
A Case of Double Standard: Sex Differences in Multiple Sclerosis Risk Factors

 , , ,
, , , 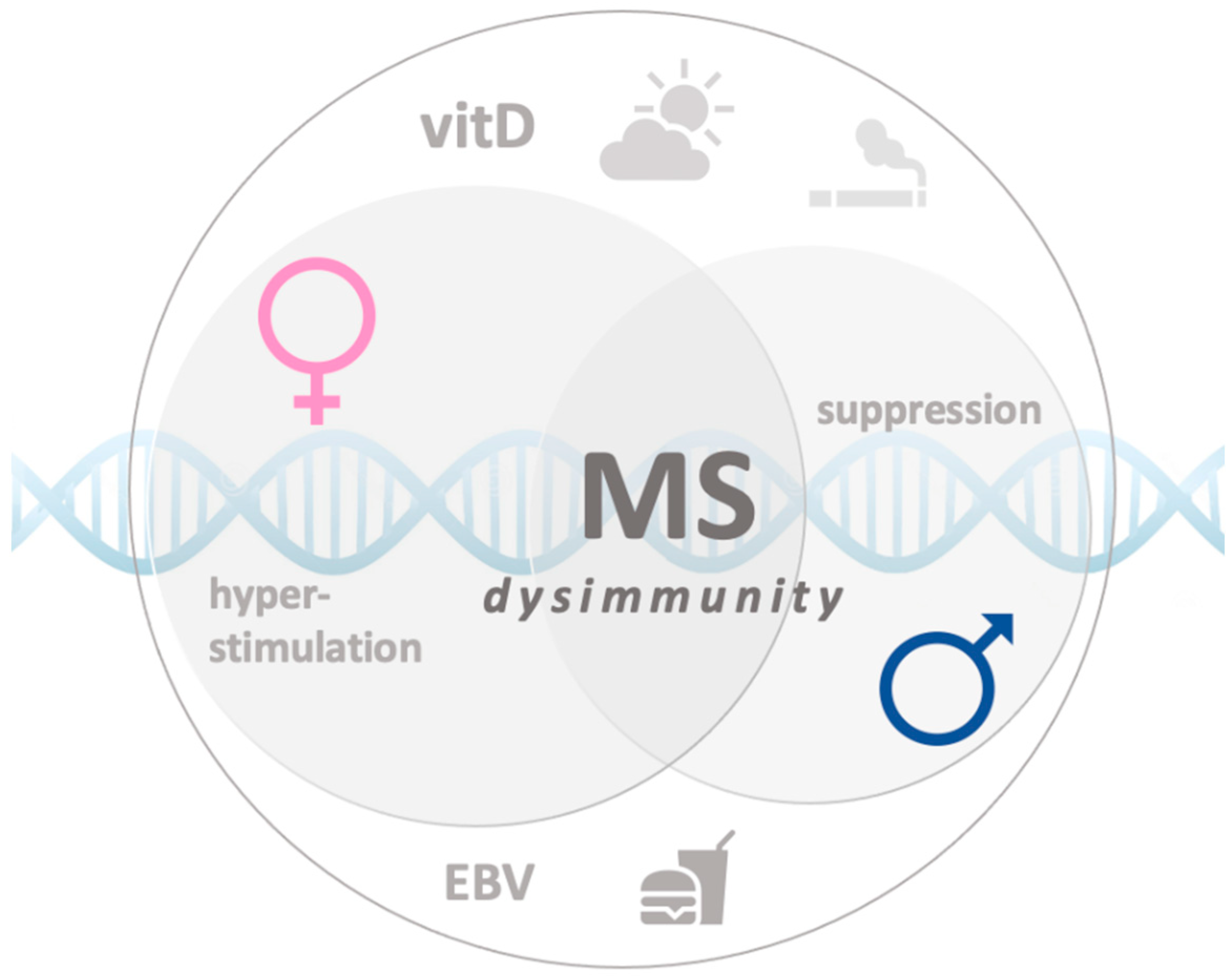{kind=link}
Abstract
1. Introduction
2. Risk Factors
2.1. Environmental Factors
2.1.1. Epstein–Barr Virus
2.1.2. Cigarette Smoke
2.1.3. Lack of Sun Exposure and Low Vitamin D
2.1.4. Obesity
2.2. Genetic Factors
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Baecher-Allan, C.; Kaskow, B.J.; Weiner, H.L. Multiple Sclerosis: Mechanisms and Immunotherapy. Neuron 2018, 97, 742–768. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.; Short, S.E. Gender Differences in Biological Function in Young Adulthood: An Intragenerational Perspective. RSF 2018, 4, 98–119. [Google Scholar] [CrossRef]
- Sadovnick, A.D. European Charcot Foundation Lecture: The natural history of multiple sclerosis and gender. J. Neurol. Sci. 2009, 286, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Ramagopalan, S.V.; Byrnes, J.K.; Orton, S.M.; Dyment, D.A.; Guimond, C.; Yee, I.M.; Ebers, G.C.; Sadovnick, A.D. Sex ratio of multiple sclerosis and clinical phenotype. Eur. J. Neurol. 2010, 17, 634–637. [Google Scholar] [CrossRef] [PubMed]
- Ysrraelit, M.C.; Correale, J. Impact of sex hormones on immune function and multiple sclerosis development. Immunology 2019, 156, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Gilli, F.; DiSano, K.D.; Pachner, A.R. SeXX Matters in Multiple Sclerosis. Front. Neurol. 2020, 11, 616. [Google Scholar] [CrossRef] [PubMed]
- Chitnis, T. Role of puberty in multiple sclerosis risk and course. Clin. Immunol. 2013, 149, 192–200. [Google Scholar] [CrossRef]
- D’hooghe, M.B.; Haentjens, P.; Nagels, G.; D’Hooghe, T.; De Keyser, J. Menarche, oral contraceptives, pregnancy and progression of disability in relapsing onset and progressive onset multiple sclerosis. J. Neurol. 2012, 259, 855–861. [Google Scholar] [CrossRef]
- Sloka, J.S.; Pryse-Phillips, W.E.; Stefanelli, M. The relation between menarche and the age of first symptoms in a multiple sclerosis cohort. Mult. Scler. 2006, 12, 333–339. [Google Scholar] [CrossRef]
- Kalincik, T.; Vivek, V.; Jokubaitis, V.; Lechner-Scott, J.; Trojano, M.; Izquierdo, G.; Lugaresi, A.; Grand’maison, F.; Hupperts, R.; Oreja-Guevara, C.; et al. Sex as a determinant of relapse incidence and progressive course of multiple sclerosis. Brain 2013, 136, 3609–3617. [Google Scholar] [CrossRef]
- Pozzilli, C.; Tomassini, V.; Marinelli, F.; Paolillo, A.; Gasperini, C.; Bastianello, S. ’Gender gap’ in multiple sclerosis: Magnetic resonance imaging evidence. Eur. J. Neurol. 2003, 10, 95–97. [Google Scholar] [CrossRef]
- Schoonheim, M.M.; Popescu, V.; Rueda Lopes, F.C.; Wiebenga, O.T.; Vrenken, H.; Douw, L.; Polman, C.H.; Geurts, J.J.; Barkhof, F. Subcortical atrophy and cognition: Sex effects in multiple sclerosis. Neurology 2012, 79, 1754–1761. [Google Scholar] [CrossRef]
- Dimitrijević, M.; Arsenović-Ranin, N.; Kosec, D.; Bufan, B.; Nacka-Aleksić, M.; Pilipović, I.; Leposavić, G. Sex differences in Tfh cell help to B cells contribute to sexual dimorphism in severity of rat collagen-induced arthritis. Sci. Rep. 2020, 10, 1214. [Google Scholar] [CrossRef] [PubMed]
- Kovats, S. Estrogen receptors regulate innate immune cells and signaling pathways. Cell. Immunol. 2015, 294, 63–69. [Google Scholar] [CrossRef]
- Airas, L. Hormonal and gender-related immune changes in multiple sclerosis. Acta Neurol. Scand. 2015, 132, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Gold, S.M.; Voskuhl, R.R. Pregnancy and multiple sclerosis: From molecular mechanisms to clinical application. Semin. Immunopathol. 2016, 38, 709–718. [Google Scholar] [CrossRef] [PubMed]
- Patas, K.; Engler, J.B.; Friese, M.A.; Gold, S.M. Pregnancy and multiple sclerosis: Feto-maternal immune cross talk and its implications for disease activity. J. Reprod. Immunol. 2013, 97, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Jalkanen, A.; Alanen, A.; Airas, L.; Group, F.M.S.a.P.S. Pregnancy outcome in women with multiple sclerosis: Results from a prospective nationwide study in Finland. Mult. Scler. 2010, 16, 950–955. [Google Scholar] [CrossRef]
- Vukusic, S.; Hutchinson, M.; Hours, M.; Moreau, T.; Cortinovis-Tourniaire, P.; Adeleine, P.; Confavreux, C.; Group, P.I.M.S. Pregnancy and multiple sclerosis (the PRIMS study): Clinical predictors of post-partum relapse. Brain 2004, 127, 1353–1360. [Google Scholar] [CrossRef] [PubMed]
- Handel, A.E.; Williamson, A.J.; Disanto, G.; Handunnetthi, L.; Giovannoni, G.; Ramagopalan, S.V. An updated meta-analysis of risk of multiple sclerosis following infectious mononucleosis. PLoS ONE 2010, 5. [Google Scholar] [CrossRef]
- Ascherio, A.; Munger, K.L. Environmental risk factors for multiple sclerosis. Part I: The role of infection. Ann. Neurol. 2007, 61, 288–299. [Google Scholar] [CrossRef]
- Nielsen, T.R.; Rostgaard, K.; Nielsen, N.M.; Koch-Henriksen, N.; Haahr, S.; Sørensen, P.S.; Hjalgrim, H. Multiple sclerosis after infectious mononucleosis. Arch. Neurol. 2007, 64, 72–75. [Google Scholar] [CrossRef] [PubMed]
- Levin, L.I.; Munger, K.L.; O’Reilly, E.J.; Falk, K.I.; Ascherio, A. Primary infection with the Epstein-Barr virus and risk of multiple sclerosis. Ann. Neurol. 2010, 67, 824–830. [Google Scholar] [CrossRef] [PubMed]
- Biström, M.; Jons, D.; Engdahl, E.; Gustafsson, R.; Huang, J.; Brenner, N.; Butt, J.; Alonso-Magdalena, L.; Gunnarsson, M.; Vrethem, M.; et al. Epstein-Barr virus infection after adolescence and human herpesvirus 6A as risk factors for multiple sclerosis. Eur. J. Neurol. 2021, 28, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Sundström, P.; Nyström, M.; Ruuth, K.; Lundgren, E. Antibodies to specific EBNA-1 domains and HLA DRB1*1501 interact as risk factors for multiple sclerosis. J. Neuroimmunol. 2009, 215, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Mescheriakova, J.Y.; van Nierop, G.P.; van der Eijk, A.A.; Kreft, K.L.; Hintzen, R.Q. EBNA-1 titer gradient in families with multiple sclerosis indicates a genetic contribution. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7. [Google Scholar] [CrossRef] [PubMed]
- Beecham, A.H.; Patsopoulos, N.A.; Xifara, D.K.; Davis, M.F.; Kemppinen, A.; Cotsapas, C.; Shah, T.S.; Spencer, C.; Booth, D.; Goris, A.; et al. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat. Genet. 2013, 45, 1353–1360. [Google Scholar] [CrossRef]
- Ricigliano, V.A.; Handel, A.E.; Sandve, G.K.; Annibali, V.; Ristori, G.; Mechelli, R.; Cader, M.Z.; Salvetti, M. EBNA2 binds to genomic intervals associated with multiple sclerosis and overlaps with vitamin D receptor occupancy. PLoS ONE 2015, 10, e0119605. [Google Scholar] [CrossRef]
- Harley, J.B.; Chen, X.; Pujato, M.; Miller, D.; Maddox, A.; Forney, C.; Magnusen, A.F.; Lynch, A.; Chetal, K.; Yukawa, M.; et al. Transcription factors operate across disease loci, with EBNA2 implicated in autoimmunity. Nat. Genet. 2018, 50, 699–707. [Google Scholar] [CrossRef]
- Mechelli, R.; Manzari, C.; Policano, C.; Annese, A.; Picardi, E.; Umeton, R.; Fornasiero, A.; D’Erchia, A.M.; Buscarinu, M.C.; Agliardi, C.; et al. Epstein-Barr virus genetic variants are associated with multiple sclerosis. Neurology 2015, 84, 1362–1368. [Google Scholar] [CrossRef]
- Hauser, S.L.; Waubant, E.; Arnold, D.L.; Vollmer, T.; Antel, J.; Fox, R.J.; Bar-Or, A.; Panzara, M.; Sarkar, N.; Agarwal, S.; et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N. Engl. J. Med. 2008, 358, 676–688. [Google Scholar] [CrossRef] [PubMed]
- Klein, G.; Klein, E.; Kashuba, E. Interaction of Epstein-Barr virus (EBV) with human B-lymphocytes. Biochem. Biophys. Res. Commun. 2010, 396, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Bengali, Z.H.; Das, S.P.; Middleton, M.B.; Levine, P.H. Seroepidemiology of Epstein--Barr virus-associated diseases--I. A pilot evaluation using a radiometric quantitative complement fixation test. Comp. Immunol. Microbiol. Infect. Dis. 1979, 2, 213–220. [Google Scholar] [CrossRef]
- Ford, J.L.; Stowe, R.P. Racial-ethnic differences in Epstein-Barr virus antibody titers among U.S. children and adolescents. Ann. Epidemiol. 2013, 23, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Wagner, H.J.; Hornef, M.; Teichert, H.M.; Kirchner, H. Sex difference in the serostatus of adults to the Epstein-Barr virus. Immunobiology 1994, 190, 424–429. [Google Scholar] [CrossRef]
- Butterworth, M.; McClellan, B.; Allansmith, M. Influence of sex in immunoglobulin levels. Nature 1967, 214, 1224–1225. [Google Scholar] [CrossRef]
- Sue, K. The science behind “man flu”. BMJ 2017, 359, j5560. [Google Scholar] [CrossRef]
- Voskuhl, R. Sex differences in autoimmune diseases. Biol. Sex Differ. 2011, 2, 1. [Google Scholar] [CrossRef]
- Jones, B.G.; Sealy, R.E.; Penkert, R.R.; Surman, S.L.; Maul, R.W.; Neale, G.; Xu, B.; Gearhart, P.J.; Hurwitz, J.L. Complex sex-biased antibody responses: Estrogen receptors bind estrogen response elements centered within immunoglobulin heavy chain gene enhancers. Int. Immunol. 2019, 31, 141–156. [Google Scholar] [CrossRef]
- Hernán, M.A.; Jick, S.S.; Logroscino, G.; Olek, M.J.; Ascherio, A.; Jick, H. Cigarette smoking and the progression of multiple sclerosis. Brain 2005, 128, 1461–1465. [Google Scholar] [CrossRef]
- Alrouji, M.; Manouchehrinia, A.; Gran, B.; Constantinescu, C.S. Effects of cigarette smoke on immunity, neuroinflammation and multiple sclerosis. J. Neuroimmunol. 2019, 329, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Öckinger, J.; Hagemann-Jensen, M.; Kullberg, S.; Engvall, B.; Eklund, A.; Grunewald, J.; Piehl, F.; Olsson, T.; Wahlström, J. T-cell activation and HLA-regulated response to smoking in the deep airways of patients with multiple sclerosis. Clin. Immunol. 2016, 169, 114–120. [Google Scholar] [CrossRef]
- Petecchia, L.; Sabatini, F.; Varesio, L.; Camoirano, A.; Usai, C.; Pezzolo, A.; Rossi, G.A. Bronchial airway epithelial cell damage following exposure to cigarette smoke includes disassembly of tight junction components mediated by the extracellular signal-regulated kinase 1/2 pathway. Chest 2009, 135, 1502–1512. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Xie, J.; Wu, C.; Xiao, G. Correlation Between Smoking and Passive Smoking with Multiple Sclerosis and the Underlying Molecular Mechanisms. Med Sci. Monit. 2019, 25, 893–902. [Google Scholar] [CrossRef] [PubMed]
- Sundström, P.; Nyström, L.; Hallmans, G. Smoke exposure increases the risk for multiple sclerosis. Eur. J. Neurol. 2008, 15, 579–583. [Google Scholar] [CrossRef] [PubMed]
- Mooney, L.A.; Perera, F.P.; Van Bennekum, A.M.; Blaner, W.S.; Karkoszka, J.; Covey, L.; Hsu, Y.; Cooper, T.B.; Frenkel, K. Gender differences in autoantibodies to oxidative DNA base damage in cigarette smokers. Cancer Epidemiol. Biomark. Prev. 2001, 10, 641–648. [Google Scholar]
- Exley, C. Combined effects of smoking, anti-EBNA antibodies, and HLA-DRB1 1501 on multiple sclerosis risk. Neurology 2010, 75, 752. [Google Scholar] [CrossRef]
- Handel, A.E.; Williamson, A.J.; Disanto, G.; Dobson, R.; Giovannoni, G.; Ramagopalan, S.V. Smoking and multiple sclerosis: An updated meta-analysis. PLoS ONE 2011, 6, e16149. [Google Scholar] [CrossRef]
- Westerlind, H.; Boström, I.; Stawiarz, L.; Landtblom, A.M.; Almqvist, C.; Hillert, J. New data identify an increasing sex ratio of multiple sclerosis in Sweden. Mult. Scler. 2014, 20, 1578–1583. [Google Scholar] [CrossRef]
- Palacios, N.; Alonso, A.; Brønnum-Hansen, H.; Ascherio, A. Smoking and increased risk of multiple sclerosis: Parallel trends in the sex ratio reinforce the evidence. Ann. Epidemiol. 2011, 21, 536–542. [Google Scholar] [CrossRef]
- Bikle, D.D. Vitamin D metabolism, mechanism of action, and clinical applications. Chem. Biol. 2014, 21, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Bivona, G.; Agnello, L.; Ciaccio, M. The immunological implication of the new vitamin D metabolism. Cent. Eur. J. Immunol. 2018, 43, 331–334. [Google Scholar] [CrossRef]
- Lemire, J.M.; Adams, J.S.; Sakai, R.; Jordan, S.C. 1 alpha,25-dihydroxyvitamin D3 suppresses proliferation and immunoglobulin production by normal human peripheral blood mononuclear cells. J. Clin. Investig. 1984, 74, 657–661. [Google Scholar] [CrossRef] [PubMed]
- Ascherio, A.; Munger, K.L.; White, R.; Köchert, K.; Simon, K.C.; Polman, C.H.; Freedman, M.S.; Hartung, H.P.; Miller, D.H.; Montalbán, X.; et al. Vitamin D as an early predictor of multiple sclerosis activity and progression. JAMA Neurol. 2014, 71, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Verdoia, M.; Schaffer, A.; Barbieri, L.; Di Giovine, G.; Marino, P.; Suryapranata, H.; De Luca, G.; (NAS), N.A.S.G. Impact of gender difference on vitamin D status and its relationship with the extent of coronary artery disease. Nutr. Metab. Cardiovasc. Dis. 2015, 25, 464–470. [Google Scholar] [CrossRef] [PubMed]
- Lips, P. Vitamin D deficiency and secondary hyperparathyroidism in the elderly: Consequences for bone loss and fractures and therapeutic implications. Endocr. Rev. 2001, 22, 477–501. [Google Scholar] [CrossRef]
- Dong, J.; Wong, S.L.; Lau, C.W.; Lee, H.K.; Ng, C.F.; Zhang, L.; Yao, X.; Chen, Z.Y.; Vanhoutte, P.M.; Huang, Y. Calcitriol protects renovascular function in hypertension by down-regulating angiotensin II type 1 receptors and reducing oxidative stress. Eur. Hear. J. 2012, 33, 2980–2990. [Google Scholar] [CrossRef]
- Hossein-nezhad, A.; Holick, M.F. Vitamin D for health: A global perspective. Mayo Clin. Proc. 2013, 88, 720–755. [Google Scholar] [CrossRef] [PubMed]
- Bivona, G.; Agnello, L.; Bellia, C.; Iacolino, G.; Scazzone, C.; Lo Sasso, B.; Ciaccio, M. Non-Skeletal Activities of Vitamin D: From Physiology to Brain Pathology. Medicina (Kaunas) 2019, 55, 341. [Google Scholar] [CrossRef]
- Harmon, Q.E.; Umbach, D.M.; Baird, D.D. Use of Estrogen-Containing Contraception Is Associated With Increased Concentrations of 25-Hydroxy Vitamin D. J. Clin. Endocrinol. Metab. 2016, 101, 3370–3377. [Google Scholar] [CrossRef]
- Merhi, Z.; Doswell, A.; Krebs, K.; Cipolla, M. Vitamin D alters genes involved in follicular development and steroidogenesis in human cumulus granulosa cells. J. Clin. Endocrinol. Metab. 2014, 99, E1137–E1145. [Google Scholar] [CrossRef]
- Adams, J.S.; Hewison, M. Extrarenal expression of the 25-hydroxyvitamin D-1-hydroxylase. Arch. Biochem. Biophys. 2012, 523, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Parikh, G.; Varadinova, M.; Suwandhi, P.; Araki, T.; Rosenwaks, Z.; Poretsky, L.; Seto-Young, D. Vitamin D regulates steroidogenesis and insulin-like growth factor binding protein-1 (IGFBP-1) production in human ovarian cells. Horm. Metab. Res. 2010, 42, 754–757. [Google Scholar] [CrossRef] [PubMed]
- Harmon, Q.E.; Kissell, K.; Jukic, A.M.Z.; Kim, K.; Sjaarda, L.; Perkins, N.J.; Umbach, D.M.; Schisterman, E.F.; Baird, D.D.; Mumford, S.L. Vitamin D and Reproductive Hormones Across the Menstrual Cycle. Hum. Reprod. 2020, 35, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Scazzone, C.; Agnello, L.; Bivona, G.; Lo Sasso, B.; Ciaccio, M. Vitamin D and Genetic Susceptibility to Multiple Sclerosis. Biochem. Genet. 2021, 59, 1–30. [Google Scholar] [CrossRef]
- Stone, T.W.; McPherson, M.; Gail Darlington, L. Obesity and Cancer: Existing and New Hypotheses for a Causal Connection. EBioMedicine 2018, 30, 14–28. [Google Scholar] [CrossRef] [PubMed]
- Bibiloni, M.e.M.; Pons, A.; Tur, J.A. Prevalence of overweight and obesity in adolescents: A systematic review. ISRN Obes. 2013, 2013, 392747. [Google Scholar] [CrossRef]
- Inoue, Y.; Qin, B.; Poti, J.; Sokol, R.; Gordon-Larsen, P. Epidemiology of Obesity in Adults: Latest Trends. Curr. Obes. Rep. 2018, 7, 276–288. [Google Scholar] [CrossRef]
- Brown, L.M.; Gent, L.; Davis, K.; Clegg, D.J. Metabolic impact of sex hormones on obesity. Brain Res. 2010, 1350, 77–85. [Google Scholar] [CrossRef]
- Bralić, I.; Tahirović, H.; Matanić, D.; Vrdoljak, O.; Stojanović-Spehar, S.; Kovacić, V.; Blazeković-Milaković, S. Association of early menarche age and overweight/obesity. J. Pediatr. Endocrinol. Metab. 2012, 25, 57–62. [Google Scholar] [CrossRef]
- Adami, F.; Benedet, J.; Takahashi, L.A.R.; da Silva Lopes, A.; da Silva Paiva, L.; de Vasconcelos, F.A.G. Association between pubertal development stages and body adiposity in children and adolescents. Health Qual. Life Outcomes 2020, 18, 93. [Google Scholar] [CrossRef]
- Munger, K.L.; Chitnis, T.; Ascherio, A. Body size and risk of MS in two cohorts of US women. Neurology 2009, 73, 1543–1550. [Google Scholar] [CrossRef]
- De Rosa, V.; Procaccini, C.; Calì, G.; Pirozzi, G.; Fontana, S.; Zappacosta, S.; La Cava, A.; Matarese, G. A key role of leptin in the control of regulatory T cell proliferation. Immunity 2007, 26, 241–255. [Google Scholar] [CrossRef]
- Link, J.C.; Reue, K. Genetic Basis for Sex Differences in Obesity and Lipid Metabolism. Annu. Rev. Nutr. 2017, 37, 225–245. [Google Scholar] [CrossRef]
- Matarese, G.; Di Giacomo, A.; Sanna, V.; Lord, G.M.; Howard, J.K.; Di Tuoro, A.; Bloom, S.R.; Lechler, R.I.; Zappacosta, S.; Fontana, S. Requirement for leptin in the induction and progression of autoimmune encephalomyelitis. J. Immunol. 2001, 166, 5909–5916. [Google Scholar] [CrossRef]
- Park, H.K.; Ahima, R.S. Physiology of leptin: Energy homeostasis, neuroendocrine function and metabolism. Metabolism 2015, 64, 24–34. [Google Scholar] [CrossRef]
- Martínez-Sánchez, N. There and Back Again: Leptin Actions in White Adipose Tissue. Int. J. Mol. Sci. 2020, 21, 6039. [Google Scholar] [CrossRef] [PubMed]
- Hedström, A.K.; Olsson, T.; Alfredsson, L. High body mass index before age 20 is associated with increased risk for multiple sclerosis in both men and women. Mult. Scler. 2012, 18, 1334–1336. [Google Scholar] [CrossRef] [PubMed]
- Karastergiou, K.; Smith, S.R.; Greenberg, A.S.; Fried, S.K. Sex differences in human adipose tissues-the biology of pear shape. Biol. Sex Differ. 2012, 3, 13. [Google Scholar] [CrossRef] [PubMed]
- Manolopoulos, K.N.; Karpe, F.; Frayn, K.N. Gluteofemoral body fat as a determinant of metabolic health. Int. J. Obes. 2010, 34, 949–959. [Google Scholar] [CrossRef] [PubMed]
- Fish, E.N. The X-files in immunity: Sex-based differences predispose immune responses. Nat. Rev. Immunol. 2008, 8, 737–744. [Google Scholar] [CrossRef]
- Souyris, M.; Cenac, C.; Azar, P.; Daviaud, D.; Canivet, A.; Grunenwald, S.; Pienkowski, C.; Chaumeil, J.; Mejía, J.E.; Guéry, J.C. TLR7 escapes X chromosome inactivation in immune cells. Sci. Immunol. 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Tukiainen, T.; Villani, A.C.; Yen, A.; Rivas, M.A.; Marshall, J.L.; Satija, R.; Aguirre, M.; Gauthier, L.; Fleharty, M.; Kirby, A.; et al. Landscape of X chromosome inactivation across human tissues. Nature 2017, 550, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y.; Golden, L.C.; Itoh, N.; Matsukawa, M.A.; Ren, E.; Tse, V.; Arnold, A.P.; Voskuhl, R.R. The X-linked histone demethylase Kdm6a in CD4+ T lymphocytes modulates autoimmunity. J. Clin. Investig. 2019, 129, 3852–3863. [Google Scholar] [CrossRef] [PubMed]
- Smith-Bouvier, D.L.; Divekar, A.A.; Sasidhar, M.; Du, S.; Tiwari-Woodruff, S.K.; King, J.K.; Arnold, A.P.; Singh, R.R.; Voskuhl, R.R. A role for sex chromosome complement in the female bias in autoimmune disease. J. Exp. Med. 2008, 205, 1099–1108. [Google Scholar] [CrossRef]
- Teuscher, C.; Noubade, R.; Spach, K.; McElvany, B.; Bunn, J.Y.; Fillmore, P.D.; Zachary, J.F.; Blankenhorn, E.P. Evidence that the Y chromosome influences autoimmune disease in male and female mice. Proc. Natl. Acad. Sci. USA 2006, 103, 8024–8029. [Google Scholar] [CrossRef]
- Palaszynski, K.M.; Smith, D.L.; Kamrava, S.; Burgoyne, P.S.; Arnold, A.P.; Voskuhl, R.R. A yin-yang effect between sex chromosome complement and sex hormones on the immune response. Endocrinology 2005, 146, 3280–3285. [Google Scholar] [CrossRef]
- Felderhoff-Mueser, U.; Schmidt, O.I.; Oberholzer, A.; Bührer, C.; Stahel, P.F. IL-18: A key player in neuroinflammation and neurodegeneration? Trends Neurosci. 2005, 28, 487–493. [Google Scholar] [CrossRef]
- Losy, J.; Niezgoda, A. IL-18 in patients with multiple sclerosis. Acta Neurol. Scand. 2001, 104, 171–173. [Google Scholar] [CrossRef]
- Gregg, C.; Zhang, J.; Butler, J.E.; Haig, D.; Dulac, C. Sex-specific parent-of-origin allelic expression in the mouse brain. Science 2010, 329, 682–685. [Google Scholar] [CrossRef]
- Zeft, A.; Shear, E.S.; Thompson, S.D.; Glass, D.N.; Prahalad, S. Familial autoimmunity: Maternal parent-of-origin effect in juvenile idiopathic arthritis. Clin. Rheumatol. 2008, 27, 241–244. [Google Scholar] [CrossRef]
- Barbé, L.; Lanni, S.; López-Castel, A.; Franck, S.; Spits, C.; Keymolen, K.; Seneca, S.; Tomé, S.; Miron, I.; Letourneau, J.; et al. CpG Methylation, a Parent-of-Origin Effect for Maternal-Biased Transmission of Congenital Myotonic Dystrophy. Am. J. Hum. Genet. 2017, 100, 488–505. [Google Scholar] [CrossRef] [PubMed]
- Ramagopalan, S.V.; Yee, I.M.; Dyment, D.A.; Orton, S.M.; Marrie, R.A.; Sadovnick, A.D.; Ebers, G.C.; Group, C.C.S. Parent-of-origin effect in multiple sclerosis: Observations from interracial matings. Neurology 2009, 73, 602–605. [Google Scholar] [CrossRef] [PubMed]
- Herrera, B.M.; Ramagopalan, S.V.; Lincoln, M.R.; Orton, S.M.; Chao, M.J.; Sadovnick, A.D.; Ebers, G.C. Parent-of-origin effects in MS: Observations from avuncular pairs. Neurology 2008, 71, 799–803. [Google Scholar] [CrossRef] [PubMed]
- Ebers, G.C.; Sadovnick, A.D.; Dyment, D.A.; Yee, I.M.; Willer, C.J.; Risch, N. Parent-of-origin effect in multiple sclerosis: Observations in half-siblings. Lancet 2004, 363, 1773–1774. [Google Scholar] [CrossRef]
- Sadovnick, A.D.; Yee, I.M.; Guimond, C.; Reis, J.; Dyment, D.A.; Ebers, G.C. Age of onset in concordant twins and other relative pairs with multiple sclerosis. Am. J. Epidemiol. 2009, 170, 289–296. [Google Scholar] [CrossRef]
- Voskuhl, R.R.; Sawalha, A.H.; Itoh, Y. Sex chromosome contributions to sex differences in multiple sclerosis susceptibility and progression. Mult. Scler. 2018, 24, 22–31. [Google Scholar] [CrossRef]
- Stürner, K.H.; Siembab, I.; Schön, G.; Stellmann, J.P.; Heidari, N.; Fehse, B.; Heesen, C.; Eiermann, T.H.; Martin, R.; Binder, T.M. Is multiple sclerosis progression associated with the HLA-DR15 haplotype? Mult. Scler. J. Exp. Transl. Clin. 2019, 5, 2055217319894615. [Google Scholar] [CrossRef]
- Bahrami, T.; Taheri, M.; Javadi, S.; Omrani, M.D.; Karimipour, M. Expression Analysis of Long Non-coding RNA Lnc-DC in HLA-DRB1*15:01-Negative Patients with Multiple Sclerosis: A Probable Cause for Gender Differences in Multiple Sclerosis Susceptibility? J. Mol. Neurosci. 2020, 71, 821–825. [Google Scholar] [CrossRef]
- Consortium, I.M.S.G. Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science 2019, 365. [Google Scholar] [CrossRef]
- Gershoni, M.; Pietrokovski, S. The landscape of sex-differential transcriptome and its consequent selection in human adults. BMC Biol. 2017, 15, 7. [Google Scholar] [CrossRef] [PubMed]
- Consortium, G. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 2020, 369, 1318–1330. [Google Scholar] [CrossRef] [PubMed]
- Lenz, K.M.; McCarthy, M.M. A starring role for microglia in brain sex differences. Neuroscientist 2015, 21, 306–321. [Google Scholar] [CrossRef] [PubMed]
- Rahimian, R.; Cordeau, P.; Kriz, J. Brain Response to Injuries: When Microglia Go Sexist. Neuroscience 2019, 405, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Saijo, K.; Glass, C.K. Microglial cell origin and phenotypes in health and disease. Nat. Rev. Immunol. 2011, 11, 775–787. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Angeloni, B.; Bigi, R.; Bellucci, G.; Mechelli, R.; Ballerini, C.; Romano, C.; Morena, E.; Pellicciari, G.; Reniè, R.; Rinaldi, V.; et al. A Case of Double Standard: Sex Differences in Multiple Sclerosis Risk Factors. Int. J. Mol. Sci. 2021, 22, 3696. https://doi.org/10.3390/ijms22073696
Angeloni B, Bigi R, Bellucci G, Mechelli R, Ballerini C, Romano C, Morena E, Pellicciari G, Reniè R, Rinaldi V, et al. A Case of Double Standard: Sex Differences in Multiple Sclerosis Risk Factors. International Journal of Molecular Sciences. 2021; 22(7):3696. https://doi.org/10.3390/ijms22073696
Chicago/Turabian StyleAngeloni, Benedetta, Rachele Bigi, Gianmarco Bellucci, Rosella Mechelli, Chiara Ballerini, Carmela Romano, Emanuele Morena, Giulia Pellicciari, Roberta Reniè, Virginia Rinaldi, and et al. 2021. "A Case of Double Standard: Sex Differences in Multiple Sclerosis Risk Factors" International Journal of Molecular Sciences 22, no. 7: 3696. https://doi.org/10.3390/ijms22073696
APA StyleAngeloni, B., Bigi, R., Bellucci, G., Mechelli, R., Ballerini, C., Romano, C., Morena, E., Pellicciari, G., Reniè, R., Rinaldi, V., Buscarinu, M. C., Romano, S., Ristori, G., & Salvetti, M. (2021). A Case of Double Standard: Sex Differences in Multiple Sclerosis Risk Factors. International Journal of Molecular Sciences, 22(7), 3696. https://doi.org/10.3390/ijms22073696