
Computational Analysis of the Interactions between the S100B Extracellular Chaperone and Its Amyloid β Peptide Client

 , ,
, ,  and
and 
Abstract
{kind=link}
{kind=link}
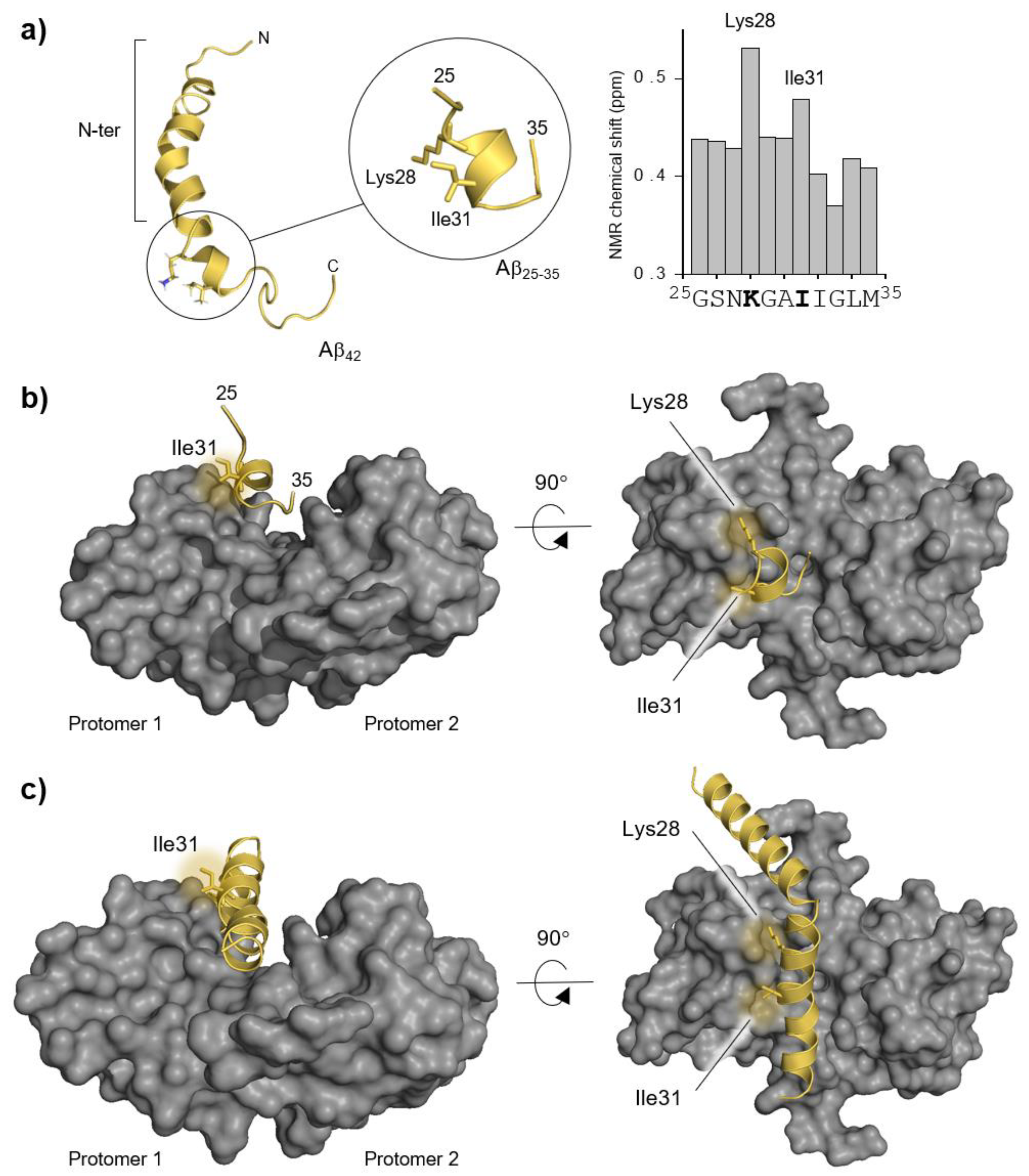
{kind=link}
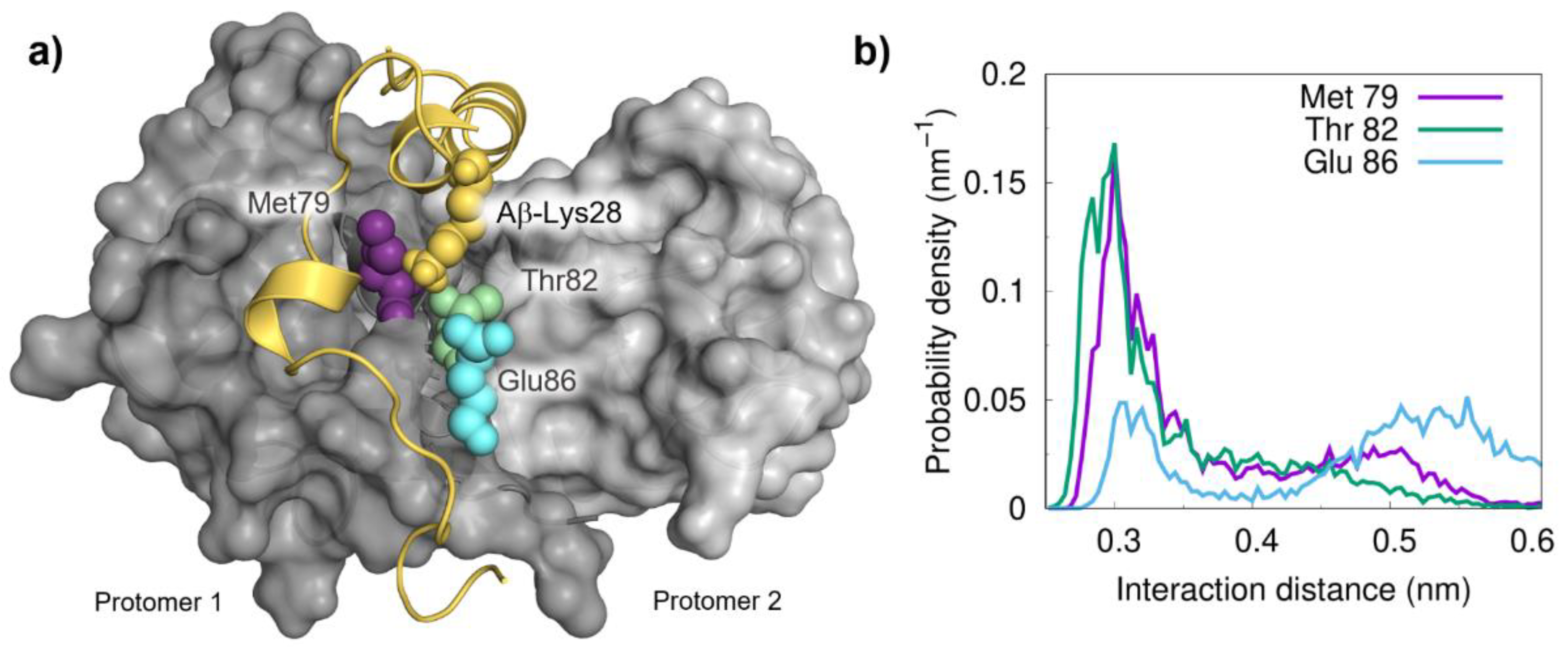{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Guided Docking of Aβ42 to S100B and Building of Interaction Complexes
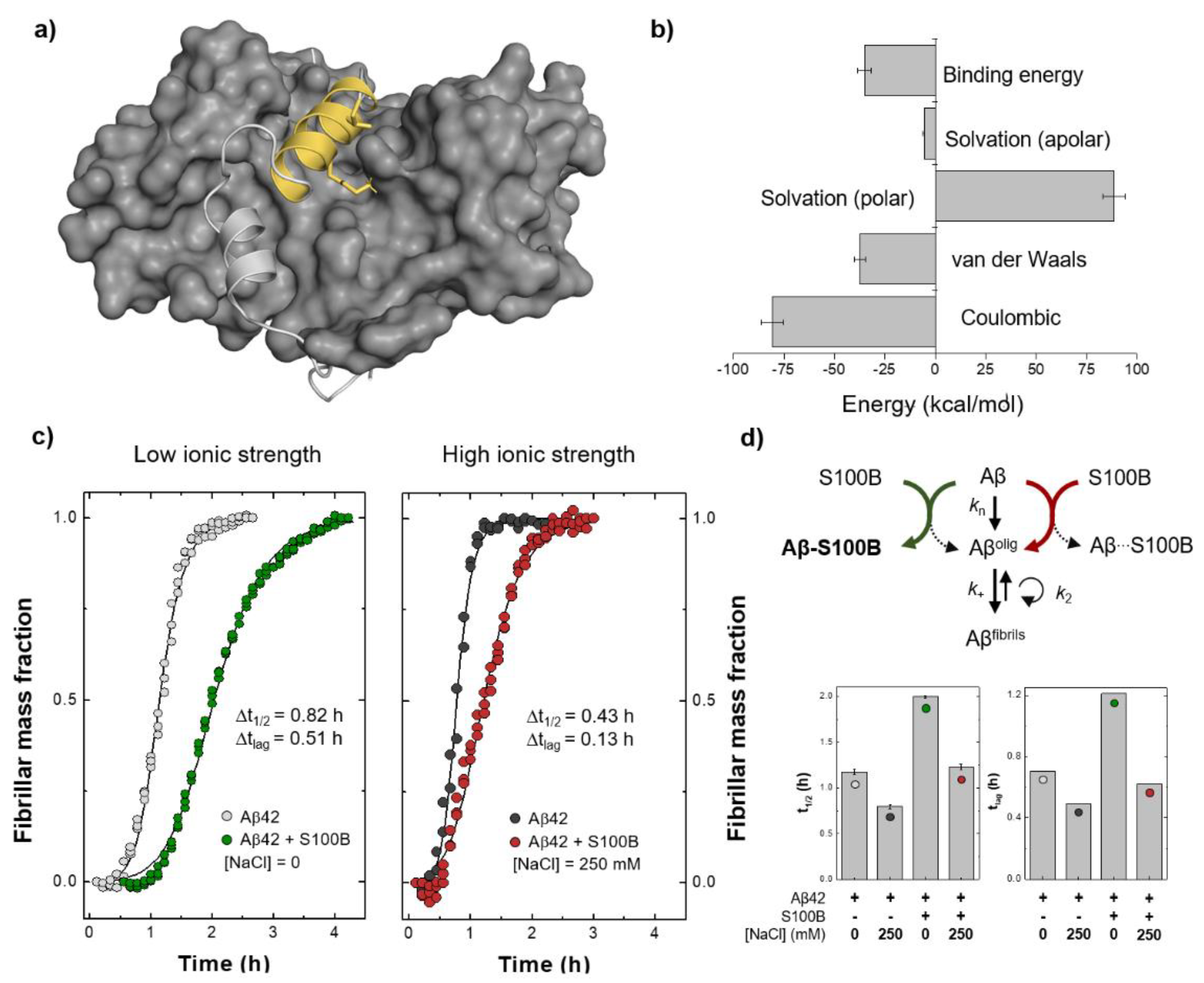
2.2. Molecular Dynamics Simulations and Mapping of Interactions
2.3. Energetics of the Interaction between Aβ and S100B and Implications in Chaperone Activity
3. Materials and Methods
3.1. Materials and Protein Purification
3.2. Aβ42 Aggregation Kinetics
3.3. Molecular Docking Settings
3.4. Molecular Mechanics/Molecular Dynamics Settings
3.5. Computational Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ciechanover, A.; Kwon, Y.T. Protein Quality Control by Molecular Chaperones in Neurodegeneration. Front. Neurosci. 2017, 11, 185. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M. Neuroinflammation in Alzheimer’s Disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Wyatt, A.R.; Yerbury, J.J.; Ecroyd, H.; Wilson, M.R. Extracellular Chaperones and Proteostasis. Annu. Rev. Biochem. 2013, 82, 295–322. [Google Scholar] [CrossRef] [PubMed]
- Lewit-Bentley, A.; Réty, S. EF-hand calcium-binding proteins. Curr. Opin. Struct. Biol. 2000, 10, 637–643. [Google Scholar] [CrossRef]
- Fritz, G.; Botelho, H.M.; Morozova-Roche, L.A.; Gomes, C.M. Natural and amyloid self-assembly of S100 proteins: Structural basis of functional diversity. FEBS J. 2010, 277, 4578–4590. [Google Scholar] [CrossRef] [PubMed]
- Cristóvão, J.S.; Figueira, A.J.; Carapeto, A.P.; Rodrigues, M.S.; Cardoso, I.; Gomes, C.M. The S100B Alarmin Is a Dual-Function Chaperone Suppressing Amyloid-β Oligomerization through Combined Zinc Chelation and Inhibition of Protein Aggregation. ACS Chem. Neurosci. 2020, 11, 2753–2760. [Google Scholar] [CrossRef]
- Cristóvão, J.S.; Moreira, G.G.; Rodrigues, F.E.P.; Carapeto, A.P.; Rodrigues, M.S.; Cardoso, I.; Ferreira, A.E.N.; Machuqueiro, M.; Fritz, G.; Gomes, C.M. Cu2+-binding to S100B triggers polymerization of disulfide cross-linked tetramers with enhanced chaperone activity against amyloid-β aggregation. Chem. Commun. 2021, 57, 379–382. [Google Scholar] [CrossRef]
- Cristóvão, J.S.; Morris, V.K.; Cardoso, I.; Leal, S.S.; Martínez, J.; Botelho, H.M.; Göbl, C.; David, R.; Kierdorf, K.; Alemi, M.; et al. The neuronal S100B protein is a calcium-tuned suppressor of amyloid-β aggregation. Sci. Adv. 2018, 4, eaaq1702. [Google Scholar] [CrossRef]
- Cristóvão, J.; Romão, M.; Gallardo, R.; Schymkowitz, J.; Rousseau, F.; Gomes, C. Targeting S100B with Peptides Encoding Intrinsic Aggregation-Prone Sequence Segments. Molecules 2021, 26, 440. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, S.B.; Botelho, H.M.; Leal, S.S.; Cardoso, I.; Fritz, G.; Gomes, C.M. Intrinsically Disordered and Aggregation Prone Regions Underlie β-Aggregation in S100 Proteins. PLoS ONE 2013, 8, e76629. [Google Scholar] [CrossRef]
- Carvalho, S.B.; Cardoso, I.; Botelho, H.M.; Yanamandra, K.; Fritz, G.; Gomes, C.M.; Morozova-Roche, L.A. Structural Heterogeneity and Bioimaging of S100 Amyloid Assemblies. In Bio-Nanoimaging; Elsevier BV: Amsterdam, The Netherlands, 2014; pp. 197–212. [Google Scholar] [CrossRef]
- Ilie, I.M.; Caflisch, A. Simulation Studies of Amyloidogenic Polypeptides and Their Aggregates. Chem. Rev. 2019, 119, 6956–6993. [Google Scholar] [CrossRef]
- Grasso, G.; Danani, A. Molecular simulations of amyloid beta assemblies. Adv. Phys. X 2020, 5, 1770627. [Google Scholar] [CrossRef]
- Van Der Munnik, N.P.; Sajib, S.J.; Moss, M.A.; Wei, T.; Uline, M.J. Determining the Potential of Mean Force for Amyloid-β Dimerization: Combining Self-Consistent Field Theory with Molecular Dynamics Simulation. J. Chem. Theory Comput. 2018, 14, 2696–2704. [Google Scholar] [CrossRef]
- Saravanan, K.M.; Zhang, H.; Zhang, H.; Xi, W.; Wei, Y. On the Conformational Dynamics of β-Amyloid Forming Peptides: A Computational Perspective. Front. Bioeng. Biotechnol. 2020, 8, 532. [Google Scholar] [CrossRef]
- Nasica-Labouze, J.; Nguyen, P.H.; Sterpone, F.; Berthoumieu, O.; Buchete, N.-V.; Coté, S.; De Simone, A.; Doig, A.J.; Faller, P.; Garcia, A.; et al. Amyloid β Protein and Alzheimer’s Disease: When Computer Simulations Complement Experimental Studies. Chem. Rev. 2015, 115, 3518–3563. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.M.; Bevan, D.R. Molecular Dynamics Simulations of Amyloid β -Peptide (1-42): Tetramer Formation and Membrane Interactions. Biophys. J. 2016, 111, 937–949. [Google Scholar] [CrossRef]
- Bhattacharyya, R.; Bhattacharjee, S.; Pathak, B.K.; Sengupta, J. Heptameric Peptide Interferes with Amyloid-β Aggregation by Structural Reorganization of the Toxic Oligomers. ACS Omega 2020, 5, 16128–16138. [Google Scholar] [CrossRef] [PubMed]
- Puig, E.; Tolchard, J.; Riera, A.; Carulla, N. Somatostatin, an In Vivo Binder to Aβ Oligomers, Binds to βPFOAβ(1–42) Tetramers. ACS Chem. Neurosci. 2020, 11, 3358–3365. [Google Scholar] [CrossRef] [PubMed]
- Owen, M.C.; Gnutt, D.; Gao, M.; Wärmländer, S.K.T.S.; Jarvet, J.; Gräslund, A.; Winter, R.; Ebbinghaus, S.; Strodel, B. Effects ofin vivoconditions on amyloid aggregation. Chem. Soc. Rev. 2019, 48, 3946–3996. [Google Scholar] [CrossRef]
- Coles, M.; Bicknell, W.; Watson, A.A.; Fairlie, D.P.; Craik, D.J. Solution Structure of Amyloid Beta-Peptide(1-40) in a Water-Micelle Environment. Is the Membrane-Spanning Domain Where We Think It Is? Biochemistry 1998, 37, 11064–11077. [Google Scholar] [CrossRef]
- Tomaselli, S.; Esposito, V.; Vangone, P.; van Nuland, N.A.J.; Bonvin, A.M.J.J.; Guerrini, R.; Tancredi, T.; Temussi, P.A.; Picone, D. The Alpha-to-Beta Conformational Transition of Alzheimer’s Abeta-(1-42) Peptide in Aqueous Media Is Reversible: A Step by Step Conformational Analysis Suggests the Location of Beta Conformation Seeding. Chembiochem 2006, 7, 257–267. [Google Scholar] [CrossRef]
- Wimley, W.C.; White, S.H. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat. Struct. Mol. Biol. 1996, 3, 842–848. [Google Scholar] [CrossRef] [PubMed]
- Arosio, P.; Knowles, T.P.; Linse, S. On the lag phase in amyloid fibril formation. Phys. Chem. Chem. Phys. 2015, 17, 7606–7618. [Google Scholar] [CrossRef] [PubMed]
- Michaels, T.C.; Šarić, A.; Habchi, J.; Chia, S.; Meisl, G.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P. Chemical Kinetics for Bridging Molecular Mechanisms and Macroscopic Measurements of Amyloid Fibril Formation. Annu. Rev. Phys. Chem. 2018, 69, 273–298. [Google Scholar] [CrossRef]
- Arosio, P.; Michaels, T.C.T.; Linse, S.; Månsson, C.; Emanuelsson, C.; Presto, J.; Johansson, J.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P.J. Kinetic analysis reveals the diversity of microscopic mechanisms through which molecular chaperones suppress amyloid formation. Nat. Commun. 2016, 7, 10948. [Google Scholar] [CrossRef]
- Walsh, D.M.; Thulin, E.; Minogue, A.M.; Gustavsson, N.; Pang, E.; Teplow, D.B.; Linse, S. A Facile Method for Expression and Purification of the Alzheimer’s Disease-associated Amyloid Β-peptide. FEBS J. 2009, 276, 1266–1281. [Google Scholar] [CrossRef]
- Botelho, H.M.; Fritz, G.; Gomes, C.M. Analysis of S100 oligomers and amyloids. In Amyloid Proteins; Springer: Berlin/Heidelberg, Germany, 2012; pp. 373–386. [Google Scholar]
- Meisl, G.; Kirkegaard, J.B.; Arosio, P.; Michaels, T.C.T.; Vendruscolo, M.; Dobson, C.M.; Linse, S.; Knowles, T.P.J. Molecular mechanisms of protein aggregation from global fitting of kinetic models. Nat. Protoc. 2016, 11, 252–272. [Google Scholar] [CrossRef]
- Meisl, G.; Yang, X.; Hellstrand, E.; Frohm, B.; Kirkegaard, J.B.; Cohen, S.I.A.; Dobson, C.M.; Linse, S.; Knowles, T.P.J. Differences in nucleation behavior underlie the contrasting aggregation kinetics of the Aβ40 and Aβ42 peptides. Proc. Natl. Acad. Sci. USA 2014, 111, 9384–9389. [Google Scholar] [CrossRef]
- Ostendorp, T.; Diez, J.; Heizmann, C.W.; Fritz, G.; Fritz, G. The crystal structures of human S100B in the zinc- and calcium-loaded state at three pH values reveal zinc ligand swapping. Biochim. Biophys. Acta (BBA) Bioenerg. 2011, 1813, 1083–1091. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Schmid, N.; Eichenberger, A.P.; Choutko, A.; Riniker, S.; Winger, M.; Mark, A.E.; Van Gunsteren, W.F. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur. Biophys. J. 2011, 40, 843–856. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Lin, Z.; Van Gunsteren, W.F. Validation of the GROMOS 54A7 Force Field with Respect to β-Peptide Folding. J. Chem. Theory Comput. 2011, 7, 1237–1243. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09. Available online: https://gaussian.com/g09citation/ (accessed on 24 March 2021).
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Batsanov, S.S. Van der Waals Radii of Elements. Inorg. Mater. 2001, 37, 871–885. [Google Scholar] [CrossRef]
- Teixeira, V.H.; Capacho, A.S.C.; Machuqueiro, M. The role of electrostatics in TrxR electron transfer mechanism: A computational approach. Proteins: Struct. Funct. Bioinform. 2016, 84, 1836–1843. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Hess, B. P-LINCS: A Parallel Linear Constraint Solver for Molecular Simulation. J. Chem. Theory Comput. 2007, 4, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Wang, C.; Nguyen, P.H.; Pham, K.; Huynh, D.; Le, T.-B.N.; Wang, H.; Ren, P.; Luo, R. Calculating protein-ligand binding affinities with MMPBSA: Method and error analysis. J. Comput. Chem. 2016, 37, 2436–2446. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodrigues, F.E.P.; Figueira, A.J.; Gomes, C.M.; Machuqueiro, M. Computational Analysis of the Interactions between the S100B Extracellular Chaperone and Its Amyloid β Peptide Client. Int. J. Mol. Sci. 2021, 22, 3629. https://doi.org/10.3390/ijms22073629
Rodrigues FEP, Figueira AJ, Gomes CM, Machuqueiro M. Computational Analysis of the Interactions between the S100B Extracellular Chaperone and Its Amyloid β Peptide Client. International Journal of Molecular Sciences. 2021; 22(7):3629. https://doi.org/10.3390/ijms22073629
Chicago/Turabian StyleRodrigues, Filipe E. P., António J. Figueira, Cláudio M. Gomes, and Miguel Machuqueiro. 2021. "Computational Analysis of the Interactions between the S100B Extracellular Chaperone and Its Amyloid β Peptide Client" International Journal of Molecular Sciences 22, no. 7: 3629. https://doi.org/10.3390/ijms22073629
APA StyleRodrigues, F. E. P., Figueira, A. J., Gomes, C. M., & Machuqueiro, M. (2021). Computational Analysis of the Interactions between the S100B Extracellular Chaperone and Its Amyloid β Peptide Client. International Journal of Molecular Sciences, 22(7), 3629. https://doi.org/10.3390/ijms22073629