 219–230. [Google Scholar] [CrossRef]
- Li, Y.F.; Ouyang, S.H.; Tu, L.F.; Wang, X.; Yuan, W.L.; Wang, G.E.; Wu, Y.P.; Duan, W.J.; Yu, H.M.; Fang, Z.Z.; et al. Caffeine Protects Skin from Oxidative Stress-Induced Senescence through the Activation of Autophagy. Theranostics. 2018, 8, 5713–5730. [Google Scholar] [CrossRef]
- Zhu, W.; Zhao, Z.; Cheng, B. The role of autophagy in skin pigmentation. Eur. J. Dermatol. 2020, 30, 655–662. [Google Scholar]
- Murase, D.; Hachiya, A.; Fullenkamp, R.; Beck, A.; Moriwaki, S.; Hase, T.; Takema, Y.; Manga, P. Variation in Hsp70-1A Expression Contributes to Skin Color Diversity. J. Investig. Dermatol. 2016, 136, 1681–1691. [Google Scholar] [CrossRef]
- Kim, N.H.; Choi, S.H.; Yi, N.; Lee, T.R.; Lee, A.Y. Arginase-2, a miR-1299 target, enhances pigmentation in melasma by reducing melanosome degradation via senescence-induced autophagy inhibition. Pigment. Cell Melanoma Res. 2017, 30, 521–530. [Google Scholar] [CrossRef]
- Cho, Y.H.; Park, J.E.; Lim, D.S.; Lee, J.S. Tranexamic acid inhibits melanogenesis by activating the autophagy system in cultured melanoma cells. J. Dermatol. Sci. 2017, 88, 96–102. [Google Scholar] [CrossRef]
- Lee, K.W.; Ryu, H.W.; Oh, S.S.; Park, S.; Madhi, H.; Yoo, J.; Park, K.H.; Kim, K.D. Depigmentation of α-melanocyte-stimulating hormone-treated melanoma cells by β-mangostin is mediated by selective autophagy. Exp. Dermatol. 2017, 26, 585–591. [Google Scholar] [CrossRef]
- Katsuyama, Y.; Taira, N.; Yoshioka, M.; Okano, Y.; Masaki, H. 3-O-Glyceryl-2-O-hexyl Ascorbate Suppresses Melanogenesis through Activation of the Autophagy System. Biol. Pharm. Bull. 2018, 41, 824–827. [Google Scholar] [CrossRef]
- Park, H.J.; Jo, D.S.; Choi, H.; Ji-Eun Bae, J.-E.; Park, N.Y.; Kim, J.B.; Choi, J.Y.; Kim, Y.H.; Oh, G.S.; Chang, J.H.; et al. Melasolv induces melanosome autophagy to inhibit pigmentation in B16F1 cells. PLoS ONE 2020, 15, e0239019. [Google Scholar] [CrossRef]
- Chen, L.; Xu, Z.; Jiang, M.; Zhang, C.; Wang, X.; Xiang, L. Light-emitting diode 585 nm photomodulation inhibiting melanin synthesis and inducing autophagy in human melanocytes. J. Dermatol. Sci. 2018, 89, 11–18. [Google Scholar] [CrossRef]
- Byrne, S.; Dionisi-Vici, C.; Smith, L.; Gautel, M.; Jungbluth, H. Vici syndrome: A review. Orphanet. J. Rare Dis. 2016, 11, 21. [Google Scholar] [CrossRef]
- Hori, I.; Otomo, T.; Nakashima, M.; Miya, F.; Negishi, Y.; Shiraishi, H.; Nonoda, Y.; Magara, S.; Tohyama, J.; Okamoto, N.; et al. Defects in autophagosome-lysosome fusion underlie Vici syndrome, a neurodevelopmental disorder with multisystem involvement. Sci. Rep. 2017, 7, 3552. [Google Scholar] [CrossRef]
- Cao, J.; Tyburczy, M.E.; Moss, J.; Darling, T.N.; Widlund, H.R.; Kwiatkowski, D.J. Tuberous sclerosis complex inactivation disrupts melanogenesis via mTORC1 activation. J. Clin. Investig. 2017, 127, 349–364. [Google Scholar] [CrossRef]
- Yang, F.; Yang, L.; Wataya-Kaneda, M.; Hasegawa, J.; Yoshimori, T.; Tanemura, A.; Tsuruta, D.; Katayama, I. Dysregulation of autophagy in melanocytes contributes to hypopigmented macules in tuberous sclerosis complex. J. Dermatol. Sci. 2018, 89, 155–164. [Google Scholar] [CrossRef]
- Yang, C.-F.; Lin, S.-P.; Chiang, C.-P.; Wu, Y.-H.; H’ng, W.S.; Chang, C.-P.; Chen, Y.-T.; Wu, J.-Y. Loss of GPNMB Causes Autosomal-Recessive Amyloidosis Cutis Dyschromica in Humans. Am. J. Hum. Genet. 2018, 102, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Hoashi, T.; Sato, S.; Yamaguchi, Y.; Passeron, T.; Tamaki, K.; Hearing, V.J. Glycoprotein nonmetastatic melanoma protein b, a melanocytic cell marker, is a melanosome-specific and proteolytically released protein. FASEB J. 2010, 24, 1616–1629. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Liu, W.; Zhu, C.; Yuan, X.; Li, D.; Gu, W.; Ma, H.; Xie, X.; Gao, T. Silencing of GPNMB by siRNA inhibits the formation of melanosomes in melanocytes in a MITF-independent fashion. PLoS ONE 2012, 7, e42955. [Google Scholar] [CrossRef] [PubMed]
- Biswas, K.B.; Takahashi, A.; Mizutani, Y.; Takayama, S.; Ishitsuka, S.; Yang, L.; Yang, F.; Iddamalgoda, A.; Katayama, I.; Inoue, S. GPNMB is expressed in human epidermal keratinocytes but disappears in the vitiligo lesional skin. Sci. Rep. 2020, 10, 4930. [Google Scholar] [CrossRef]
- Grönniger, E.; Weber, B.; Heil, O.; Peters, N.; Stäb, F.; Wenck, H.; Korn, B.; Winnefeld, M.; Lyko, F. Aging and chronic sun exposure cause distinct epigenetic changes in human skin. PLoS Genet. 2010, 6, e1000971. [Google Scholar] [CrossRef]
- Sturm, G.; Cardenas, A.; Bind, M.-A.; Horvath, S.; Wang, S.; Wang, Y.; Hägg, S.; Hirano, M.; Picard, M. Human aging DNA methylation signatures are conserved but accelerated in cultured fibroblasts. Epigenetics 2019, 14, 961–976. [Google Scholar] [CrossRef]
- Gerasymchuk, M.; Cherkasova, V.; Kovalchuk, O.; Kovalchuk, I. The Role of microRNAs in Organismal and Skin Aging. Int. J. Mol. Sci. 2020, 21, 5281. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).