
PGC-1s in the Spotlight with Parkinson’s Disease

 , ,
, ,  and
and {kind=link}
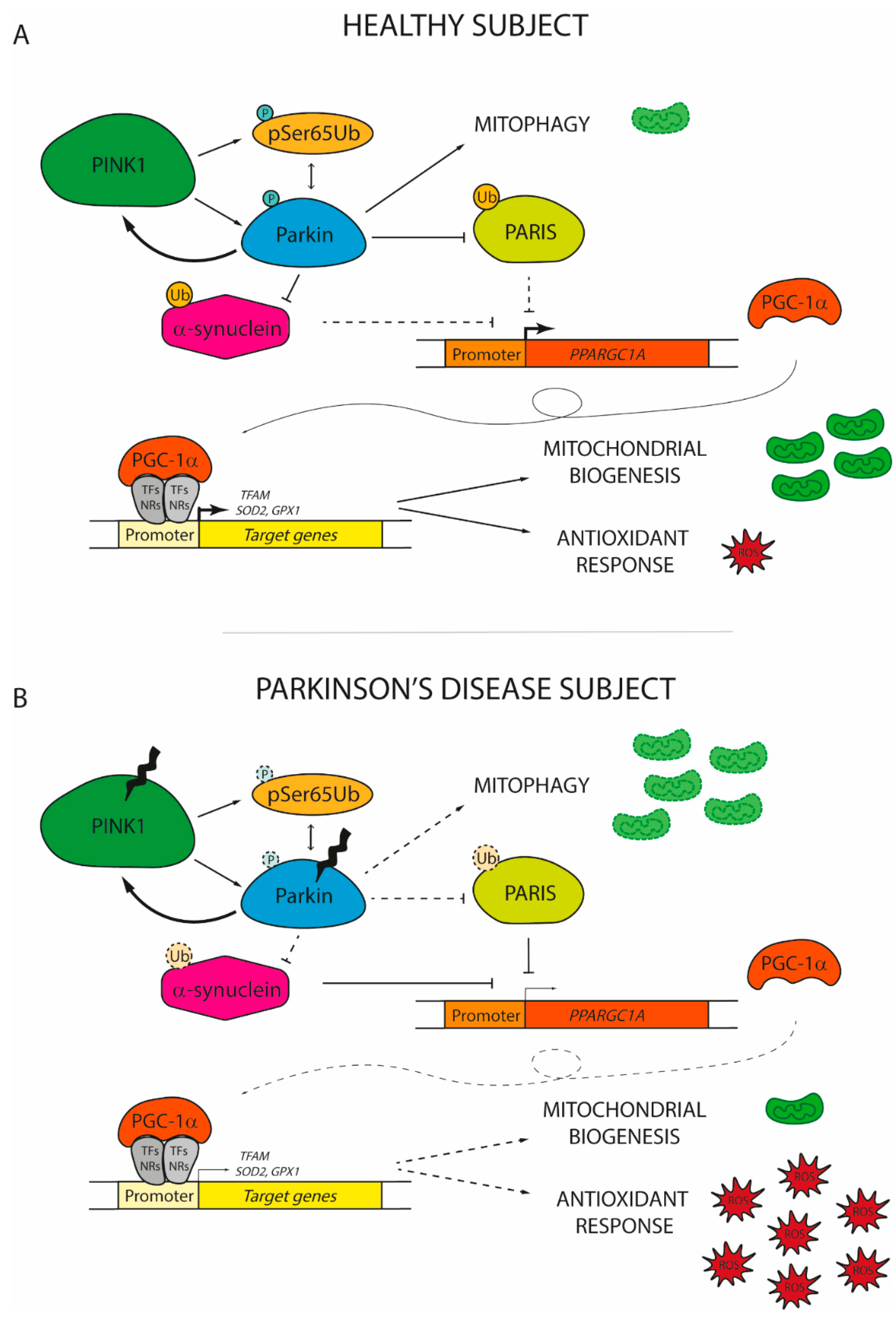{kind=link}
Abstract
1. Introduction
2. Parkinson’s Disease: An Overview
2.1. Parkinson’s Disease and Mitochondria: A Tight Bond Underlying Pathogenic Mechanism
2.1.1. mtDNA: A Neuronal Clock for Parkinson’s Disease?
2.1.2. PINK1 and Parkin: Two Players, One Axis
2.1.3. α-synuclein: Not Just A Simple Aggregation
2.1.4. LRRK2: One Protein, Different Functions
2.1.5. VPS35: The Mitochondria Traffic Warden
2.1.6. DJ-1: A Shield Against Oxidative Stress
2.1.7. CHCHD2: In Close Connection with Mitochondrial Electron Transport Chain Complex IV
3. PGC-1 Family: The Masters of Mitochondrial Biogenesis
3.1. PGC-1s’ Architecture
3.2. PGC-1s’ Activity: Boosting Mitochondrial Functions
4. PGC-1s in Parkinson’s Disease
4.1. Deepening the Role of PGC-1α in Parkinson’s Disease
4.2. PGC-1α and Parkinson’s Mutated Genes: Defining The Network Implicated in Parkinson’s Disease
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Diseases, G.B.D.; Injuries, C. Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1204–1222. [Google Scholar] [CrossRef]
- Pezzoli, G.; Cereda, E. Exposure to pesticides or solvents and risk of Parkinson disease. Neurology 2013, 80, 2035–2041. [Google Scholar] [CrossRef] [PubMed]
- Langston, J.W.; Ballard, P.A., Jr. Parkinson’s disease in a chemist working with 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. N. Engl. J. Med. 1983, 309, 310. [Google Scholar] [CrossRef] [PubMed]
- Grunewald, A.; Kumar, K.R.; Sue, C.M. New insights into the complex role of mitochondria in Parkinson’s disease. Prog. Neurobiol. 2019, 177, 73–93. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Van Houten, B. SnapShot: Mitochondrial quality control. Cell 2011, 147, 950–950 e951. [Google Scholar] [CrossRef]
- Rona-Voros, K.; Weydt, P. The role of PGC-1alpha in the pathogenesis of neurodegenerative disorders. Curr. Drug Targets 2010, 11, 1262–1269. [Google Scholar] [CrossRef] [PubMed]
- Mota, B.C.; Almpani, E.V.; Nikolaou, M.N.; Garcia-Segura, M.E.; Huang, Y.H.; Keniyopoullos, R.; Mazarakis, N.D.; Sastre, M. Investigation of the effect of PGC1A gene therapy at advanced stages of Alzheimer’s disease in an animal model of amyloid pathology. Alzheimer’s Dement. 2020, 16. [Google Scholar] [CrossRef]
- Dexter, D.T.; Jenner, P. Parkinson disease: From pathology to molecular disease mechanisms. Free. Radic. Biol. Med. 2013, 62, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M. Alzheimer’s and Parkinson’s diseases: The prion concept in relation to assembled Abeta, tau, and alpha-synuclein. Science 2015, 349, 1255555. [Google Scholar] [CrossRef]
- Shahmoradian, S.H.; Lewis, A.J.; Genoud, C.; Hench, J.; Moors, T.E.; Navarro, P.P.; Castano-Diez, D.; Schweighauser, G.; Graff-Meyer, A.; Goldie, K.N.; et al. Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat. Neurosci. 2019, 22, 1099–1109. [Google Scholar] [CrossRef]
- Hunn, B.H.; Cragg, S.J.; Bolam, J.P.; Spillantini, M.G.; Wade-Martins, R. Impaired intracellular trafficking defines early Parkinson’s disease. Trends Neurosci 2015, 38, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.A.; Gitler, A.D.; Cashikar, A.; Haynes, C.M.; Hill, K.J.; Bhullar, B.; Liu, K.; Xu, K.; Strathearn, K.E.; Liu, F.; et al. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 2006, 313, 324–328. [Google Scholar] [CrossRef]
- Chung, C.Y.; Khurana, V.; Auluck, P.K.; Tardiff, D.F.; Mazzulli, J.R.; Soldner, F.; Baru, V.; Lou, Y.; Freyzon, Y.; Cho, S.; et al. Identification and rescue of alpha-synuclein toxicity in Parkinson patient-derived neurons. Science 2013, 342, 983–987. [Google Scholar] [CrossRef]
- Mazzulli, J.R.; Zunke, F.; Isacson, O.; Studer, L.; Krainc, D. alpha-Synuclein-induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain synucleinopathy models. Proc. Natl. Acad. Sci. USA 2016, 113, 1931–1936. [Google Scholar] [CrossRef]
- Wang, X.; Becker, K.; Levine, N.; Zhang, M.; Lieberman, A.P.; Moore, D.J.; Ma, J. Pathogenic alpha-synuclein aggregates preferentially bind to mitochondria and affect cellular respiration. Acta Neuropathol. Commun. 2019, 7, 41. [Google Scholar] [CrossRef]
- Calvano, C.D.; Ventura, G.; Sardanelli, A.M.M.; Savino, L.; Losito, I.; Michele, G.; Palmisano, F.; Cataldi, T.R.I. Searching for Potential Lipid Biomarkers of Parkinson’s Disease in Parkin-Mutant Human Skin Fibroblasts by HILIC-ESI-MS/MS: Preliminary Findings. Int. J. Mol. Sci. 2019, 20, 3341. [Google Scholar] [CrossRef]
- Lobasso, S.; Tanzarella, P.; Vergara, D.; Maffia, M.; Cocco, T.; Corcelli, A. Lipid profiling of parkin-mutant human skin fibroblasts. J. Cell. Physiol. 2017, 232, 3540–3551. [Google Scholar] [CrossRef] [PubMed]
- Fanning, S.; Haque, A.; Imberdis, T.; Baru, V.; Barrasa, M.I.; Nuber, S.; Termine, D.; Ramalingam, N.; Ho, G.P.H.; Noble, T.; et al. Lipidomic Analysis of alpha-Synuclein Neurotoxicity Identifies Stearoyl CoA Desaturase as a Target for Parkinson Treatment. Mol. Cell 2019, 73, 1001–1014.e1008. [Google Scholar] [CrossRef] [PubMed]
- Imberdis, T.; Negri, J.; Ramalingam, N.; Terry-Kantor, E.; Ho, G.P.H.; Fanning, S.; Stirtz, G.; Kim, T.E.; Levy, O.A.; Young-Pearse, T.L.; et al. Cell models of lipid-rich alpha-synuclein aggregation validate known modifiers of alpha-synuclein biology and identify stearoyl-CoA desaturase. Proc. Natl. Acad. Sci. USA 2019, 116, 20760–20769. [Google Scholar] [CrossRef] [PubMed]
- Ntambi, J.M.; Miyazaki, M. Regulation of stearoyl-CoA desaturases and role in metabolism. Prog. Lipid Res. 2004, 43, 91–104. [Google Scholar] [CrossRef]
- Piccinin, E.; Cariello, M.; De Santis, S.; Ducheix, S.; Sabba, C.; Ntambi, J.M.; Moschetta, A. Role of Oleic Acid in the Gut-Liver Axis: From Diet to the Regulation of Its Synthesis via Stearoyl-CoA Desaturase 1 (SCD1). Nutrients 2019, 11, 2283. [Google Scholar] [CrossRef] [PubMed]
- Vincent, B.M.; Tardiff, D.F.; Piotrowski, J.S.; Aron, R.; Lucas, M.C.; Chung, C.Y.; Bacherman, H.; Chen, Y.; Pires, M.; Subramaniam, R.; et al. Inhibiting Stearoyl-CoA Desaturase Ameliorates alpha-Synuclein Cytotoxicity. Cell Rep. 2018, 25, 2742–2754 e2731. [Google Scholar] [CrossRef]
- Bandres-Ciga, S.; Diez-Fairen, M.; Kim, J.J.; Singleton, A.B. Genetics of Parkinson’s disease: An introspection of its journey towards precision medicine. Neurobiol. Dis. 2020, 137, 104782. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Wang, P.; Jankovic, J. The genetics of Parkinson disease. Ageing Res. Rev. 2018, 42, 72–85. [Google Scholar] [CrossRef]
- Kumar, K.R.; Djarmati-Westenberger, A.; Grunewald, A. Genetics of Parkinson’s disease. Semin. Neurol. 2011, 31, 433–440. [Google Scholar] [CrossRef]
- Kumar, K.R.; Lohmann, K.; Klein, C. Genetics of Parkinson disease and other movement disorders. Curr. Opin. Neurol. 2012, 25, 466–474. [Google Scholar] [CrossRef]
- International Parkinson Disease Genomics Consortium; Nalls, M.A.; Plagnol, V.; Hernandez, D.G.; Sharma, M.; Sheerin, U.M.; Saad, M.; Simon-Sanchez, J.; Schulte, C.; Lesage, S.; et al. Imputation of sequence variants for identification of genetic risks for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet 2011, 377, 641–649. [Google Scholar] [CrossRef]
- International Parkinson’s Disease Genomics Consortium; Wellcome Trust Case Control Consortium. A two-stage meta-analysis identifies several new loci for Parkinson’s disease. PLoS Genet. 2011, 7, e1002142. [Google Scholar] [CrossRef]
- Park, J.S.; Davis, R.L.; Sue, C.M. Mitochondrial Dysfunction in Parkinson’s Disease: New Mechanistic Insights and Therapeutic Perspectives. Curr. Neurol. Neurosci. Rep. 2018, 18, 21. [Google Scholar] [CrossRef]
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef]
- Cocco, T.; Pacelli, C.; Sgobbo, P.; Villani, G. Control of OXPHOS efficiency by complex I in brain mitochondria. Neurobiol. Aging 2009, 30, 622–629. [Google Scholar] [CrossRef]
- Davey, G.P.; Peuchen, S.; Clark, J.B. Energy thresholds in brain mitochondria. Potential involvement in neurodegeneration. J. Biol. Chem. 1998, 273, 12753–12757. [Google Scholar] [CrossRef]
- Cocco, T.; Sgobbo, P.; Clemente, M.; Lopriore, B.; Grattagliano, I.; Di Paola, M.; Villani, G. Tissue-specific changes of mitochondrial functions in aged rats: Effect of a long-term dietary treatment with N-acetylcysteine. Free. Radic. Biol. Med. 2005, 38, 796–805. [Google Scholar] [CrossRef]
- Blesa, J.; Phani, S.; Jackson-Lewis, V.; Przedborski, S. Classic and new animal models of Parkinson’s disease. J. Biomed. Biotechnol. 2012, 2012, 845618. [Google Scholar] [CrossRef]
- Tanzarella, P.; Ferretta, A.; Barile, S.N.; Ancona, M.; De Rasmo, D.; Signorile, A.; Papa, S.; Capitanio, N.; Pacelli, C.; Cocco, T. Increased Levels of cAMP by the Calcium-Dependent Activation of Soluble Adenylyl Cyclase in Parkin-Mutant Fibroblasts. Cells 2019, 8, 250. [Google Scholar] [CrossRef] [PubMed]
- Whitworth, A.J.; Pallanck, L.J. PINK1/Parkin mitophagy and neurodegeneration-what do we really know in vivo? Curr. Opin. Genet. Dev. 2017, 44, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Buneeva, O.; Fedchenko, V.; Kopylov, A.; Medvedev, A. Mitochondrial Dysfunction in Parkinson’s Disease: Focus on Mitochondrial DNA. Biomedicines 2020, 8, 591. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H.; Parks, J.K.; Miller, S.W.; Tuttle, J.B.; Trimmer, P.A.; Sheehan, J.P.; Bennett, J.P., Jr.; Davis, R.E.; Parker, W.D., Jr. Origin and functional consequences of the complex I defect in Parkinson’s disease. Ann. Neurol. 1996, 40, 663–671. [Google Scholar] [CrossRef]
- Esteves, A.R.; Domingues, A.F.; Ferreira, I.L.; Januario, C.; Swerdlow, R.H.; Oliveira, C.R.; Cardoso, S.M. Mitochondrial function in Parkinson’s disease cybrids containing an nt2 neuron-like nuclear background. Mitochondrion 2008, 8, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.; Krishnan, K.J.; Morris, C.M.; Taylor, G.A.; Reeve, A.K.; Perry, R.H.; Jaros, E.; Hersheson, J.S.; Betts, J.; Klopstock, T.; et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 2006, 38, 515–517. [Google Scholar] [CrossRef]
- Dolle, C.; Flones, I.; Nido, G.S.; Miletic, H.; Osuagwu, N.; Kristoffersen, S.; Lilleng, P.K.; Larsen, J.P.; Tysnes, O.B.; Haugarvoll, K.; et al. Defective mitochondrial DNA homeostasis in the substantia nigra in Parkinson disease. Nat. Commun. 2016, 7, 13548. [Google Scholar] [CrossRef]
- Kraytsberg, Y.; Kudryavtseva, E.; McKee, A.C.; Geula, C.; Kowall, N.W.; Khrapko, K. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat. Genet. 2006, 38, 518–520. [Google Scholar] [CrossRef]
- Filograna, R.; Mennuni, M.; Alsina, D.; Larsson, N.G. Mitochondrial DNA copy number in human disease: The more the better? FEBS Lett. 2020. [Google Scholar] [CrossRef]
- Reutzel, M.; Grewal, R.; Dilberger, B.; Silaidos, C.; Joppe, A.; Eckert, G.P. Cerebral Mitochondrial Function and Cognitive Performance during Aging: A Longitudinal Study in NMRI Mice. Oxid. Med. Cell Longev. 2020, 2020, 4060769. [Google Scholar] [CrossRef]
- Mengel-From, J.; Thinggaard, M.; Dalgard, C.; Kyvik, K.O.; Christensen, K.; Christiansen, L. Mitochondrial DNA copy number in peripheral blood cells declines with age and is associated with general health among elderly. Hum. Genet. 2014, 133, 1149–1159. [Google Scholar] [CrossRef]
- Pyle, A.; Anugrha, H.; Kurzawa-Akanbi, M.; Yarnall, A.; Burn, D.; Hudson, G. Reduced mitochondrial DNA copy number is a biomarker of Parkinson’s disease. Neurobiol. Aging 2016, 38, 216.e7–216.e10. [Google Scholar] [CrossRef] [PubMed]
- Grunewald, A.; Rygiel, K.A.; Hepplewhite, P.D.; Morris, C.M.; Picard, M.; Turnbull, D.M. Mitochondrial DNA Depletion in Respiratory Chain-Deficient Parkinson Disease Neurons. Ann. Neurol. 2016, 79, 366–378. [Google Scholar] [CrossRef] [PubMed]
- Gui, Y.X.; Xu, Z.P.; Lv, W.; Zhao, J.J.; Hu, X.Y. Evidence for polymerase gamma, POLG1 variation in reduced mitochondrial DNA copy number in Parkinson’s disease. Parkinsonism. Relat. Disord. 2015, 21, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Pyle, A.; Brennan, R.; Kurzawa-Akanbi, M.; Yarnall, A.; Thouin, A.; Mollenhauer, B.; Burn, D.; Chinnery, P.F.; Hudson, G. Reduced cerebrospinal fluid mitochondrial DNA is a biomarker for early-stage Parkinson’s disease. Ann. Neurol. 2015, 78, 1000–1004. [Google Scholar] [CrossRef]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [PubMed]
- Wauer, T.; Komander, D. Structure of the human Parkin ligase domain in an autoinhibited state. EMBO J. 2013, 32, 2099–2112. [Google Scholar] [CrossRef]
- Yamano, K.; Youle, R.J. PINK1 is degraded through the N-end rule pathway. Autophagy 2013, 9, 1758–1769. [Google Scholar] [CrossRef]
- Koyano, F.; Okatsu, K.; Kosako, H.; Tamura, Y.; Go, E.; Kimura, M.; Kimura, Y.; Tsuchiya, H.; Yoshihara, H.; Hirokawa, T.; et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 2014, 510, 162–166. [Google Scholar] [CrossRef]
- Wauer, T.; Simicek, M.; Schubert, A.; Komander, D. Mechanism of phospho-ubiquitin-induced PARKIN activation. Nature 2015, 524, 370–374. [Google Scholar] [CrossRef] [PubMed]
- Kazlauskaite, A.; Kondapalli, C.; Gourlay, R.; Campbell, D.G.; Ritorto, M.S.; Hofmann, K.; Alessi, D.R.; Knebel, A.; Trost, M.; Muqit, M.M. Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem. J. 2014, 460, 127–139. [Google Scholar] [CrossRef]
- Kane, L.A.; Lazarou, M.; Fogel, A.I.; Li, Y.; Yamano, K.; Sarraf, S.A.; Banerjee, S.; Youle, R.J. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 2014, 205, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Gladkova, C.; Maslen, S.L.; Skehel, J.M.; Komander, D. Mechanism of parkin activation by PINK1. Nature 2018, 559, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.W.; Ordureau, A.; Heo, J.M. Building and decoding ubiquitin chains for mitophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 93–108. [Google Scholar] [CrossRef]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef]
- Heo, J.M.; Ordureau, A.; Paulo, J.A.; Rinehart, J.; Harper, J.W. The PINK1-PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Mol. Cell 2015, 60, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Chen, K.; Chen, W.; Yao, Y.; Ni, S.; Ye, M.; Zhuang, G.; Hu, M.; Gao, J.; Gao, C.; et al. Paradoxical Mitophagy Regulation by PINK1 and TUFm. Mol. Cell 2020, 80, 607–620.e612. [Google Scholar] [CrossRef] [PubMed]
- Malpartida, A.B.; Williamson, M.; Narendra, D.P.; Wade-Martins, R.; Ryan, B.J. Mitochondrial Dysfunction and Mitophagy in Parkinson’s Disease: From Mechanism to Therapy. Trends Biochem. Sci. 2020. [Google Scholar] [CrossRef]
- Fiesel, F.C.; Caulfield, T.R.; Moussaud-Lamodiere, E.L.; Ogaki, K.; Dourado, D.F.; Flores, S.C.; Ross, O.A.; Springer, W. Structural and Functional Impact of Parkinson Disease-Associated Mutations in the E3 Ubiquitin Ligase Parkin. Hum. Mutat. 2015, 36, 774–786. [Google Scholar] [CrossRef]
- Lee, J.J.; Sanchez-Martinez, A.; Martinez Zarate, A.; Beninca, C.; Mayor, U.; Clague, M.J.; Whitworth, A.J. Basal mitophagy is widespread in Drosophila but minimally affected by loss of Pink1 or parkin. J. Cell Biol. 2018, 217, 1613–1622. [Google Scholar] [CrossRef]
- Pinto, M.; Nissanka, N.; Moraes, C.T. Lack of Parkin Anticipates the Phenotype and Affects Mitochondrial Morphology and mtDNA Levels in a Mouse Model of Parkinson’s Disease. J. Neurosci. 2018, 38, 1042–1053. [Google Scholar] [CrossRef]
- McWilliams, T.G.; Prescott, A.R.; Montava-Garriga, L.; Ball, G.; Singh, F.; Barini, E.; Muqit, M.M.K.; Brooks, S.P.; Ganley, I.G. Basal Mitophagy Occurs Independently of PINK1 in Mouse Tissues of High Metabolic Demand. Cell Metab. 2018, 27, 439–449.e435. [Google Scholar] [CrossRef]
- Stefanovic, A.N.; Stockl, M.T.; Claessens, M.M.; Subramaniam, V. alpha-Synuclein oligomers distinctively permeabilize complex model membranes. FEBS J. 2014, 281, 2838–2850. [Google Scholar] [CrossRef] [PubMed]
- Plotegher, N.; Gratton, E.; Bubacco, L. Number and Brightness analysis of alpha-synuclein oligomerization and the associated mitochondrial morphology alterations in live cells. Biochim. Biophys. Acta 2014, 1840, 2014–2024. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S.R.; Vergnes, L.; Franich, N.R.; Reue, K.; Chesselet, M.F. Region specific mitochondrial impairment in mice with widespread overexpression of alpha-synuclein. Neurobiol. Dis. 2014, 70, 204–213. [Google Scholar] [CrossRef]
- Ryan, S.D.; Dolatabadi, N.; Chan, S.F.; Zhang, X.; Akhtar, M.W.; Parker, J.; Soldner, F.; Sunico, C.R.; Nagar, S.; Talantova, M.; et al. Isogenic human iPSC Parkinson’s model shows nitrosative stress-induced dysfunction in MEF2-PGC1alpha transcription. Cell 2013, 155, 1351–1364. [Google Scholar] [CrossRef]
- Hsieh, C.H.; Shaltouki, A.; Gonzalez, A.E.; Bettencourt da Cruz, A.; Burbulla, L.F.; St Lawrence, E.; Schule, B.; Krainc, D.; Palmer, T.D.; Wang, X. Functional Impairment in Miro Degradation and Mitophagy Is a Shared Feature in Familial and Sporadic Parkinson’s Disease. Cell Stem Cell 2016, 19, 709–724. [Google Scholar] [CrossRef]
- Glater, E.E.; Megeath, L.J.; Stowers, R.S.; Schwarz, T.L. Axonal transport of mitochondria requires milton to recruit kinesin heavy chain and is light chain independent. J. Cell Biol. 2006, 173, 545–557. [Google Scholar] [CrossRef] [PubMed]
- Bonello, F.; Hassoun, S.M.; Mouton-Liger, F.; Shin, Y.S.; Muscat, A.; Tesson, C.; Lesage, S.; Beart, P.M.; Brice, A.; Krupp, J.; et al. LRRK2 impairs PINK1/Parkin-dependent mitophagy via its kinase activity: Pathologic insights into Parkinson’s disease. Hum. Mol. Genet. 2019, 28, 1645–1660. [Google Scholar] [CrossRef]
- Wauters, F.; Cornelissen, T.; Imberechts, D.; Martin, S.; Koentjoro, B.; Sue, C.; Vangheluwe, P.; Vandenberghe, W. LRRK2 mutations impair depolarization-induced mitophagy through inhibition of mitochondrial accumulation of RAB10. Autophagy 2020, 16, 203–222. [Google Scholar] [CrossRef]
- Zimprich, A.; Benet-Pages, A.; Struhal, W.; Graf, E.; Eck, S.H.; Offman, M.N.; Haubenberger, D.; Spielberger, S.; Schulte, E.C.; Lichtner, P.; et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am. J. Hum. Genet. 2011, 89, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Vilarino-Guell, C.; Wider, C.; Ross, O.A.; Dachsel, J.C.; Kachergus, J.M.; Lincoln, S.J.; Soto-Ortolaza, A.I.; Cobb, S.A.; Wilhoite, G.J.; Bacon, J.A.; et al. VPS35 mutations in Parkinson disease. Am. J. Hum. Genet. 2011, 89, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Hierro, A.; Rojas, A.L.; Rojas, R.; Murthy, N.; Effantin, G.; Kajava, A.V.; Steven, A.C.; Bonifacino, J.S.; Hurley, J.H. Functional architecture of the retromer cargo-recognition complex. Nature 2007, 449, 1063–1067. [Google Scholar] [CrossRef]
- Wang, W.; Ma, X.; Zhou, L.; Liu, J.; Zhu, X. A conserved retromer sorting motif is essential for mitochondrial DLP1 recycling by VPS35 in Parkinson’s disease model. Hum. Mol. Genet. 2017, 26, 781–789. [Google Scholar] [CrossRef]
- Tsika, E.; Glauser, L.; Moser, R.; Fiser, A.; Daniel, G.; Sheerin, U.M.; Lees, A.; Troncoso, J.C.; Lewis, P.A.; Bandopadhyay, R.; et al. Parkinson’s disease-linked mutations in VPS35 induce dopaminergic neurodegeneration. Hum. Mol. Genet. 2014, 23, 4621–4638. [Google Scholar] [CrossRef]
- Wang, W.; Wang, X.; Fujioka, H.; Hoppel, C.; Whone, A.L.; Caldwell, M.A.; Cullen, P.J.; Liu, J.; Zhu, X. Parkinson’s disease-associated mutant VPS35 causes mitochondrial dysfunction by recycling DLP1 complexes. Nat. Med. 2016, 22, 54–63. [Google Scholar] [CrossRef]
- Wang, H.; Song, P.; Du, L.; Tian, W.; Yue, W.; Liu, M.; Li, D.; Wang, B.; Zhu, Y.; Cao, C.; et al. Parkin ubiquitinates Drp1 for proteasome-dependent degradation: Implication of dysregulated mitochondrial dynamics in Parkinson disease. J. Biol. Chem. 2011, 286, 11649–11658. [Google Scholar] [CrossRef]
- Wang, X.; Yan, M.H.; Fujioka, H.; Liu, J.; Wilson-Delfosse, A.; Chen, S.G.; Perry, G.; Casadesus, G.; Zhu, X. LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum. Mol. Genet. 2012, 21, 1931–1944. [Google Scholar] [CrossRef]
- Buhlman, L.; Damiano, M.; Bertolin, G.; Ferrando-Miguel, R.; Lombes, A.; Brice, A.; Corti, O. Functional interplay between Parkin and Drp1 in mitochondrial fission and clearance. Biochim. Biophys. Acta 2014, 1843, 2012–2026. [Google Scholar] [CrossRef] [PubMed]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.H.; Smith, P.D.; Aleyasin, H.; Hayley, S.; Mount, M.P.; Pownall, S.; Wakeham, A.; You-Ten, A.J.; Kalia, S.K.; Horne, P.; et al. Hypersensitivity of DJ-1-deficient mice to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyrindine (MPTP) and oxidative stress. Proc. Natl. Acad. Sci. USA 2005, 102, 5215–5220. [Google Scholar] [CrossRef] [PubMed]
- Larsen, N.J.; Ambrosi, G.; Mullett, S.J.; Berman, S.B.; Hinkle, D.A. DJ-1 knock-down impairs astrocyte mitochondrial function. Neuroscience 2011, 196, 251–264. [Google Scholar] [CrossRef][Green Version]
- Krebiehl, G.; Ruckerbauer, S.; Burbulla, L.F.; Kieper, N.; Maurer, B.; Waak, J.; Wolburg, H.; Gizatullina, Z.; Gellerich, F.N.; Woitalla, D.; et al. Reduced basal autophagy and impaired mitochondrial dynamics due to loss of Parkinson’s disease-associated protein DJ-1. PLoS ONE 2010, 5, e9367. [Google Scholar] [CrossRef] [PubMed]
- Irrcher, I.; Aleyasin, H.; Seifert, E.L.; Hewitt, S.J.; Chhabra, S.; Phillips, M.; Lutz, A.K.; Rousseaux, M.W.; Bevilacqua, L.; Jahani-Asl, A.; et al. Loss of the Parkinson’s disease-linked gene DJ-1 perturbs mitochondrial dynamics. Hum. Mol. Genet. 2010, 19, 3734–3746. [Google Scholar] [CrossRef]
- Hao, L.Y.; Giasson, B.I.; Bonini, N.M. DJ-1 is critical for mitochondrial function and rescues PINK1 loss of function. Proc. Natl. Acad. Sci. USA 2010, 107, 9747–9752. [Google Scholar] [CrossRef]
- Junn, E.; Jang, W.H.; Zhao, X.; Jeong, B.S.; Mouradian, M.M. Mitochondrial localization of DJ-1 leads to enhanced neuroprotection. J. Neurosci. Res. 2009, 87, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Canet-Aviles, R.M.; Wilson, M.A.; Miller, D.W.; Ahmad, R.; McLendon, C.; Bandyopadhyay, S.; Baptista, M.J.; Ringe, D.; Petsko, G.A.; Cookson, M.R. The Parkinson’s disease protein DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization. Proc. Natl. Acad. Sci. USA 2004, 101, 9103–9108. [Google Scholar] [CrossRef]
- Biosa, A.; Sandrelli, F.; Beltramini, M.; Greggio, E.; Bubacco, L.; Bisaglia, M. Recent findings on the physiological function of DJ-1: Beyond Parkinson’s disease. Neurobiol. Dis. 2017, 108, 65–72. [Google Scholar] [CrossRef]
- Dolgacheva, L.P.; Berezhnov, A.V.; Fedotova, E.I.; Zinchenko, V.P.; Abramov, A.Y. Role of DJ-1 in the mechanism of pathogenesis of Parkinson’s disease. J. Bioenerg. Biomembr. 2019, 51, 175–188. [Google Scholar] [CrossRef]
- Strobbe, D.; Robinson, A.A.; Harvey, K.; Rossi, L.; Ferraina, C.; de Biase, V.; Rodolfo, C.; Harvey, R.J.; Campanella, M. Distinct Mechanisms of Pathogenic DJ-1 Mutations in Mitochondrial Quality Control. Front. Mol. Neurosci. 2018, 11, 68. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Park, H.A.; Mnatsakanyan, N.; Niu, Y.; Licznerski, P.; Wu, J.; Miranda, P.; Graham, M.; Tang, J.; Boon, A.J.W.; et al. Parkinson’s disease protein DJ-1 regulates ATP synthase protein components to increase neuronal process outgrowth. Cell Death Dis. 2019, 10, 469. [Google Scholar] [CrossRef]
- Funayama, M.; Ohe, K.; Amo, T.; Furuya, N.; Yamaguchi, J.; Saiki, S.; Li, Y.; Ogaki, K.; Ando, M.; Yoshino, H.; et al. CHCHD2 mutations in autosomal dominant late-onset Parkinson’s disease: A genome-wide linkage and sequencing study. Lancet Neurol. 2015, 14, 274–282. [Google Scholar] [CrossRef]
- Aras, S.; Bai, M.; Lee, I.; Springett, R.; Huttemann, M.; Grossman, L.I. MNRR1 (formerly CHCHD2) is a bi-organellar regulator of mitochondrial metabolism. Mitochondrion 2015, 20, 43–51. [Google Scholar] [CrossRef]
- Meng, H.; Yamashita, C.; Shiba-Fukushima, K.; Inoshita, T.; Funayama, M.; Sato, S.; Hatta, T.; Natsume, T.; Umitsu, M.; Takagi, J.; et al. Loss of Parkinson’s disease-associated protein CHCHD2 affects mitochondrial crista structure and destabilizes cytochrome c. Nat. Commun. 2017, 8, 15500. [Google Scholar] [CrossRef] [PubMed]
- Villani, G.; Greco, M.; Papa, S.; Attardi, G. Low reserve of cytochrome c oxidase capacity in vivo in the respiratory chain of a variety of human cell types. J. Biol. Chem. 1998, 273, 31829–31836. [Google Scholar] [CrossRef]
- Villani, G.; Attardi, G. In vivo control of respiration by cytochrome c oxidase in human cells. Free. Radic. Biol. Med. 2000, 29, 202–210. [Google Scholar] [CrossRef]
- Pacelli, C.; Latorre, D.; Cocco, T.; Capuano, F.; Kukat, C.; Seibel, P.; Villani, G. Tight control of mitochondrial membrane potential by cytochrome c oxidase. Mitochondrion 2011, 11, 334–341. [Google Scholar] [CrossRef]
- Villena, J.A. New insights into PGC-1 coactivators: Redefining their role in the regulation of mitochondrial function and beyond. FEBS J. 2015, 282, 647–672. [Google Scholar] [CrossRef]
- Yoshioka, T.; Inagaki, K.; Noguchi, T.; Sakai, M.; Ogawa, W.; Hosooka, T.; Iguchi, H.; Watanabe, E.; Matsuki, Y.; Hiramatsu, R.; et al. Identification and characterization of an alternative promoter of the human PGC-1alpha gene. Biochem. Biophys. Res. Commun. 2009, 381, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Soyal, S.M.; Felder, T.K.; Auer, S.; Hahne, P.; Oberkofler, H.; Witting, A.; Paulmichl, M.; Landwehrmeyer, G.B.; Weydt, P.; Patsch, W.; et al. A greatly extended PPARGC1A genomic locus encodes several new brain-specific isoforms and influences Huntington disease age of onset. Hum. Mol. Genet. 2012, 21, 3461–3473. [Google Scholar] [CrossRef]
- Zhang, Y.; Huypens, P.; Adamson, A.W.; Chang, J.S.; Henagan, T.M.; Boudreau, A.; Lenard, N.R.; Burk, D.; Klein, J.; Perwitz, N.; et al. Alternative mRNA splicing produces a novel biologically active short isoform of PGC-1alpha. J. Biol. Chem. 2009, 284, 32813–32826. [Google Scholar] [CrossRef] [PubMed]
- Monsalve, M.; Wu, Z.; Adelmant, G.; Puigserver, P.; Fan, M.; Spiegelman, B.M. Direct coupling of transcription and mRNA processing through the thermogenic coactivator PGC-1. Mol.Cell 2000, 6, 307–316. [Google Scholar] [CrossRef]
- Puigserver, P.; Adelmant, G.; Wu, Z.; Fan, M.; Xu, J.; O’Malley, B.; Spiegelman, B.M. Activation of PPARgamma coactivator-1 through transcription factor docking. Science 1999, 286, 1368–1371. [Google Scholar] [CrossRef] [PubMed]
- Wallberg, A.E.; Yamamura, S.; Malik, S.; Spiegelman, B.M.; Roeder, R.G. Coordination of p300-mediated chromatin remodeling and TRAP/mediator function through coactivator PGC-1alpha. Mol. Cell 2003, 12, 1137–1149. [Google Scholar] [CrossRef]
- Li, S.; Liu, C.; Li, N.; Hao, T.; Han, T.; Hill, D.E.; Vidal, M.; Lin, J.D. Genome-wide coactivation analysis of PGC-1alpha identifies BAF60a as a regulator of hepatic lipid metabolism. Cell Metab. 2008, 8, 105–117. [Google Scholar] [CrossRef]
- Borgius, L.J.; Steffensen, K.R.; Gustafsson, J.A.; Treuter, E. Glucocorticoid signaling is perturbed by the atypical orphan receptor and corepressor SHP. J. Biol. Chem. 2002, 277, 49761–49766. [Google Scholar] [CrossRef]
- Lin, J.; Wu, H.; Tarr, P.T.; Zhang, C.Y.; Wu, Z.; Boss, O.; Michael, L.F.; Puigserver, P.; Isotani, E.; Olson, E.N.; et al. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature 2002, 418, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Kressler, D.; Schreiber, S.N.; Knutti, D.; Kralli, A. The PGC-1-related protein PERC is a selective coactivator of estrogen receptor alpha. J. Biol. Chem. 2002, 277, 13918–13925. [Google Scholar] [CrossRef] [PubMed]
- Andersson, U.; Scarpulla, R.C. Pgc-1-related coactivator, a novel, serum-inducible coactivator of nuclear respiratory factor 1-dependent transcription in mammalian cells. Mol. Cell Biol. 2001, 21, 3738–3749. [Google Scholar] [CrossRef] [PubMed]
- Lerin, C.; Rodgers, J.T.; Kalume, D.E.; Kim, S.H.; Pandey, A.; Puigserver, P. GCN5 acetyltransferase complex controls glucose metabolism through transcriptional repression of PGC-1alpha. Cell Metab. 2006, 3, 429–438. [Google Scholar] [CrossRef]
- Kelly, T.J.; Lerin, C.; Haas, W.; Gygi, S.P.; Puigserver, P. GCN5-mediated transcriptional control of the metabolic coactivator PGC-1beta through lysine acetylation. J. Biol. Chem. 2009, 284, 19945–19952. [Google Scholar] [CrossRef]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 2005, 434, 113–118. [Google Scholar] [CrossRef]
- Puigserver, P.; Rhee, J.; Lin, J.; Wu, Z.; Yoon, J.C.; Zhang, C.Y.; Krauss, S.; Mootha, V.K.; Lowell, B.B.; Spiegelman, B.M. Cytokine stimulation of energy expenditure through p38 MAP kinase activation of PPARgamma coactivator-1. Mol. Cell 2001, 8, 971–982. [Google Scholar] [CrossRef]
- Liang, H.L.; Dhar, S.S.; Wong-Riley, M.T. p38 mitogen-activated protein kinase and calcium channels mediate signaling in depolarization-induced activation of peroxisome proliferator-activated receptor gamma coactivator-1alpha in neurons. J. Neurosci. Res. 2010, 88, 640–649. [Google Scholar] [CrossRef]
- Irrcher, I.; Ljubicic, V.; Kirwan, A.F.; Hood, D.A. AMP-activated protein kinase-regulated activation of the PGC-1alpha promoter in skeletal muscle cells. PLoS ONE 2008, 3, e3614. [Google Scholar] [CrossRef]
- Jager, S.; Handschin, C.; St-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef]
- Li, X.; Monks, B.; Ge, Q.; Birnbaum, M.J. Akt/PKB regulates hepatic metabolism by directly inhibiting PGC-1alpha transcription coactivator. Nature 2007, 447, 1012–1016. [Google Scholar] [CrossRef]
- Teyssier, C.; Ma, H.; Emter, R.; Kralli, A.; Stallcup, M.R. Activation of nuclear receptor coactivator PGC-1alpha by arginine methylation. Genes Dev. 2005, 19, 1466–1473. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, J.T.; Lerin, C.; Gerhart-Hines, Z.; Puigserver, P. Metabolic adaptations through the PGC-1 alpha and SIRT1 pathways. FEBS Lett. 2008, 582, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Puigserver, P.; Wu, Z.; Park, C.W.; Graves, R.; Wright, M.; Spiegelman, B.M. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 1998, 92, 829–839. [Google Scholar] [CrossRef]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef]
- Schreiber, S.N.; Emter, R.; Hock, M.B.; Knutti, D.; Cardenas, J.; Podvinec, M.; Oakeley, E.J.; Kralli, A. The estrogen-related receptor alpha (ERRalpha) functions in PPARgamma coactivator 1alpha (PGC-1alpha)-induced mitochondrial biogenesis. Proc. Natl. Acad. Sci. USA 2004, 101, 6472–6477. [Google Scholar] [CrossRef]
- Scarpulla, R.C. Nuclear control of respiratory chain expression by nuclear respiratory factors and PGC-1-related coactivator. Ann. N. Y. Acad Sci. 2008, 1147, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Larsson, N.G.; Wang, J.; Wilhelmsson, H.; Oldfors, A.; Rustin, P.; Lewandoski, M.; Barsh, G.S.; Clayton, D.A. Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat. Genet. 1998, 18, 231–236. [Google Scholar] [CrossRef]
- Scarpulla, R.C. Nuclear activators and coactivators in mammalian mitochondrial biogenesis. Biochim. Biophys. Acta 2002, 1576, 1–14. [Google Scholar] [CrossRef]
- Scarpulla, R.C. Transcriptional activators and coactivators in the nuclear control of mitochondrial function in mammalian cells. Gene 2002, 286, 81–89. [Google Scholar] [CrossRef]
- Aquilano, K.; Baldelli, S.; Pagliei, B.; Cannata, S.M.; Rotilio, G.; Ciriolo, M.R. p53 orchestrates the PGC-1alpha-mediated antioxidant response upon mild redox and metabolic imbalance. Antioxid. Redox Signal. 2013, 18, 386–399. [Google Scholar] [CrossRef]
- Lin, J.; Tarr, P.T.; Yang, R.; Rhee, J.; Puigserver, P.; Newgard, C.B.; Spiegelman, B.M. PGC-1beta in the regulation of hepatic glucose and energy metabolism. J. Biol. Chem. 2003, 278, 30843–30848. [Google Scholar] [CrossRef]
- Puigserver, P.; Rhee, J.; Donovan, J.; Walkey, C.J.; Yoon, J.C.; Oriente, F.; Kitamura, Y.; Altomonte, J.; Dong, H.; Accili, D.; et al. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature 2003, 423, 550–555. [Google Scholar] [CrossRef]
- Herzig, S.; Long, F.; Jhala, U.S.; Hedrick, S.; Quinn, R.; Bauer, A.; Rudolph, D.; Schutz, G.; Yoon, C.; Puigserver, P.; et al. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature 2001, 413, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Rhee, J.; Inoue, Y.; Yoon, J.C.; Puigserver, P.; Fan, M.; Gonzalez, F.J.; Spiegelman, B.M. Regulation of hepatic fasting response by PPARgamma coactivator-1alpha (PGC-1): Requirement for hepatocyte nuclear factor 4alpha in gluconeogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 4012–4017. [Google Scholar] [CrossRef] [PubMed]
- Leone, T.C.; Lehman, J.J.; Finck, B.N.; Schaeffer, P.J.; Wende, A.R.; Boudina, S.; Courtois, M.; Wozniak, D.F.; Sambandam, N.; Bernal-Mizrachi, C.; et al. PGC-1alpha deficiency causes multi-system energy metabolic derangements: Muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol. 2005, 3, e101. [Google Scholar] [CrossRef] [PubMed]
- Koo, S.H.; Satoh, H.; Herzig, S.; Lee, C.H.; Hedrick, S.; Kulkarni, R.; Evans, R.M.; Olefsky, J.; Montminy, M. PGC-1 promotes insulin resistance in liver through PPAR-alpha-dependent induction of TRB-3. Nat. Med. 2004, 10, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Sonoda, J.; Mehl, I.R.; Chong, L.W.; Nofsinger, R.R.; Evans, R.M. PGC-1beta controls mitochondrial metabolism to modulate circadian activity, adaptive thermogenesis, and hepatic steatosis. Proc. Natl. Acad. Sci. USA 2007, 104, 5223–5228. [Google Scholar] [CrossRef] [PubMed]
- Uldry, M.; Yang, W.; St-Pierre, J.; Lin, J.; Seale, P.; Spiegelman, B.M. Complementary action of the PGC-1 coactivators in mitochondrial biogenesis and brown fat differentiation. Cell Metab. 2006, 3, 333–341. [Google Scholar] [CrossRef]
- Lin, J.; Yang, R.; Tarr, P.T.; Wu, P.H.; Handschin, C.; Li, S.; Yang, W.; Pei, L.; Uldry, M.; Tontonoz, P.; et al. Hyperlipidemic effects of dietary saturated fats mediated through PGC-1beta coactivation of SREBP. Cell 2005, 120, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Piccinin, E.; Peres, C.; Bellafante, E.; Ducheix, S.; Pinto, C.; Villani, G.; Moschetta, A. Hepatic peroxisome proliferator-activated receptor gamma coactivator 1beta drives mitochondrial and anabolic signatures that contribute to hepatocellular carcinoma progression in mice. Hepatology 2018, 67, 884–898. [Google Scholar] [CrossRef]
- Bellafante, E.; Murzilli, S.; Salvatore, L.; Latorre, D.; Villani, G.; Moschetta, A. Hepatic-specific activation of peroxisome proliferator-activated receptor gamma coactivator-1beta protects against steatohepatitis. Hepatology 2013, 57, 1343–1356. [Google Scholar] [CrossRef]
- Piccinin, E.; Villani, G.; Moschetta, A. Metabolic aspects in NAFLD, NASH and hepatocellular carcinoma: The role of PGC1 coactivators. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 160–174. [Google Scholar] [CrossRef]
- Lin, J.; Wu, P.H.; Tarr, P.T.; Lindenberg, K.S.; St-Pierre, J.; Zhang, C.Y.; Mootha, V.K.; Jager, S.; Vianna, C.R.; Reznick, R.M.; et al. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell 2004, 119, 121–135. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jager, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006, 127, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Ciron, C.; Zheng, L.; Bobela, W.; Knott, G.W.; Leone, T.C.; Kelly, D.P.; Schneider, B.L. PGC-1alpha activity in nigral dopamine neurons determines vulnerability to alpha-synuclein. Acta Neuropathol. Commun. 2015, 3, 16. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Kang, S.U.; Zhang, S.; Karuppagounder, S.; Xu, J.; Lee, Y.K.; Kang, B.G.; Lee, Y.; Zhang, J.; Pletnikova, O.; et al. Adult Conditional Knockout of PGC-1alpha Leads to Loss of Dopamine Neurons. eNeuro 2016, 3. [Google Scholar] [CrossRef]
- Ekstrand, M.I.; Terzioglu, M.; Galter, D.; Zhu, S.; Hofstetter, C.; Lindqvist, E.; Thams, S.; Bergstrand, A.; Hansson, F.S.; Trifunovic, A.; et al. Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc. Natl. Acad. Sci. USA 2007, 104, 1325–1330. [Google Scholar] [CrossRef]
- Wareski, P.; Vaarmann, A.; Choubey, V.; Safiulina, D.; Liiv, J.; Kuum, M.; Kaasik, A. PGC-1{alpha} and PGC-1{beta} regulate mitochondrial density in neurons. J. Biol. Chem. 2009, 284, 21379–21385. [Google Scholar] [CrossRef]
- Clark, J.; Reddy, S.; Zheng, K.; Betensky, R.A.; Simon, D.K. Association of PGC-1alpha polymorphisms with age of onset and risk of Parkinson’s disease. BMC Med Genet. 2011, 12, 69. [Google Scholar] [CrossRef]
- Soyal, S.M.; Zara, G.; Ferger, B.; Felder, T.K.; Kwik, M.; Nofziger, C.; Dossena, S.; Schwienbacher, C.; Hicks, A.A.; Pramstaller, P.P.; et al. The PPARGC1A locus and CNS-specific PGC-1alpha isoforms are associated with Parkinson’s Disease. Neurobiol. Dis. 2019, 121, 34–46. [Google Scholar] [CrossRef]
- Su, X.; Chu, Y.; Kordower, J.H.; Li, B.; Cao, H.; Huang, L.; Nishida, M.; Song, L.; Wang, D.; Federoff, H.J. PGC-1alpha Promoter Methylation in Parkinson’s Disease. PLoS ONE 2015, 10, e0134087. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.D.; Qian, Y.W.; Xu, S.Q.; Wan, D.Y.; Sun, F.H.; Chen, S.D.; Xiao, Q. Expression of the gene coading for PGC-1alpha in peripheral blood leukocytes and related gene variants in patients with Parkinson’s disease. Parkinsonism. Relat. Disord. 2018, 51, 30–35. [Google Scholar] [CrossRef]
- Yang, X.; Xu, S.; Qian, Y.; He, X.; Chen, S.; Xiao, Q. Hypermethylation of the Gene Coding for PGC-1alpha in Peripheral Blood Leukocytes of Patients With Parkinson’s Disease. Front. Neurosci. 2020, 14, 97. [Google Scholar] [CrossRef]
- Eschbach, J.; von Einem, B.; Muller, K.; Bayer, H.; Scheffold, A.; Morrison, B.E.; Rudolph, K.L.; Thal, D.R.; Witting, A.; Weydt, P.; et al. Mutual exacerbation of peroxisome proliferator-activated receptor gamma coactivator 1alpha deregulation and alpha-synuclein oligomerization. Ann. Neurol. 2015, 77, 15–32. [Google Scholar] [CrossRef]
- Zheng, B.; Liao, Z.; Locascio, J.J.; Lesniak, K.A.; Roderick, S.S.; Watt, M.L.; Eklund, A.C.; Zhang-James, Y.; Kim, P.D.; Hauser, M.A.; et al. PGC-1alpha, a potential therapeutic target for early intervention in Parkinson’s disease. Sci. Transl. Med. 2010, 2, 52ra73. [Google Scholar] [CrossRef]
- Pacelli, C.; De Rasmo, D.; Signorile, A.; Grattagliano, I.; di Tullio, G.; D’Orazio, A.; Nico, B.; Comi, G.P.; Ronchi, D.; Ferranini, E.; et al. Mitochondrial defect and PGC-1alpha dysfunction in parkin-associated familial Parkinson’s disease. Biochim. Biophys. Acta 2011, 1812, 1041–1053. [Google Scholar] [CrossRef] [PubMed]
- Antony, P.M.A.; Kondratyeva, O.; Mommaerts, K.; Ostaszewski, M.; Sokolowska, K.; Baumuratov, A.S.; Longhino, L.; Poulain, J.F.; Grossmann, D.; Balling, R.; et al. Fibroblast mitochondria in idiopathic Parkinson’s disease display morphological changes and enhanced resistance to depolarization. Sci. Rep. 2020, 10, 1569. [Google Scholar] [CrossRef] [PubMed]
- Logroscino, G.; Marder, K.; Cote, L.; Tang, M.X.; Shea, S.; Mayeux, R. Dietary lipids and antioxidants in Parkinson’s disease: A population-based, case-control study. Ann. Neurol. 1996, 39, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhang, S.M.; Hernan, M.A.; Willett, W.C.; Ascherio, A. Dietary intakes of fat and risk of Parkinson’s disease. Am. J. Epidemiol. 2003, 157, 1007–1014. [Google Scholar] [CrossRef]
- Morris, J.K.; Bomhoff, G.L.; Stanford, J.A.; Geiger, P.C. Neurodegeneration in an animal model of Parkinson’s disease is exacerbated by a high-fat diet. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 299, R1082–R1090. [Google Scholar] [CrossRef]
- Merzetti, E.M.; Staveley, B.E. spargel, the PGC-1alpha homologue, in models of Parkinson disease in Drosophila melanogaster. BMC Neurosci. 2015, 16, 70. [Google Scholar] [CrossRef]
- Ng, C.H.; Basil, A.H.; Hang, L.; Tan, R.; Goh, K.L.; O’Neill, S.; Zhang, X.; Yu, F.; Lim, K.L. Genetic or pharmacological activation of the Drosophila PGC-1alpha ortholog spargel rescues the disease phenotypes of genetic models of Parkinson’s disease. Neurobiol. Aging 2017, 55, 33–37. [Google Scholar] [CrossRef]
- Ye, Q.; Huang, W.; Li, D.; Si, E.; Wang, J.; Wang, Y.; Chen, C.; Chen, X. Overexpression of PGC-1alpha Influences Mitochondrial Signal Transduction of Dopaminergic Neurons. Mol. Neurobiol. 2016, 53, 3756–3770. [Google Scholar] [CrossRef] [PubMed]
- Ferretta, A.; Gaballo, A.; Tanzarella, P.; Piccoli, C.; Capitanio, N.; Nico, B.; Annese, T.; Di Paola, M.; Dell’aquila, C.; De Mari, M.; et al. Effect of resveratrol on mitochondrial function: Implications in parkin-associated familiar Parkinson’s disease. Biochim. Biophys. Acta 2014, 1842, 902–915. [Google Scholar] [CrossRef]
- Mudo, G.; Makela, J.; Di Liberto, V.; Tselykh, T.V.; Olivieri, M.; Piepponen, P.; Eriksson, O.; Malkia, A.; Bonomo, A.; Kairisalo, M.; et al. Transgenic expression and activation of PGC-1alpha protect dopaminergic neurons in the MPTP mouse model of Parkinson’s disease. Cell. Mol. Life Sci. 2012, 69, 1153–1165. [Google Scholar] [CrossRef]
- Clark, J.; Silvaggi, J.M.; Kiselak, T.; Zheng, K.; Clore, E.L.; Dai, Y.; Bass, C.E.; Simon, D.K. Pgc-1alpha overexpression downregulates Pitx3 and increases susceptibility to MPTP toxicity associated with decreased Bdnf. PLoS ONE 2012, 7, e48925. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Ko, H.S.; Kang, H.; Lee, Y.; Lee, Y.I.; Pletinkova, O.; Troconso, J.C.; Dawson, V.L.; Dawson, T.M. PARIS (ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson’s disease. Cell 2011, 144, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Stevens, D.A.; Kang, S.U.; Jiang, H.; Lee, Y.I.; Ko, H.S.; Scarffe, L.A.; Umanah, G.E.; Kang, H.; Ham, S.; et al. PINK1 Primes Parkin-Mediated Ubiquitination of PARIS in Dopaminergic Neuronal Survival. Cell Rep. 2017, 18, 918–932. [Google Scholar] [CrossRef]
- Kumar, M.; Acevedo-Cintron, J.; Jhaldiyal, A.; Wang, H.; Andrabi, S.A.; Eacker, S.; Karuppagounder, S.S.; Brahmachari, S.; Chen, R.; Kim, H.; et al. Defects in Mitochondrial Biogenesis Drive Mitochondrial Alterations in PARKIN-Deficient Human Dopamine Neurons. Stem Cell Rep. 2020, 15, 629–645. [Google Scholar] [CrossRef]
- Pirooznia, S.K.; Yuan, C.; Khan, M.R.; Karuppagounder, S.S.; Wang, L.; Xiong, Y.; Kang, S.U.; Lee, Y.; Dawson, V.L.; Dawson, T.M. PARIS induced defects in mitochondrial biogenesis drive dopamine neuron loss under conditions of parkin or PINK1 deficiency. Mol. Neurodegener. 2020, 15, 17. [Google Scholar] [CrossRef]
- Stevens, D.A.; Lee, Y.; Kang, H.C.; Lee, B.D.; Lee, Y.I.; Bower, A.; Jiang, H.; Kang, S.U.; Andrabi, S.A.; Dawson, V.L.; et al. Parkin loss leads to PARIS-dependent declines in mitochondrial mass and respiration. Proc. Natl. Acad. Sci. USA 2015, 112, 11696–11701. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Bernard-Marissal, N.; Moullan, N.; D’Amico, D.; Auwerx, J.; Moore, D.J.; Knott, G.; Aebischer, P.; Schneider, B.L. Parkin functionally interacts with PGC-1alpha to preserve mitochondria and protect dopaminergic neurons. Hum. Mol. Genet. 2017, 26, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Sterky, F.H.; Mourier, A.; Terzioglu, M.; Cullheim, S.; Olson, L.; Larsson, N.G. Mitofusin 2 is necessary for striatal axonal projections of midbrain dopamine neurons. Hum. Mol. Genet. 2012, 21, 4827–4835. [Google Scholar] [CrossRef]
- Zhong, N.; Xu, J. Synergistic activation of the human MnSOD promoter by DJ-1 and PGC-1alpha: Regulation by SUMOylation and oxidation. Hum. Mol. Genet. 2008, 17, 3357–3367. [Google Scholar] [CrossRef]
- Siddiqui, A.; Chinta, S.J.; Mallajosyula, J.K.; Rajagopolan, S.; Hanson, I.; Rane, A.; Melov, S.; Andersen, J.K. Selective binding of nuclear alpha-synuclein to the PGC1alpha promoter under conditions of oxidative stress may contribute to losses in mitochondrial function: Implications for Parkinson’s disease. Free. Radic. Biol. Med. 2012, 53, 993–1003. [Google Scholar] [CrossRef]
- Ebrahim, A.S.; Ko, L.W.; Yen, S.H. Reduced expression of peroxisome-proliferator activated receptor gamma coactivator-1alpha enhances alpha-synuclein oligomerization and down regulates AKT/GSK3beta signaling pathway in human neuronal cells that inducibly express alpha-synuclein. Neurosci. Lett. 2010, 473, 120–125. [Google Scholar] [CrossRef]
- Shimura, H.; Schlossmacher, M.G.; Hattori, N.; Frosch, M.P.; Trockenbacher, A.; Schneider, R.; Mizuno, Y.; Kosik, K.S.; Selkoe, D.J. Ubiquitination of a new form of alpha-synuclein by parkin from human brain: Implications for Parkinson’s disease. Science 2001, 293, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Norris, K.L.; Hao, R.; Chen, L.F.; Lai, C.H.; Kapur, M.; Shaughnessy, P.J.; Chou, D.; Yan, J.; Taylor, J.P.; Engelender, S.; et al. Convergence of Parkin, PINK1, and alpha-Synuclein on Stress-induced Mitochondrial Morphological Remodeling. J. Biol. Chem. 2015, 290, 13862–13874. [Google Scholar] [CrossRef]
- Handschin, C. The biology of PGC-1alpha and its therapeutic potential. Trends Pharmacol. Sci. 2009, 30, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Fanibunda, S.E.; Deb, S.; Maniyadath, B.; Tiwari, P.; Ghai, U.; Gupta, S.; Figueiredo, D.; Weisstaub, N.; Gingrich, J.A.; Vaidya, A.D.B.; et al. Serotonin regulates mitochondrial biogenesis and function in rodent cortical neurons via the 5-HT2A receptor and SIRT1-PGC-1alpha axis. Proc. Natl. Acad. Sci. USA 2019, 116, 11028–11037. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piccinin, E.; Sardanelli, A.M.; Seibel, P.; Moschetta, A.; Cocco, T.; Villani, G. PGC-1s in the Spotlight with Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 3487. https://doi.org/10.3390/ijms22073487
Piccinin E, Sardanelli AM, Seibel P, Moschetta A, Cocco T, Villani G. PGC-1s in the Spotlight with Parkinson’s Disease. International Journal of Molecular Sciences. 2021; 22(7):3487. https://doi.org/10.3390/ijms22073487
Chicago/Turabian StylePiccinin, Elena, Anna Maria Sardanelli, Peter Seibel, Antonio Moschetta, Tiziana Cocco, and Gaetano Villani. 2021. "PGC-1s in the Spotlight with Parkinson’s Disease" International Journal of Molecular Sciences 22, no. 7: 3487. https://doi.org/10.3390/ijms22073487
APA StylePiccinin, E., Sardanelli, A. M., Seibel, P., Moschetta, A., Cocco, T., & Villani, G. (2021). PGC-1s in the Spotlight with Parkinson’s Disease. International Journal of Molecular Sciences, 22(7), 3487. https://doi.org/10.3390/ijms22073487