
The RARγ Oncogene: An Achilles Heel for Some Cancers


Abstract
1. Introduction
2. The Concept of the Cancer Stem Cell
3. Targeted Therapies for Cancer
4. RARγ Is a Fundamental Control on Stem Cell Behavior
5. RARγ Is an Oncogene for Some Cancers
6. Antagonising RARγ Kills Cancer Stem Cells
7. The Oncogenic Action of RARγ
8. Concluding Remarks
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Dick, J.E. Stem cell concepts renew cancer research. Blood 2008, 112, 4793–4807. [Google Scholar] [CrossRef]
- Greaves, M.F.; Maia, A.T.; Wiemels, J.L.; Ford, A.M. Leukemia in twins: Lessons in natural history. Blood 2003, 102, 2321–2333. [Google Scholar] [CrossRef] [PubMed]
- Holyoake, T.; Jiang, X.; Eaves, C.; Eaves, A. Isolation of a highly quiescent subpopulation of primitive leukemic cells in chronic myeloid leukemia. Blood 1999, 94, 2056–2064. [Google Scholar] [CrossRef] [PubMed]
- Corbin, A.S.; Agarwal, A.; Loriaux, M.; Cortes, J.; Deininger, M.W.; Druker, B.J. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J. Clin. Investig. 2011, 121, 396–409. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.M.; Jorgensen, H.G.; Allan, E.; Pearson, C.; Alcorn, M.J.; Richmond, L.; Holyoake, T.L. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood 2002, 99, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Lim, Z.; Brand, R.; Martino, R.; van Biezen, A.; Finke, J.; Bacigalupo, A.; Beelen, D.; Devergie, A.; Alessandrino, E.; Willemze, R.; et al. Allogeneic hematopoietic stem-cell transplantation for patients 50 years or older with myelodysplastic syndromes or secondary acute myeloid leukemia. J. Clin. Oncol. 2010, 28, 405–411. [Google Scholar] [CrossRef]
- Wilkinson, A.C.; Igarashi, K.J.; Nakauchi, H. Haematopoietic stem cell self-renewal in vivo and ex vivo. Nat. Rev. Genet. 2020, 21, 541–554. [Google Scholar] [CrossRef]
- Vetrie, D.; Helgason, G.V.; Copland, M. The leukaemia stem cell: Similarities, differences and clinical prospects in CML and AML. Nat. Rev. Cancer 2020, 20, 158–173. [Google Scholar] [CrossRef]
- Robey, P.G.; Kuznetsov, S.A.; Bianco, P.; Riminucci, M. Bone marrow stromal cell assays: In vitro and in vivo. Methods Mol. Biol. 2021, 2230, 379–396. [Google Scholar]
- Sabbath, K.D.; Ball, E.D.; Larcom, P.; Davis, R.B.; Griffin, J.D. Heterogeneity of clonogenic cells in acute myeloblastic leukemia. J. Clin. Investig. 1985, 75, 746–753. [Google Scholar] [CrossRef]
- Lemieux, M.E.; Rebel, V.I.; Lansdorp, P.M.; Eaves, C.J. Characterization and purification of a primitive hematopoietic cell type in adult mouse marrow capable of lymphomyeloid differentiation in long-term marrow “switch” cultures. Blood 1995, 86, 1339–1347. [Google Scholar] [CrossRef] [PubMed]
- Rongvaux, A.; Takizawa, H.; Strowig, T.; Willinger, T.; Eynon, E.E.; Flavell, R.A.; Manz, M.G. Human hemato-lymphoid system mice: Current use and future potential for medicine. Annu. Rev. Immunol. 2013, 31, 635–674. [Google Scholar] [CrossRef] [PubMed]
- Gbyli, R.; Song, Y.; Halene, S. Humanized mice as preclinical models for myeloid malignancies. Biochem. Pharmacol. 2020, 174, 113794. [Google Scholar] [CrossRef]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef]
- Ailles, L.E.; Weissman, I.L. Cancer stem cells in solid tumors. Curr. Opin. Biotechnol. 2007, 18, 460–466. [Google Scholar] [CrossRef]
- Dalerba, P.; Dylla, S.J.; Park, I.K.; Liu, R.; Wang, X.; Cho, R.W.; Hoey, T.; Gurney, A.; Huang, E.H.; Simeone, D.M.; et al. Phenotypic characterization of human colorectal cancer stem cells. Proc. Natl. Acad. Sci. USA 2007, 104, 10158–10163. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Prince, M.E.; Sivanandan, R.; Kaczorowski, A.; Wolf, G.T.; Kaplan, M.J.; Dalerba, P.; Weissman, I.L.; Clarke, M.F.; Ailles, L.E. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 2007, 104, 973–978. [Google Scholar] [CrossRef]
- Quintana, E.; Shackleton, M.; Sabel, M.S.; Fullen, D.R.; Johnson, T.M.; Morrison, S.J. Efficient tumour formation by single human melanoma cells. Nature 2008, 456, 593–598. [Google Scholar] [CrossRef]
- Fang, D.; Nguyen, T.K.; Leishear, K.; Finko, R.; Kulp, A.N.; Hotz, S.; Van Belle, P.A.; Xu, X.; Elder, D.E.; Herlyn, M. A tumorigenic subpopulation with stem cell properties in melanomas. Cancer Res. 2005, 65, 9328–9337. [Google Scholar] [CrossRef]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Liu, R.; Wang, X.; Chen, G.Y.; Dalerba, P.; Gurney, A.; Hoey, T.; Sherlock, G.; Lewicki, J.; Shedden, K.; Clarke, M.F. The prognostic role of a gene signature from tumorigenic breast-cancer cells. N. Engl. J. Med. 2007, 356, 217–226. [Google Scholar] [CrossRef]
- Baudino, T.A. Targeted cancer therapy: The next generation of cancer treatment. Curr. Drug Discov. Technol. 2015, 12, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Costello, R.T.; Mallet, F.; Gaugler, B.; Sainty, D.; Arnoulet, C.; Gastaut, J.A.; Olive, D. Human acute myeloid leukemia CD34+/CD38- progenitor cells have decreased sensitivity to chemotherapy and Fas-induced apoptosis, reduced immunogenicity, and impaired dendritic cell transformation capacities. Cancer Res. 2000, 60, 4403–4411. [Google Scholar] [PubMed]
- Guzman, M.L.; Neering, S.J.; Upchurch, D.; Grimes, B.; Howard, D.S.; Rizzieri, D.A.; Luger, S.M.; Jordan, C.T. Nuclear factor-kappaB is constitutively activated in primitive human acute myelogenous leukemia cells. Blood 2001, 98, 2301–2307. [Google Scholar] [CrossRef] [PubMed]
- Matthews, W.; Jordan, C.T.; Wiegand, G.W.; Pardoll, D.; Lemischka, I.R. A receptor tyrosine kinase specific to hematopoietic stem and progenitor cell-enriched populations. Cell 1991, 65, 1143–1152. [Google Scholar] [CrossRef]
- Small, D.; Levenstein, M.; Kim, E.; Carow, C.; Amin, S.; Rockwell, P.; Witte, L.; Burrow, C.; Ratajczak, M.Z.; Gewirtz, A.M.; et al. STK-1, the human homolog of Flk-2/Flt-3, is selectively expressed in CD34+ human bone marrow cells and is involved in the proliferation of early progenitor/stem cells. Proc. Natl. Acad. Sci. USA 1994, 91, 459–463. [Google Scholar] [CrossRef]
- Tsapogas, P.; Swee, L.K.; Nusser, A.; Nuber, N.; Kreuzaler, M.; Capoferri, G.; Rolink, H.; Ceredig, R.; Rolink, A. In vivo evidence for an instructive role of fms-like tyrosine kinase-3 (FLT3) ligand in hematopoietic development. Haematologica 2014, 99, 638–646. [Google Scholar] [CrossRef]
- Small, D. Targeting FLT3 for the treatment of leukemia. Semin. Hematol. 2008, 45 (Suppl. S2), S17–S21. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.; Schlenk, R.F.; Russell, N.H.; Levis, M.J. Targeting FLT3 mutations in AML: Review of current knowledge and evidence. Leukemia 2019, 33, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Petrie, K.; Zelent, A.; Waxman, S. Differentiation therapy of acute myeloid leukemia: Past, present and future. Curr. Opin. Hematol. 2009, 16, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Edwards, R.H.; Wasik, M.A.; Finan, J.; Rodriguez, R.; Moore, J.; Kamoun, M.; Rennert, H.; Bird, J.; Nowell, P.C.; Salhany, K.E. Evidence for early hematopoietic progenitor cell involvement in acute promyelocytic leukemia. Am. J. Clin. Pathol. 1999, 112, 819–827. [Google Scholar] [CrossRef]
- Grimwade, D.; Mistry, A.R.; Solomon, E.; Guidez, F. Acute promyelocytic leukemia: A paradigm for differentiation therapy. Cancer Treat. Res. 2010, 145, 219–235. [Google Scholar]
- Sanz, M.A.; Fenaux, P.; Tallman, M.S.; Estey, E.H.; Lowenberg, B.; Naoe, T.; Lengfelder, E.; Dohner, H.; Burnett, A.K.; Chen, S.J.; et al. Management of acute promyelocytic leukemia: Updated recommendations from an expert panel of the European LeukemiaNet. Blood 2019, 133, 1630–1643. [Google Scholar] [CrossRef]
- Martens, J.H.; Brinkman, A.B.; Simmer, F.; Francoijs, K.J.; Nebbioso, A.; Ferrara, F.; Altucci, L.; Stunnenberg, H.G. PML-RARalpha/RXR alters the epigenetic landscape in acute promyelocytic Leukemia. Cancer Cell 2010, 17, 173–185. [Google Scholar] [CrossRef]
- Huynh, T.T.; Sultan, M.; Vidovic, D.; Dean, C.A.; Cruickshank, B.M.; Lee, K.; Loung, C.Y.; Holloway, R.W.; Hoskin, D.W.; Waisman, D.M.; et al. Retinoic acid and arsenic trioxide induce lasting differentiation and demethylation of target genes in APL cells. Sci. Rep. 2019, 9, 9414. [Google Scholar] [CrossRef]
- Wass, M.; Gollner, S.; Besenbeck, B.; Schlenk, R.F.; Mundmann, P.; Gothert, J.R.; Noppeney, R.; Schliemann, C.; Mikesch, J.H.; Lenz, G.; et al. A proof of concept phase I/II pilot trial of LSD1 inhibition by tranylcypromine combined with ATRA in refractory/relapsed AML patients not eligible for intensive therapy. Leukemia 2021, 35, 701–711. [Google Scholar] [CrossRef]
- Schenk, T.; Chen, W.C.; Gollner, S.; Howell, L.; Jin, L.; Hebestreit, K.; Klein, H.U.; Popescu, A.C.; Burnett, A.; Mills, K.; et al. Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat. Med. 2012, 18, 605–611. [Google Scholar] [CrossRef]
- Marcu, L.G.; Marcu, D. In silico modelling of a cancer stem cell-targeting agent and its effects on tumour control during radiotherapy. Sci. Rep. 2016, 6, 32332. [Google Scholar] [CrossRef]
- Garcia, J.; Hurwitz, H.I.; Sandler, A.B.; Miles, D.; Coleman, R.L.; Deurloo, R.; Chinot, O.L. Bevacizumab (Avastin(R)) in cancer treatment: A review of 15 years of clinical experience and future outlook. Cancer Treat. Rev. 2020, 86, 102017. [Google Scholar] [CrossRef]
- Wagner, A.D.; Arnold, D.; Grothey, A.A.; Haerting, J.; Unverzagt, S. Anti-angiogenic therapies for metastatic colorectal cancer. Cochrane Database Syst. Rev. 2009, CD005392. [Google Scholar] [CrossRef]
- Arnold, D.; Stein, A. New developments in the second-line treatment of metastatic colorectal cancer: Potential place in therapy. Drugs 2013, 73, 883–891. [Google Scholar] [CrossRef]
- Jaszai, J.; Schmidt, M.H.H. Trends and challenges in tumor anti-angiogenic therapies. Cells 2019, 8, 1102. [Google Scholar] [CrossRef] [PubMed]
- Sartor, O. Androgen deprivation—Continuous, intermittent, or none at all? N. Engl. J. Med. 2012, 367, 945–946. [Google Scholar] [CrossRef]
- Schroder, F.; Crawford, E.D.; Axcrona, K.; Payne, H.; Keane, T.E. Androgen deprivation therapy: Past, present and future. BJU Int. 2012, 109 (Suppl. S6), 1–12. [Google Scholar] [CrossRef] [PubMed]
- Awad, D.; Pulliam, T.L.; Lin, C.; Wilkenfeld, S.R.; Frigo, D.E. Delineation of the androgen-regulated signaling pathways in prostate cancer facilitates the development of novel therapeutic approaches. Curr. Opin. Pharmacol. 2018, 41, 1–11. [Google Scholar] [CrossRef]
- Schubert, M.; Gibert, Y. Retinoids in embryonic development. Biomolecules 2020, 10, 1278. [Google Scholar] [CrossRef] [PubMed]
- Rhinn, M.; Dolle, P. Retinoic acid signalling during development. Development 2012, 139, 843–858. [Google Scholar] [CrossRef]
- Shannon, S.R.; Moise, A.R.; Trainor, P.A. New insights and changing paradigms in the regulation of vitamin A metabolism in development. Wiley Interdiscip. Rev. Dev. Biol. 2017, 6, e264. [Google Scholar] [CrossRef]
- Rochette-Egly, C. Retinoic acid signaling and mouse embryonic stem cell differentiation: Cross talk between genomic and non-genomic effects of RA. Biochim. Biophys. Acta 2015, 1851, 66–75. [Google Scholar] [CrossRef]
- Mendoza-Parra, M.A.; Walia, M.; Sankar, M.; Gronemeyer, H. Dissecting the retinoid-induced differentiation of F9 embryonal stem cells by integrative genomics. Mol. Syst. Biol. 2011, 7, 538. [Google Scholar] [CrossRef] [PubMed]
- Purton, L.E.; Dworkin, S.; Olsen, G.H.; Walkley, C.R.; Fabb, S.A.; Collins, S.J.; Chambon, P. RARgamma is critical for maintaining a balance between hematopoietic stem cell self-renewal and differentiation. J. Exp. Med. 2006, 203, 1283–1293. [Google Scholar] [CrossRef] [PubMed]
- Chee, L.C.; Hendy, J.; Purton, L.E.; McArthur, G.A. The granulocyte-colony stimulating factor receptor (G-CSFR) interacts with retinoic acid receptors (RARs) in the regulation of myeloid differentiation. J. Leukoc. Biol. 2013, 93, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Green, A.C.; Poulton, I.J.; Vrahnas, C.; Hausler, K.D.; Walkley, C.R.; Wu, J.Y.; Martin, T.J.; Gillespie, M.T.; Chandraratna, R.A.; Quinn, J.M.; et al. RARgamma is a negative regulator of osteoclastogenesis. J. Steroid Biochem. Mol. Biol. 2015, 150, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Wai, H.A.; Kawakami, K.; Wada, H.; Muller, F.; Vernallis, A.B.; Brown, G.; Johnson, W.E. The development and growth of tissues derived from cranial neural crest and primitive mesoderm is dependent on the ligation status of retinoic acid receptor gamma: Evidence that retinoic acid receptor gamma functions to maintain stem/progenitor cells in the absence of retinoic acid. Stem Cells Dev. 2015, 24, 507–519. [Google Scholar]
- Boylan, J.F.; Lohnes, D.; Taneja, R.; Chambon, P.; Gudas, L.J. Loss of retinoic acid receptor gamma function in F9 cells by gene disruption results in aberrant Hoxa-1 expression and differentiation upon retinoic acid treatment. Proc. Natl. Acad. Sci. USA 1993, 90, 9601–9605. [Google Scholar] [CrossRef]
- Chatagnon, A.; Veber, P.; Morin, V.; Bedo, J.; Triqueneaux, G.; Semon, M.; Laudet, V.; d’Alche-Buc, F.; Benoit, G. RAR/RXR binding dynamics distinguish pluripotency from differentiation associated cis-regulatory elements. Nucleic Acids Res. 2015, 43, 4833–4854. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, V.; Laursen, K.B.; Brenet, F.; Viale, A.J.; Scandura, J.M.; Gudas, L.J. RARgamma is essential for retinoic acid induced chromatin remodeling and transcriptional activation in embryonic stem cells. J. Cell Sci. 2013, 126 Pt 4, 999–1008. [Google Scholar] [CrossRef]
- Garcia-Ramirez, I.; Bhatia, S.; Rodriguez-Hernandez, G.; Gonzalez-Herrero, I.; Walter, C.; Gonzalez de Tena-Davila, S.; Parvin, S.; Haas, O.; Woessmann, W.; Stanulla, M.; et al. Lmo2 expression defines tumor cell identity during T-cell leukemogenesis. EMBO J. 2018, 37, e98783. [Google Scholar] [CrossRef]
- Martin-Lorenzo, A.; Auer, F.; Chan, L.N.; Garcia-Ramirez, I.; Gonzalez-Herrero, I.; Rodriguez-Hernandez, G.; Bartenhagen, C.; Dugas, M.; Gombert, M.; Ginzel, S.; et al. Loss of Pax5 Exploits Sca1-BCR-ABL(p190) susceptibility to confer the metabolic shift essential for pB-ALL. Cancer Res. 2018, 78, 2669–2679. [Google Scholar] [CrossRef]
- Perez-Caro, M.; Cobaleda, C.; Gonzalez-Herrero, I.; Vicente-Duenas, C.; Bermejo-Rodriguez, C.; Sanchez-Beato, M.; Orfao, A.; Pintado, B.; Flores, T.; Sanchez-Martin, M.; et al. Cancer induction by restriction of oncogene expression to the stem cell compartment. EMBO J. 2009, 28, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Kellaway, S.G.; Keane, P.; Edginton-White, B.; Regha, K.; Kennett, E.; Bonifer, C. Different mutant RUNX1 oncoproteins program alternate haematopoietic differentiation trajectories. Life Sci. Alliance 2021, 4. [Google Scholar] [CrossRef]
- Qian, P.; De Kumar, B.; He, X.C.; Nolte, C.; Gogol, M.; Ahn, Y.; Chen, S.; Li, Z.; Xu, H.; Perry, J.M.; et al. Retinoid-sensitive epigenetic regulation of the hoxb cluster maintains normal hematopoiesis and inhibits leukemogenesis. Cell Stem Cell 2018, 22, 740–754.e7. [Google Scholar] [CrossRef] [PubMed]
- Conserva, M.R.; Redavid, I.; Anelli, L.; Zagaria, A.; Specchia, G.; Albano, F. RARG gene dysregulation in acute myeloid leukemia. Front. Mol. Biosci. 2019, 6, 114. [Google Scholar] [CrossRef]
- Watts, J.M.; Perez, A.; Pereira, L.; Fan, Y.S.; Brown, G.; Vega, F.; Petrie, K.; Swords, R.T.; Zelent, A. A Case of AML characterized by a novel t(4;15)(q31;q22) translocation that confers a growth-stimulatory response to retinoid-based therapy. Int. J. Mol. Sci. 2017, 18, 1492. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.L.; Song, W.; Zhou, P.; Fu, Q.R.; Lin, C.L.; Chen, Q.X.; Shen, D.Y. Oncogenic retinoic acid receptor gamma knockdown reverses multi-drug resistance of human colorectal cancer via Wnt/beta-catenin pathway. Cell Cycle 2017, 16, 685–692. [Google Scholar] [CrossRef]
- Huang, G.L.; Luo, Q.; Rui, G.; Zhang, W.; Zhang, Q.Y.; Chen, Q.X.; Shen, D.Y. Oncogenic activity of retinoic acid receptor gamma is exhibited through activation of the Akt/NF-kappaB and Wnt/beta-catenin pathways in cholangiocarcinoma. Mol. Cell. Biol. 2013, 33, 3416–3425. [Google Scholar] [CrossRef]
- Yan, T.D.; Wu, H.; Zhang, H.P.; Lu, N.; Ye, P.; Yu, F.H.; Zhou, H.; Li, W.G.; Cao, X.; Lin, Y.Y.; et al. Oncogenic potential of retinoic acid receptor-gamma in hepatocellular carcinoma. Cancer Res. 2010, 70, 2285–2295. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, Q.; Zhang, Q.; Liu, L. MicroRNA-30a-5p suppresses proliferation, invasion and tumor growth of hepatocellular cancer cells via targeting FOXA1. Oncol. Lett. 2017, 14, 5018–5026. [Google Scholar] [CrossRef]
- Kudryavtseva, A.V.; Nyushko, K.M.; Zaretsky, A.R.; Shagin, D.A.; Kaprin, A.D.; Alekseev, B.Y.; Snezhkina, A.V. Upregulation of Rarb, rarg, and rorc genes in clear cell renal cell carcinoma. Biomed. Pharmacol. J. 2016, 9, 967–975. [Google Scholar] [CrossRef]
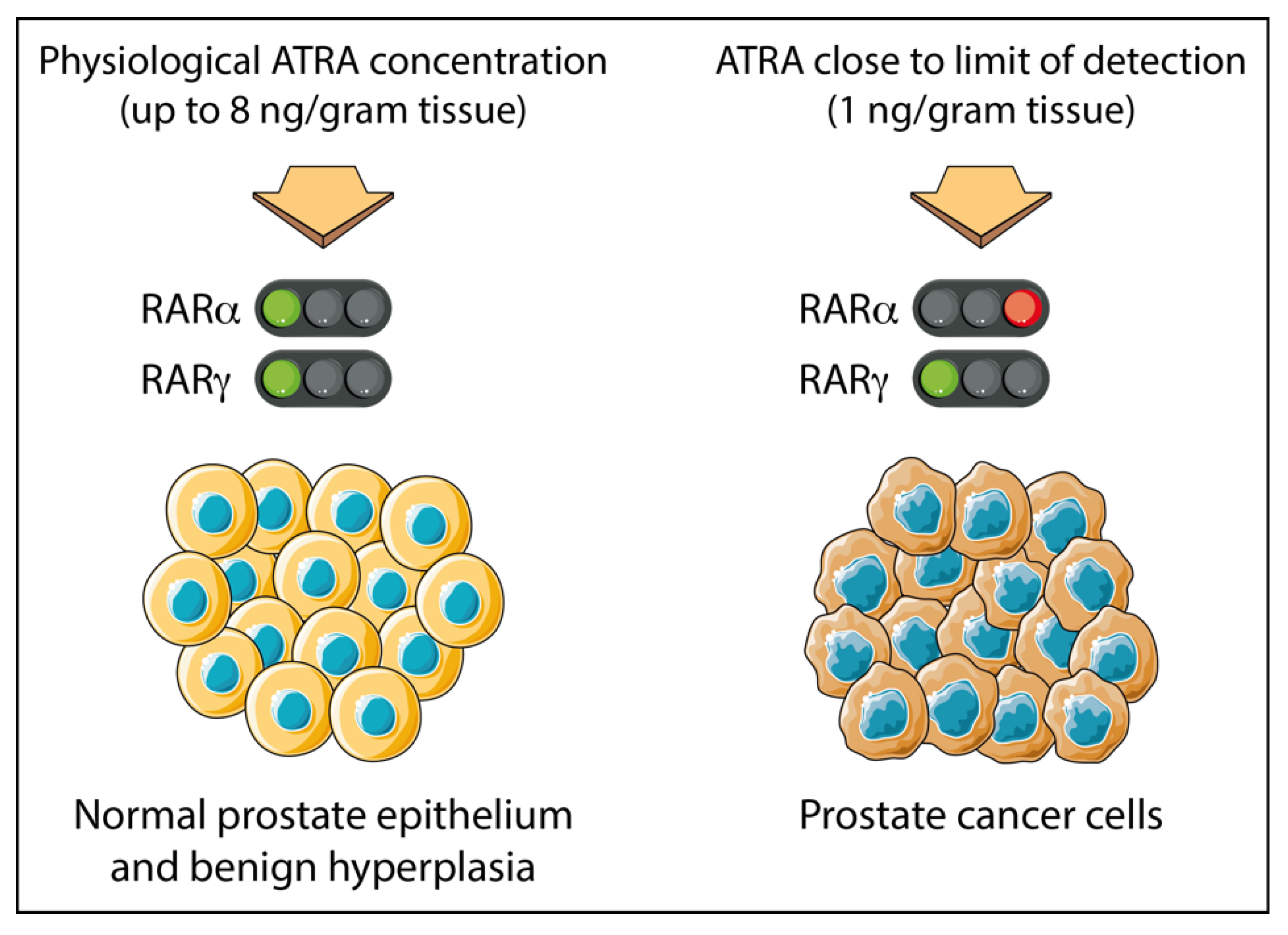
- Pasquali, D.; Thaller, C.; Eichele, G. Abnormal level of retinoic acid in prostate cancer tissues. J. Clin. Endocrinol. Metab. 1996, 81, 2186–2191. [Google Scholar] [PubMed]
- Keedwell, R.G.; Zhao, Y.; Hammond, L.A.; Wen, K.; Qin, S.; Atangan, L.I.; Shurland, D.L.; Wallace, D.M.; Bird, R.; Reitmair, A.; et al. An antagonist of retinoic acid receptors more effectively inhibits growth of human prostate cancer cells than normal prostate epithelium. Br. J. Cancer 2004, 91, 580–588. [Google Scholar] [CrossRef]
- Petrie, K.; Urban-Wojciuk, Z.; Sbirkov, Y.; Graham, A.; Hamann, A.; Brown, G. Retinoic acid receptor gamma is a therapeutically targetable driver of growth and survival in prostate cancer. Cancer Rep. 2020, 3, e1284. [Google Scholar]
- Zhao, H.; Lai, X.; Zhang, W.; Zhu, H.; Zhang, S.; Wu, W.; Wang, S.; Tang, M.; Deng, Z.; Tan, J. MiR-30a-5p frequently downregulated in prostate cancer inhibits cell proliferation via targeting PCLAF. Artif. Cells Nanomed. Biotechnol. 2019, 47, 278–289. [Google Scholar] [CrossRef]
- Chiu, H.J.; Fischman, D.A.; Hammerling, U. Vitamin A depletion causes oxidative stress, mitochondrial dysfunction, and PARP-1-dependent energy deprivation. FASEB J. 2008, 22, 3878–3887. [Google Scholar] [CrossRef]
- Gong, Y.; Fan, Z.; Luo, G.; Yang, C.; Huang, Q.; Fan, K.; Cheng, H.; Jin, K.; Ni, Q.; Yu, X.; et al. The role of necroptosis in cancer biology and therapy. Mol. Cancer 2019, 18, 100. [Google Scholar] [CrossRef]
- Lee, D.D.; Stojadinovic, O.; Krzyzanowska, A.; Vouthounis, C.; Blumenberg, M.; Tomic-Canic, M. Retinoid-responsive transcriptional changes in epidermal keratinocytes. J. Cell. Physiol. 2009, 220, 427–439. [Google Scholar] [CrossRef]
- Xu, Q.; Jitkaew, S.; Choksi, S.; Kadigamuwa, C.; Qu, J.; Choe, M.; Jang, J.; Liu, C.; Liu, Z.G. The cytoplasmic nuclear receptor RARgamma controls RIP1 initiated cell death when cIAP activity is inhibited. Nat. Commun. 2017, 8, 425. [Google Scholar] [CrossRef]
- Kadigamuwa, C.; Choksi, S.; Xu, Q.; Cataisson, C.; Greenbaum, S.S.; Yuspa, S.H.; Liu, Z.G. Role of retinoic acid receptor-gamma in DNA damage-induced necroptosis. iScience 2019, 17, 74–86. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Malignancy | RARγ Expression | Impact on Disease |
|---|---|---|
| Acute myeloid leukemia | RARγ chimeric protein | Not responsive to retinoid-based therapy |
| Colorectal carcinoma | Overexpression | Resistance to chemotherapeutics |
| Cholangiocarcinoma | Overexpression | Poor prognosis and drug resistance |
| Hepatocellular carcinoma | Overexpression | Promotes growth of xenografts |
| Renal cell carcinoma | Overexpression | May contribute to disease progression |
| Compound | Kd, nM | IC50, nM | ||||
|---|---|---|---|---|---|---|
| RARα | RARβ | RARγ | DU145 | LNCaP | PC3 | |
| Pan-RAR antagonist (AGN194310) | 4.3 | 5 | 2 | 34 | 16 | 18 |
| RARγ antagonist (AGN205728) | 2400 | 4248 | 3 | 6 | 3 | 5 |
| RARβγ antagonist (AGN194431) | 300 | 6 | 70 | 88 | 99 | 103 |
| RARα antagonist (AGN196996) | 3.9 | 4036 | >10 K | 201 | 203 | 235 |
| Pan-RAR agonist (ATRA) | ND | ND | ND | 402 | 344 | 419 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brown, G.; Petrie, K. The RARγ Oncogene: An Achilles Heel for Some Cancers. Int. J. Mol. Sci. 2021, 22, 3632. https://doi.org/10.3390/ijms22073632
Brown G, Petrie K. The RARγ Oncogene: An Achilles Heel for Some Cancers. International Journal of Molecular Sciences. 2021; 22(7):3632. https://doi.org/10.3390/ijms22073632
Chicago/Turabian StyleBrown, Geoffrey, and Kevin Petrie. 2021. "The RARγ Oncogene: An Achilles Heel for Some Cancers" International Journal of Molecular Sciences 22, no. 7: 3632. https://doi.org/10.3390/ijms22073632
APA StyleBrown, G., & Petrie, K. (2021). The RARγ Oncogene: An Achilles Heel for Some Cancers. International Journal of Molecular Sciences, 22(7), 3632. https://doi.org/10.3390/ijms22073632