
Exercise—A Panacea of Metabolic Dysregulation in Cancer: Physiological and Molecular Insights


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Cancer Survival Depends on Better Metabolism Management Strategies
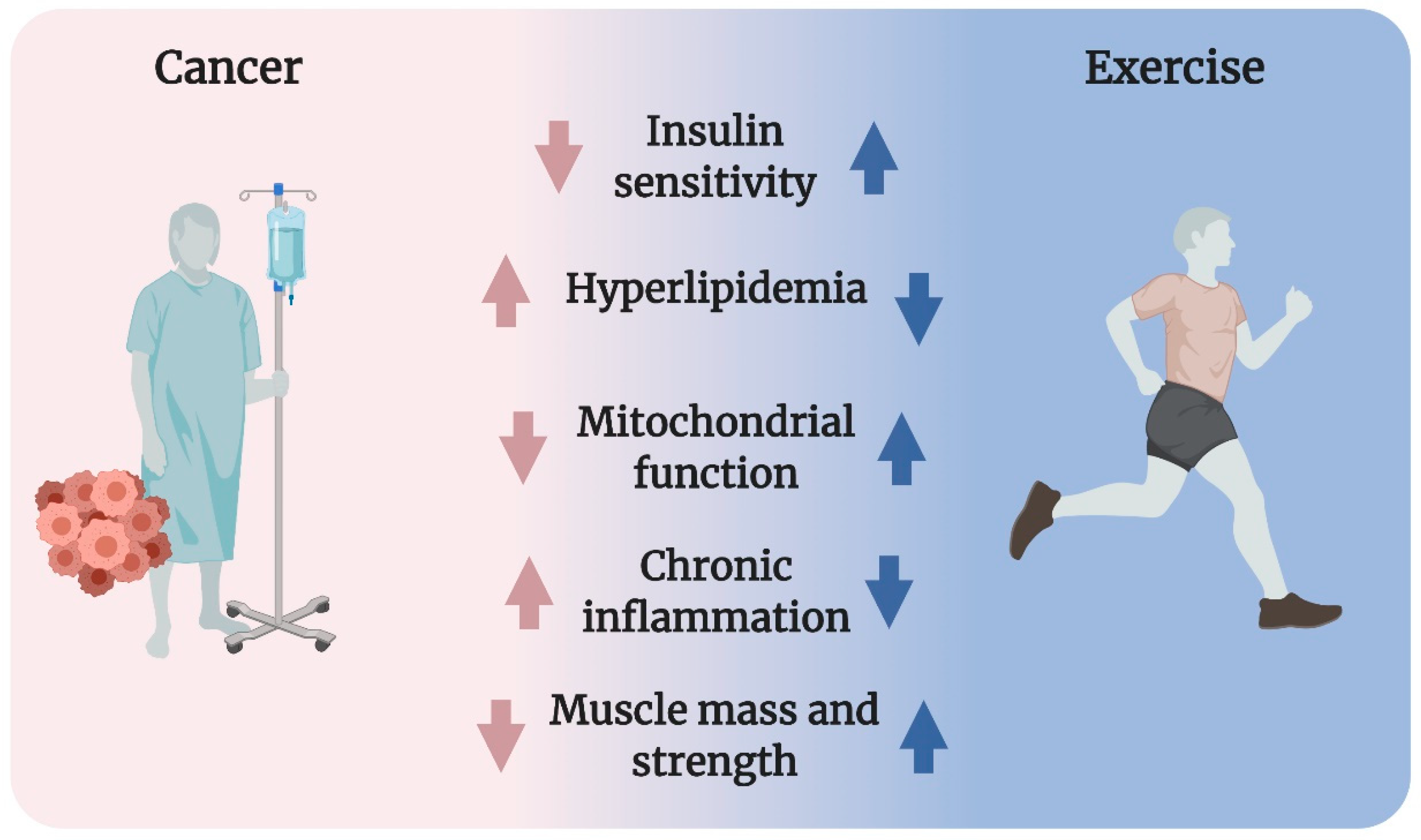
2. Exercise as a Panacea in Metabolic Dysregulation in Cancer
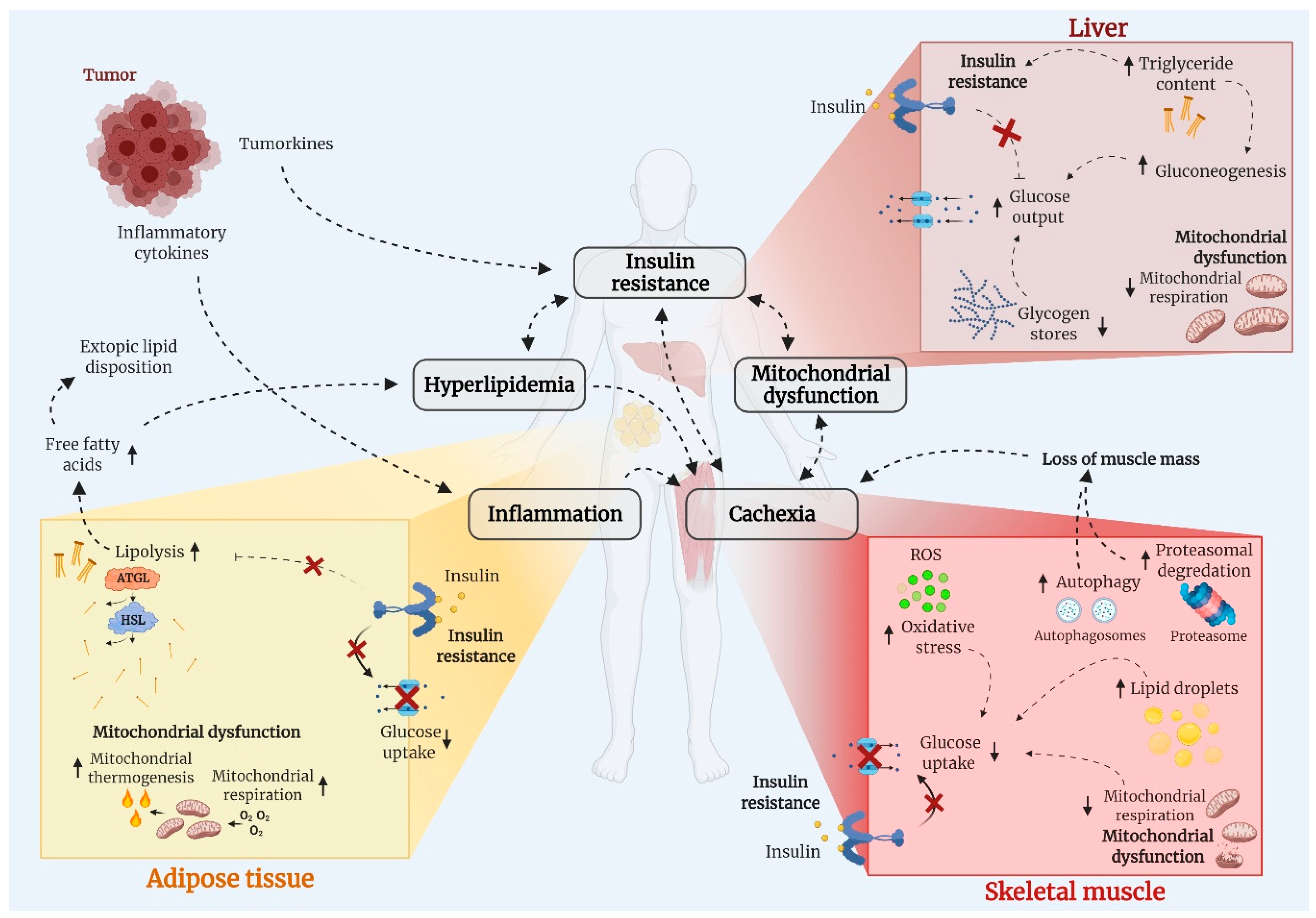
3. Potential Mechanism Underlying Cancer-Associated Metabolic Dysregulation
3.1. Insulin Resistance
3.2. Hyperlipidemia
3.3. Mitochondrial Dysfunction
3.4. Chronic Inflammation
3.5. Cancer-Associated Cachexia
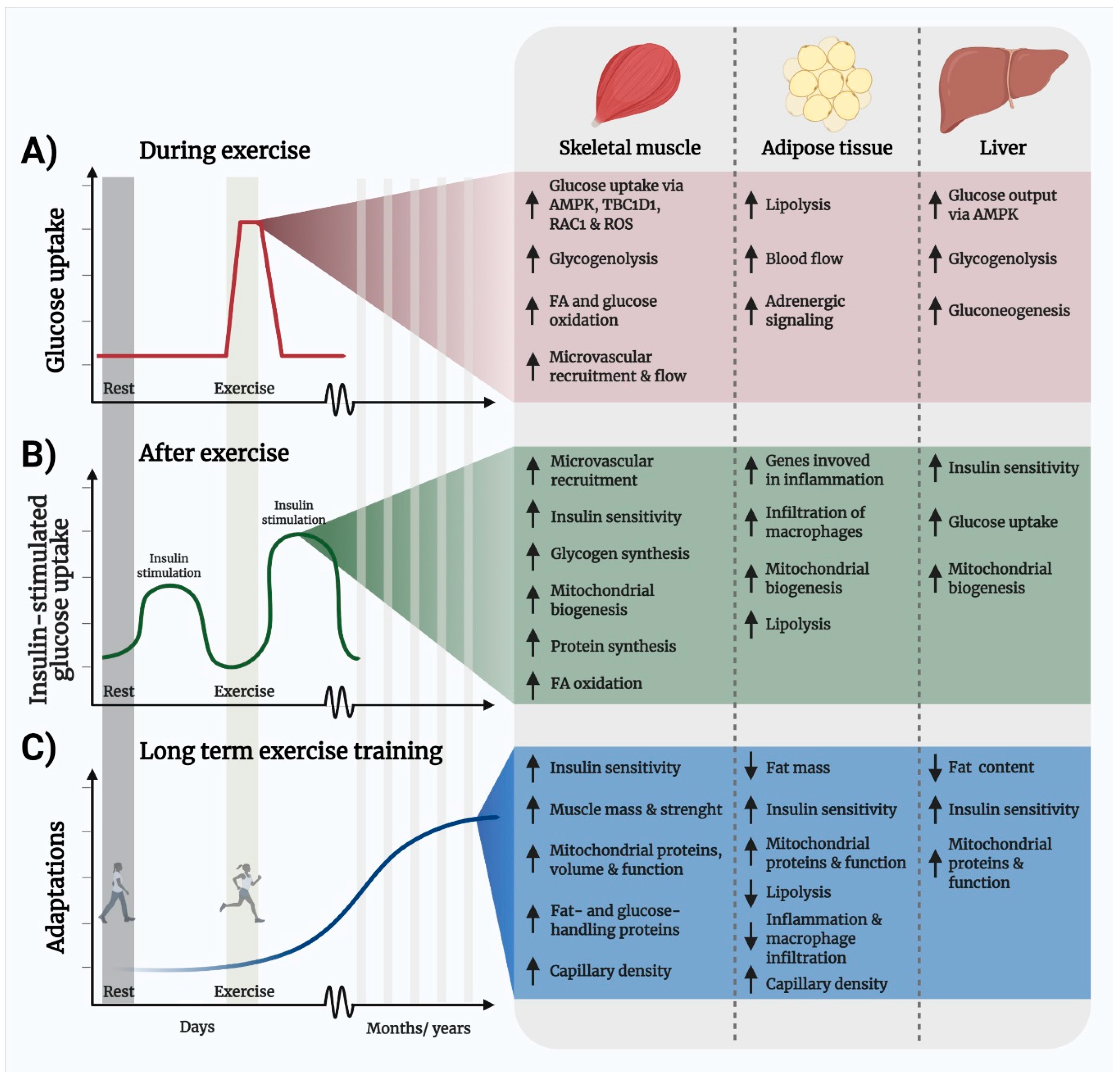
4. Exercise Is the Most Powerful Means to Improve Metabolic Regulation
4.1. Exercise Improves Insulin Sensitivity and Glucose Disposal
Exercise and Insulin Sensitivity in the Context of Cancer
4.2. Hyperlipidemia Can Be Managed by Exercise
Exercise and Hyperlipidemia in the Context of Cancer
4.3. Skeletal Muscle Mitochondrial Volume and Function Is Upregulated by Exercise
Exercise-Induced Mitochondrial Improvements in the Context of Cancer
4.4. Exercise Lowers Chronic Inflammation
Exercise Lowers Chronic Inflammation in the Context of Cancer
4.5. Exercise Increases Skeletal Muscle Mass and Strength
Exercise Enhances Muscle Mass and Strength in the Context of Cancer
5. Unanswered Questions
- The optimal exercise regimen for benefitting metabolic regulation in cancer remains to be established.
- The appropriate implementation of exercise into the oncological treatment of cancer must be determined.
- It is important to establish whether an acute exercise bout fully stimulates insulin-independent glucose uptake into the exercising muscles in cancer patients. This will be vital information in the daily life for cancer patients, as improved glycemic control is associated with the effectiveness of cancer treatment and improved survival.
- It would have real-life patient benefits to determine whether the insulin-sensitizing effect of one bout of exercise exists in patients with cancer and can be exploited in relation to the timing between exercise and meals to maximize glucose disposal, reduce hyperinsulinemia, and elevate muscle protein synthesis.
- For cancer patients with cancer cachexia, it is important to determine whether exercise can treat the loss of muscle mass and improve strength and which exercise regimen is most effective.
- Deeper knowledge of the molecular mechanisms by which exercise benefits metabolic regulation is needed to identify novel therapeutically drug targets. This is especially important in patients with cancer cachexia who are unlikely to easily exercise [260].
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rohdenburg, G.L.; Bernhard, A.; Krehbiel, O. Sugar tolerance in cancer. J. Am. Med. Assoc. 1919, 72, 1528. [Google Scholar] [CrossRef]
- Jasani, B.; Donaldson, L.J.; Ratcliffe, J.G.; Sokhi, G.S. Mechanism of impaired glucose tolerance in patients with neoplasia. Br. J. Cancer 1978, 38, 287–292. [Google Scholar] [CrossRef]
- Bishop, J.S.; Marks, P.A. Studies on carbohydrate metabolism in patients with neoplastic disease. II. Response to insulin administration. J. Clin. Investig. 1959, 38, 668–672. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Luque, R.M.; López-Sánchez, L.M.; Villa-Osaba, A.; Luque, I.M.; Santos-Romero, A.L.; Yubero-Serrano, E.M.; Cara-García, M.; Álvarez-Benito, M.; López-Miranda, J.; Gahete, M.D.; et al. Breast cancer is associated to impaired glucose/insulin homeostasis in premenopausal obese/overweight patients. Oncotarget 2017, 8, 81462–81474. [Google Scholar] [CrossRef]
- Permert, J.; Adrian, T.E.; Jacobsson, P.; Jorfelt, L.; Fruin, A.B.; Larsson, J. Is profound peripheral insulin resistance in patients with pancreatic cancer caused by a tumor-associated factor? Am. J. Surg. 1993, 165, 61–67. [Google Scholar] [CrossRef]
- Winter, A.; MacAdams, J.; Chevalier, S. Normal protein anabolic response to hyperaminoacidemia in insulin-resistant patients with lung cancer cachexia. Clin. Nutr. 2012, 31, 765–773. [Google Scholar] [CrossRef]
- Yoshikawa, T.; Noguchi, Y.; Matsumoto, A. Effects of tumor removal and body weight loss on insulin resistance in patients with cancer. Surgery 1994, 116, 62–66. [Google Scholar]
- Yoshikawa, T.; Noguchi, Y.; Doi, C.; Makino, T.; Nomura, K. Insulin resistance in patients with cancer: Relationships with tumor site, tumor stage, body-weight loss, acute-phase response, and energy expenditure. Nutrition 2001, 17, 590–593. [Google Scholar] [CrossRef]
- Pisters, P.W.; Cersosimo, E.; Rogatko, A.; Brennan, M.F. Insulin action on glucose and branched-chain amino acid metabolism in cancer cachexia: Differential effects of insulin. Surgery 1992, 111, 301–310. [Google Scholar]
- Copeland, G.P.; Leinster, S.J.; Davis, J.C.; Hipkin, L.J. Insulin resistance in patients with colorectal cancer. Br. J. Surg. 1987, 74, 1031–1035. [Google Scholar] [CrossRef]
- Lipscombe, L.L.; Chan, W.W.; Yun, L.; Austin, P.C.; Anderson, G.M.; Rochon, P.A. Incidence of diabetes among postmenopausal breast cancer survivors. Diabetologia 2013, 56, 476–483. [Google Scholar] [CrossRef]
- Singh, S.; Earle, C.C.; Bae, S.J.; Fischer, H.D.; Yun, L.; Austin, P.C.; Rochon, P.A.; Anderson, G.M.; Lipscombe, L. Incidence of Diabetes in Colorectal Cancer Survivors. J. Natl. Cancer Inst. 2016, 108, djv402. [Google Scholar] [CrossRef]
- Schoen, R.E.; Tangen, C.M.; Kuller, L.H.; Burke, G.L.; Cushman, M.; Tracy, R.P.; Dobs, A.; Savage, P.J. Increased blood glucose and insulin, body size, and incident colorectal cancer. J. Natl. Cancer Inst. 1999, 91, 1147–1154. [Google Scholar] [CrossRef] [PubMed]
- Stattin, P.; Björ, O.; Ferrari, P.; Lukanova, A.; Lenner, P.; Lindahl, B.; Hallmans, G.; Kaaks, R. Prospective study of hyperglycemia and cancer risk. Diabetes Care 2007, 30, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Giovannucci, E.; Michaud, D. The Role of Obesity and Related Metabolic Disturbances in Cancers of the Colon, Prostate, and Pancreas. Gastroenterology 2007, 132, 2208–2225. [Google Scholar] [CrossRef]
- Rapp, K.; Schroeder, J.; Klenk, J.; Ulmer, H.; Concin, H.; Diem, G.; Oberaigner, W.; Weiland, S.K. Fasting blood glucose and cancer risk in a cohort of more than 140,000 adults in Austria. Diabetologia 2006, 49, 945–952. [Google Scholar] [CrossRef] [PubMed]
- Renehan, A.G.; Tyson, M.; Egger, M.; Heller, R.F.; Zwahlen, M. Body-mass index and incidence of cancer: A systematic review and meta-analysis of prospective observational studies. Lancet 2008, 371, 569–578. [Google Scholar] [CrossRef]
- Saydah, S.H.; Loria, C.M.; Eberhardt, M.S.; Brancati, F.L. Abnormal glucose tolerance and the risk of cancer death in the United States. Am. J. Epidemiol. 2003, 157, 1092–1100. [Google Scholar] [CrossRef]
- Sun, H.J.; Ohrr, H.; Sull, J.W.; Yun, J.E.; Ji, M.; Samet, J.M. Fasting serum glucose level and cancer risk in Korean men and women. J. Am. Med. Assoc. 2005, 293, 194–202. [Google Scholar] [CrossRef]
- Bhaskaran, K.; Douglas, I.; Forbes, H.; Dos-Santos-Silva, I.; Leon, D.A.; Smeeth, L. Body-mass index and risk of 22 specific cancers: A population-based cohort study of 5·24 million UK adults. Lancet 2014, 384, 755–765. [Google Scholar] [CrossRef]
- Calle, E.E.; Rodriguez, C.; Walker-Thurmond, K.; Thun, M.J. Overweight, Obesity, and Mortality from Cancer in a Prospectively Studied Cohort of U.S. Adults. N. Engl. J. Med. 2003, 348, 1625–1638. [Google Scholar] [CrossRef] [PubMed]
- Campbell, P.T.; Newton, C.C.; Patel, A.V.; Jacobs, E.J.; Gapstur, S.M. Diabetes and cause-specific mortality in a prospective cohort of one million U.S. adults. Diabetes Care 2012, 35, 1835–1844. [Google Scholar] [CrossRef]
- Harding, J.L.; Andes, L.J.; Gregg, E.W.; Cheng, Y.J.; Weir, H.K.; Bullard, K.M.; Burrows, N.R.; Imperatore, G. Trends in cancer mortality among people with vs without diabetes in the USA, 1988-2015. Diabetologia 2020, 63, 75–84. [Google Scholar] [CrossRef]
- Calip, G.S.; Malone, K.E.; Gralow, J.R.; Stergachis, A.; Hubbard, R.A.; Boudreau, D.M. Metabolic syndrome and outcomes following early-stage breast cancer. Breast Cancer Res. Treat. 2014, 148, 363–377. [Google Scholar] [CrossRef] [PubMed]
- Currie, C.J.; Poole, C.D.; Jenkins-Jones, S.; Gale, E.A.M.; Johnson, J.A.; Morgan, C.L. Mortality after incident cancer in people with and without type 2 diabetes: Impact of metformin on survival. Diabetes Care 2012, 35, 299–304. [Google Scholar] [CrossRef]
- Bowker, S.L.; Majumdar, S.R.; Veugelers, P.; Johnson, J.A. Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin. Diabetes Care 2006, 29, 254–258. [Google Scholar] [CrossRef]
- Pearson-Stuttard, J.; Bennett, J.; Cheng, Y.J.; Vamos, E.P.; Cross, A.J.; Ezzati, M.; Gregg, E.W. Trends in predominant causes of death in individuals with and without diabetes in England from 2001 to 2018: An epidemiological analysis of linked primary care records. Lancet Diabetes Endocrinol. 2021, 9, 165–173. [Google Scholar] [CrossRef]
- Mills, K.T.; Bellows, C.F.; Hoffman, A.E.; Kelly, T.N.; Gagliardi, G. Diabetes mellitus and colorectal cancer prognosis: A meta-analysis. Dis. Colon Rectum 2013, 56, 1304–1319. [Google Scholar] [CrossRef]
- Schmitz, K.H.; Campbell, A.M.; Stuiver, M.M.; Pinto, B.M.; Schwartz, A.L.; Morris, G.S.; Ligibel, J.A.; Cheville, A.; Galvão, D.A.; Alfano, C.M.; et al. Exercise is medicine in oncology: Engaging clinicians to help patients move through cancer. CA Cancer J. Clin. 2019, 69, 468–484. [Google Scholar] [CrossRef]
- Friedenreich, C.M.; Neilson, H.K.; Farris, M.S.; Courneya, K.S. Physical activity and cancer outcomes: A precision medicine approach. Clin. Cancer Res. 2016, 22, 4766–4775. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Gunnarsson, R.; Björkman, O.; Olsson, M.; Wahren, J. Effects of insulin on peripheral and splanchnic glucose metabolism in noninsulin-dependent (type II) diabetes mellitus. J. Clin. Investig. 1985, 76, 149–155. [Google Scholar] [CrossRef]
- Ferrannini, E.; Bjorkman, O.; Reichard, G.A.; Pilo, A.; Olsson, M.; Wahren, J.; DeFronzo, R.A. The disposal of an oral glucose load in healthy subjects. A quantitative study. Diabetes 1985, 34, 580–588. [Google Scholar] [CrossRef] [PubMed]
- Booth, F.W.; Roberts, C.K.; Laye, M.J. Lack of exercise is a major cause of chronic diseases. Compr. Physiol. 2012, 2, 1143–1211. [Google Scholar] [CrossRef] [PubMed]
- Neufer, P.D.; Bamman, M.M.; Muoio, D.M.; Bouchard, C.; Cooper, D.M.; Goodpaster, B.H.; Booth, F.W.; Kohrt, W.M.; Gerszten, R.E.; Mattson, M.P.; et al. Understanding the Cellular and Molecular Mechanisms of Physical Activity-Induced Health Benefits. Cell Metab. 2015, 22, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Sylow, L.; Tokarz, V.; Richter, E.A.; Klip, A. The many actions of insulin in skeletal muscle, the paramount tissue determining glycemia. Cell Metab. 2021. [Google Scholar] [CrossRef]
- Röder, P.V.; Wu, B.; Liu, Y.; Han, W. Pancreatic regulation of glucose homeostasis. Exp. Mol. Med. 2016, 48, e219. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. The metabolism of carcinoma cells 1. J. Cancer Res. 1925, 9, 148–163. [Google Scholar] [CrossRef]
- Ye, H.; Adane, B.; Khan, N.; Alexeev, E.; Nusbacher, N.; Minhajuddin, M.; Stevens, B.M.; Winters, A.C.; Lin, X.; Ashton, J.M.; et al. Subversion of Systemic Glucose Metabolism as a Mechanism to Support the Growth of Leukemia Cells. Cancer Cell 2018, 34, 659–673.e6. [Google Scholar] [CrossRef]
- Vigneri, R.; Sciacca, L.; Vigneri, P. Rethinking the Relationship between Insulin and Cancer. Trends Endocrinol. Metab. 2020, 31, 551–560. [Google Scholar] [CrossRef]
- Han, X.; Raun, S.H.; Carlsson, M.; Sjøberg, K.A.; Henriquez-Olguín, C.; Ali, M.; Lundsgaard, A.; Fritzen, A.M.; Møller, L.L.V.; Li, Z.; et al. Cancer causes metabolic perturbations associated with reduced insulin-stimulated glucose uptake in peripheral tissues and impaired muscle microvascular perfusion. Metabolism 2020, 105, 154169. [Google Scholar] [CrossRef]
- Asp, M.L.; Tian, M.; Wendel, A.A.; Belury, M.A. Evidence for the contribution of insulin resistance to the development of cachexia in tumor-bearing mice. Int. J. cancer 2010, 126, 756–763. [Google Scholar] [CrossRef]
- Lang, C.H.; Skrepnik, N.; Dobrescu, C.; Burns, A.H. Impairment of insulin action on peripheral glucose uptake and hepatic glucose production in tumor-bearing rats. Am. J. Physiol. Integr. Comp. Physiol. 2017, 265, R356–R364. [Google Scholar] [CrossRef] [PubMed]
- Puppa, M.J.; White, J.P.; Sato, S.; Cairns, M.; Baynes, J.W.; Carson, J.A. Gut barrier dysfunction in the ApcMin/+ mouse model of colon cancer cachexia. Biochim. Biophys. Acta Mol. Basis Dis. 2011, 1812, 1601–1606. [Google Scholar] [CrossRef]
- Keske, M.A.; Clerk, L.H.; Price, W.J.; Jahn, L.A.; Barrett, E.J. Obesity Blunts Microvascular Recruitment in Human Forearm Muscle After a Mixed Meal. Diabetes Care 2009, 32, 1672–1677. [Google Scholar] [CrossRef]
- Sylow, L.; Jensen, T.E.; Kleinert, M.; Højlund, K.; Kiens, B.; Wojtaszewski, J.; Prats, C.; Schjerling, P.; Richter, E.A. Rac1 signaling is required for insulin-stimulated glucose uptake and is dysregulated in insulin-resistant murine and human skeletal muscle. Diabetes 2013, 62, 1865–1875. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, N.; Gavin, M.G.; Quinn, W.J.; Luongo, T.S.; Gelfer, R.G.; Baur, J.A.; Titchenell, P.M. The role of skeletal muscle Akt in the regulation of muscle mass and glucose homeostasis. Mol. Metab. 2019, 28, 1–13. [Google Scholar] [CrossRef]
- Sylow, L.; Kleinert, M.; Pehmøller, C.; Prats, C.; Chiu, T.T.; Klip, A.; Richter, E.A.; Jensen, T.E. Akt and Rac1 signaling are jointly required for insulin-stimulated glucose uptake in skeletal muscle and downregulated in insulin resistance. Cell. Signal. 2014, 26, 323–331. [Google Scholar] [CrossRef]
- Chadt, A.; Immisch, A.; De Wendt, C.; Springer, C.; Zhou, Z.; Stermann, T.; Holman, G.D.; Loffing-Cueni, D.; Loffing, J.; Joost, H.G.; et al. Deletion of both rab-GTPase-activating proteins TBC1D1 and TBC1D4 in mice eliminates insulin- and AICAR-stimulated glucose transport. Diabetes 2015, 64, 746–759. [Google Scholar] [CrossRef] [PubMed]
- Timmers, S.; De Vogel-Van Den Bosch, J.; Towler, M.C.; Schaart, G.; Moonen-Kornips, E.; Mensink, R.P.; Hesselink, M.K.; Hardie, G.D.; Schrauwen, P. Prevention of high-fat diet-induced muscular lipid accumulation in rats by α lipoic acid is not mediated by ampk activation. J. Lipid Res. 2010, 51, 352–359. [Google Scholar] [CrossRef]
- Kim, Y.B.; Nikoulina, S.E.; Ciaraldi, T.P.; Henry, R.R.; Kahn, B.B. Normal insulin-dependent activation of Akt/protein kinase B, with diminished activation of phosphoinositide 3-kinase, in muscle in type 2 diabetes. J. Clin. Investig. 1999, 104, 733–741. [Google Scholar] [CrossRef]
- Shao, J.; Yamashita, H.; Qiao, L.; Friedman, J. Decreased Akt kinase activity and insulin resistance C57BL/KsJ-Lepr(db/db) mice. J. Endocrinol. 2000, 167, 107–115. [Google Scholar] [CrossRef]
- Karlsson, H.K.R.; Zierath, J.R.; Kane, S.; Krook, A.; Lienhard, G.E.; Wallberg-Henriksson, H. Insulin-stimulated phosphorylation of the Akt substrate AS160 is impaired in skeletal muscle of type 2 diabetic subjects. Diabetes 2005, 54, 1692–1697. [Google Scholar] [CrossRef]
- Leij-Halfwerk, S.; Dagnelie, P.C.; Van Den Berg, J.W.O.; Wattimena, J.D.L.; Hordijk-Luijk, C.H.; Wilson, J.H.P. Weight loss and elevated gluconeogenesis from alanine in lung cancer patients. Am. J. Clin. Nutr. 2000, 71, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Leij-Halfwerk, S.; Van Den Berg, J.W.O.; Sijens, P.E.; Wilson, J.H.P.; Oudkerk, M.; Dagnelie, P.C. Altered hepatic gluconeogenesis during L-alanine infusion in weight- losing lung cancer patients as observed by phosphorus magnetic resonance spectroscopy and turnover measurements. Cancer Res. 2000, 60, 618–623. [Google Scholar]
- Goncalves, M.D.; Hwang, S.-K.; Pauli, C.; Murphy, C.J.; Cheng, Z.; Hopkins, B.D.; Wu, D.; Loughran, R.M.; Emerling, B.M.; Zhang, G.; et al. Fenofibrate prevents skeletal muscle loss in mice with lung cancer. Proc. Natl. Acad. Sci. USA 2018, 115, E743–E752. [Google Scholar] [CrossRef] [PubMed]
- Pötgens, S.A.; Thibaut, M.M.; Joudiou, N.; Sboarina, M.; Neyrinck, A.M.; Cani, P.D.; Claus, S.P.; Delzenne, N.M.; Bindels, L.B. Multi-compartment metabolomics and metagenomics reveal major hepatic and intestinal disturbances in cancer cachectic mice. J. Cachexia Sarcopenia Muscle 2021. [Google Scholar] [CrossRef]
- Raza, U.; Asif, M.R.; Bin Rehman, A.; Sheikh, A. Hyperlipidemia and hyper glycaemia in Breast Cancer Patients is related to disease stage. Pakistan J. Med. Sci. 2018, 34. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Wen, P.; Su, J.; Li, Q.; Ren, Y.; Liu, Y.; Shen, R.; Ren, J. Elevated serum triglyceride and low-density lipoprotein cholesterol promotes the formation of colorectal polyps. BMC Gastroenterol. 2019, 19, 195. [Google Scholar] [CrossRef]
- Shah, F.D.; Shukla, S.N.; Shah, P.M.; Patel, H.R.H. Prabhudas Shankerbhai Patel Significance of Alterations in Plasma Lipid Profile Levels in Breast Cancer. Integr. Cancer Ther. 2008, 7, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Fiorenza, A.M.; Branchi, A.; Sommariva, D. Serum lipoprotein profile in patients with cancer. A comparison with non-cancer subjects. Int. J. Clin. Lab. Res. 2000, 30, 141–145. [Google Scholar] [CrossRef]
- Das, S.K.; Eder, S.; Schauer, S.; Diwoky, C.; Temmel, H.; Guertl, B.; Gorkiewicz, G.; Tamilarasan, K.P.; Kumari, P.; Trauner, M.; et al. Adipose triglyceride lipase contributes to cancer-associated cachexia. Science 2011, 333, 233–238. [Google Scholar] [CrossRef]
- López-Soriano, J.; Llovera, M.; Carbó, N.; García-Martínez, C.; López-Soriano, F.J.; Argiles, J.M. Lipid metabolism in tumour-bearing mice: Studies with knockout mice for tumour necrosis factor receptor 1 protein. Mol. Cell. Endocrinol. 1997, 132, 93–99. [Google Scholar] [CrossRef]
- Donatto, F.F.; Neves, R.X.; Rosa, F.O.; Camargo, R.G.; Ribeiro, H.; Matos-Neto, E.M.; Seelaender, M. Resistance exercise modulates lipid plasma profile and cytokine content in the adipose tissue of tumour-bearing rats. Cytokine 2013, 61, 426–432. [Google Scholar] [CrossRef]
- Huang, J.; Li, L.; Lian, J.; Schauer, S.; Vesely, P.W.; Kratky, D.; Hoefler, G.; Lehner, R. Tumor-Induced Hyperlipidemia Contributes to Tumor Growth. Cell Rep. 2016, 15, 336–348. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, T.M.; Ardeshirpour, F.; Asher, S.A.; Winnike, J.H.; Yin, X.; George, J.; Guttridge, D.C.; He, W.; Wysong, A.; Willis, M.S.; et al. Metabolomic analysis of cancer cachexia reveals distinct lipid and glucose alterations. Metabolomics 2008, 4, 216–225. [Google Scholar] [CrossRef]
- Silvério, R.; Lira, F.S.; Oyama, L.M.; Oller Do Nascimento, C.M.; Otoch, J.P.; Alcântara, P.S.M.; Batista, M.L.; Seelaender, M. Lipases and lipid droplet-associated protein expression in subcutaneous white adipose tissue of cachectic patients with cancer. Lipids Health Dis. 2017, 16. [Google Scholar] [CrossRef] [PubMed]
- Manley, G. Public Access NIH Public Access. Hear. Metab. 2013, 71, 233–236. [Google Scholar] [CrossRef]
- Hoeg, L.D.; Sjoberg, K.A.; Jeppesen, J.; Jensen, T.E.; Frosig, C.; Birk, J.B.; Bisiani, B.; Hiscock, N.; Pilegaard, H.; Wojtaszewski, J.F.P.; et al. Lipid-induced insulin resistance affects women less than men and is not accompanied by inflammation or impaired proximal insulin signaling. Diabetes 2011, 60, 64–73. [Google Scholar] [CrossRef]
- Reaven, G.M.; Hollenbeck, C.; Jeng, C.Y.; Wu, M.S.; Chen, Y.D.I. Measurement of plasma glucose, free fatty acid, lactate, and insulin for 24 h in patients with NIDDM. Diabetes 1988, 37, 1020–1024. [Google Scholar] [CrossRef] [PubMed]
- Bódis, K.; Roden, M. Energy metabolism of white adipose tissue and insulin resistance in humans. Eur. J. Clin. Investig. 2018, 48, e13017. [Google Scholar] [CrossRef] [PubMed]
- Morigny, P.; Zuber, J.; Haid, M.; Kaltenecker, D.; Riols, F.; Lima, J.D.C.; Simoes, E.; Otoch, J.P.; Schmidt, S.F.; Herzig, S.; et al. High levels of modified ceramides are a defining feature of murine and human cancer cachexia. J. Cachexia Sarcopenia Muscle 2020, 11, 1459–1475. [Google Scholar] [CrossRef]
- Stephens, N.A.; Skipworth, R.J.E.; MacDonald, A.J.; Greig, C.A.; Ross, J.A.; Fearon, K.C.H. Intramyocellular lipid droplets increase with progression of cachexia in cancer patients. J. Cachexia Sarcopenia Muscle 2011, 2, 111–117. [Google Scholar] [CrossRef]
- Fukawa, T.; Yan-Jiang, B.C.; Min-Wen, J.C.; Jun-Hao, E.T.; Huang, D.; Qian, C.N.; Ong, P.; Li, Z.; Chen, S.; Mak, S.Y.; et al. Excessive fatty acid oxidation induces muscle atrophy in cancer cachexia. Nat. Med. 2016, 22, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Diaz, M.B.; Krones-Herzig, A.; Metzger, D.; Ziegler, A.; Vegiopoulos, A.; Klingenspor, M.; Müller-Decker, K.; Herzig, S. Nuclear receptor cofactor receptor interacting protein 140 controls hepatic triglyceride metabolism during wasting in mice. Hepatology 2008, 48, 782–791. [Google Scholar] [CrossRef]
- Petersen, M.C.; Shulman, G.I. Mechan isms of insulin action and insulin resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef] [PubMed]
- Fazakerley, D.J.; Krycer, J.R.; Kearney, A.L.; Hocking, S.L.; James, D.E. Muscle and adipose tissue insulin resistance: Malady without mechanism? J. Lipid Res. 2019, 60, 1720–1732. [Google Scholar] [CrossRef] [PubMed]
- Romanello, V.; Sandri, M. Mitochondrial quality control and muscle mass maintenance. Front. Physiol. 2016, 6, 422. [Google Scholar] [CrossRef]
- Ebhardt, H.A.; Degen, S.; Tadini, V.; Schilb, A.; Johns, N.; Greig, C.A.; Fearon, K.C.H.; Aebersold, R.; Jacobi, C. Comprehensive proteome analysis of human skeletal muscle in cachexia and sarcopenia: A pilot study. J. Cachexia Sarcopenia Muscle 2017, 8, 567–582. [Google Scholar] [CrossRef] [PubMed]
- Wilson, H.E.; Stanton, D.A.; Montgomery, C.; Infante, A.M.; Taylor, M.; Hazard-Jenkins, H.; Pugacheva, E.N.; Pistilli, E.E. Skeletal muscle reprogramming by breast cancer regardless of treatment history or tumor molecular subtype. npj Breast Cancer 2020, 6, 18. [Google Scholar] [CrossRef]
- Barreto, R.; Mandili, G.; Witzmann, F.A.; Novelli, F.; Zimmers, T.A.; Bonetto, A. Cancer and chemotherapy contribute to muscle loss by activating common signaling pathways. Front. Physiol. 2016, 7, 1–13. [Google Scholar] [CrossRef]
- Shum, A.M.Y.; Poljak, A.; Bentley, N.L.; Turner, N.; Tan, T.C.; Polly, P. Proteomic profiling of skeletal and cardiac muscle in cancer cachexia: Alterations in sarcomeric and mitochondrial protein expression. Oncotarget 2018, 9, 22001–22022. [Google Scholar] [CrossRef]
- Kunzke, T.; Buck, A.; Prade, V.M.; Feuchtinger, A.; Prokopchuk, O.; Martignoni, M.E.; Heisz, S.; Hauner, H.; Janssen, K.; Walch, A.; et al. Derangements of amino acids in cachectic skeletal muscle are caused by mitochondrial dysfunction. J. Cachexia Sarcopenia Muscle 2020, 11, 226–240. [Google Scholar] [CrossRef] [PubMed]
- Montero-Bullon, J.-F.; Melo, T.; Ferreira, R.; Padrão, A.I.; Oliveira, P.A.; Domingues, M.R.M.; Domingues, P. Exercise training counteracts urothelial carcinoma-induced alterations in skeletal muscle mitochondria phospholipidome in an animal model. Sci. Rep. 2019, 9, 13423. [Google Scholar] [CrossRef]
- Brown, J.L.; Rosa-Caldwell, M.E.; Lee, D.E.; Blackwell, T.A.; Brown, L.A.; Perry, R.A.; Haynie, W.S.; Hardee, J.P.; Carson, J.A.; Wiggs, M.P.; et al. Mitochondrial degeneration precedes the development of muscle atrophy in progression of cancer cachexia in tumour-bearing mice. J. Cachexia Sarcopenia Muscle 2017, 8, 926–938. [Google Scholar] [CrossRef] [PubMed]
- Fontes-Oliveira, C.C.; Busquets, S.; Toledo, M.; Penna, F.; Aylwin, M.P.; Sirisi, S.; Silva, A.P.; Orpí, M.; García, A.; Sette, A.; et al. Mitochondrial and sarcoplasmic reticulum abnormalities in cancer cachexia: Altered energetic efficiency? Biochim. Biophys. Acta - Gen. Subj. 2013, 1830, 2770–2778. [Google Scholar] [CrossRef]
- Halle, J.L.; Pena, G.S.; Paez, H.G.; Castro, A.J.; Rossiter, H.B.; Visavadiya, N.P.; Whitehurst, M.A.; Khamoui, A.V. Tissue-specific dysregulation of mitochondrial respiratory capacity and coupling control in colon-26 tumor-induced cachexia. Am. J. Physiol. Integr. Comp. Physiol. 2019, 317, R68–R82. [Google Scholar] [CrossRef] [PubMed]
- Shum, A.M.Y.; Mahendradatta, T.; Taylor, R.J.; Painter, A.B.; Moore, M.M.; Tsoli, M.; Tan, T.C.; Clarke, S.J.; Robertson, G.R.; Polly, P. Disruption of MEF2C signaling and loss of sarcomeric and mitochondrial integrity in cancer-induced skeletal muscle wasting. Aging 2012, 4, 133–143. [Google Scholar] [CrossRef]
- Hardee, J.P.; Mangum, J.E.; Gao, S.; Sato, S.; Hetzler, K.L.; Puppa, M.J.; Fix, D.K.; Carson, J.A. Eccentric contraction-induced myofiber growth in tumor-bearing mice. J. Appl. Physiol. 2016, 120, 29–37. [Google Scholar] [CrossRef]
- White, J.P.; Puppa, M.J.; Sato, S.; Gao, S.; Price, R.L.; Baynes, J.W.; Kostek, M.C.; Matesic, L.E.; Carson, J.A. IL-6 regulation on skeletal muscle mitochondrial remodeling during cancer cachexia in the ApcMin/+ mouse. Skelet. Muscle 2012, 2, 14. [Google Scholar] [CrossRef]
- Figueroa-Clarevega, A.; Bilder, D. Malignant drosophila tumors interrupt insulin signaling to induce cachexia-like wasting. Dev. Cell 2015, 33, 47–55. [Google Scholar] [CrossRef]
- de Castro, G.S.; Simoes, E.; Lima, J.D.C.C.; Ortiz-Silva, M.; Festuccia, W.T.; Tokeshi, F.; Alcântara, P.S.; Otoch, J.P.; Coletti, D.; Seelaender, M. Human Cachexia Induces Changes in Mitochondria, Autophagy and Apoptosis in the Skeletal Muscle. Cancers 2019, 11, 1264. [Google Scholar] [CrossRef] [PubMed]
- Guigni, B.A.; Callahan, D.M.; Tourville, T.W.; Miller, M.S.; Fiske, B.; Voigt, T.; Korwin-Mihavics, B.; Anathy, V.; Dittus, K.; Toth, M.J. Skeletal muscle atrophy and dysfunction in breast cancer patients: Role for chemotherapy-derived oxidant stress. Am. J. Physiol. Physiol. 2018, 315, C744–C756. [Google Scholar] [CrossRef] [PubMed]
- Green, K.; Brand, M.D.; Murphy, M.P. Prevention of Mitochondrial Oxidative Damage As A Therapeutic Strategy in Diabetes. Diabetes 2004, 53, S110–S118. [Google Scholar] [CrossRef]
- Koves, T.R.; Ussher, J.R.; Noland, R.C.; Slentz, D.; Mosedale, M.; Ilkayeva, O.; Bain, J.; Stevens, R.; Dyck, J.R.B.; Newgard, C.B.; et al. Mitochondrial Overload and Incomplete Fatty Acid Oxidation Contribute to Skeletal Muscle Insulin Resistance. Cell Metab. 2008, 7, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Alves, C.R.R.; das Neves, W.; de Almeida, N.R.; Eichelberger, E.J.; Jannig, P.R.; Voltarelli, V.A.; Tobias, G.C.; Bechara, L.R.G.; de Paula Faria, D.; Alves, M.J.N.; et al. Exercise training reverses cancer-induced oxidative stress and decrease in muscle COPS2/TRIP15/ALIEN. Mol. Metab. 2020, 39, 101012. [Google Scholar] [CrossRef]
- Padilha, C.S.; Borges, F.H.; Costa Mendes da Silva, L.E.; Frajacomo, F.T.T.; Jordao, A.A.; Duarte, J.A.; Cecchini, R.; Guarnier, F.A.; Deminice, R. Resistance exercise attenuates skeletal muscle oxidative stress, systemic pro-inflammatory state, and cachexia in Walker-256 tumor-bearing rats. Appl. Physiol. Nutr. Metab. 2017, 42, 916–923. [Google Scholar] [CrossRef]
- Ballarò, R.; Penna, F.; Pin, F.; Gómez-Cabrera, M.; Viña, J.; Costelli, P. Moderate Exercise Improves Experimental Cancer Cachexia by Modulating the Redox Homeostasis. Cancers 2019, 11, 285. [Google Scholar] [CrossRef]
- Dumas, J.-F.; Goupille, C.; Julienne, C.M.; Pinault, M.; Chevalier, S.; Bougnoux, P.; Servais, S.; Couet, C. Efficiency of oxidative phosphorylation in liver mitochondria is decreased in a rat model of peritoneal carcinosis. J. Hepatol. 2011, 54, 320–327. [Google Scholar] [CrossRef]
- Khamoui, A.V.; Tokmina-Roszyk, D.; Rossiter, H.B.; Fields, G.B.; Visavadiya, N.P. Hepatic proteome analysis reveals altered mitochondrial metabolism and suppressed acyl-CoA synthetase-1 in colon-26 tumor-induced cachexia. Physiol. Genomics 2020, 52, 203–216. [Google Scholar] [CrossRef]
- Visavadiya, N.P.; Pena, G.S.; Khamoui, A.V. Mitochondrial dynamics and quality control are altered in a hepatic cell culture model of cancer cachexia. Mol. Cell. Biochem. 2020, 476, 23–34. [Google Scholar] [CrossRef]
- Petruzzelli, M.; Schweiger, M.; Schreiber, R.; Campos-Olivas, R.; Tsoli, M.; Allen, J.; Swarbrick, M.; Rose-John, S.; Rincon, M.; Robertson, G.; et al. A Switch from White to Brown Fat Increases Energy Expenditure in Cancer-Associated Cachexia. Cell Metab. 2014, 20, 433–447. [Google Scholar] [CrossRef]
- Becker, A.S.; Zellweger, C.; Bacanovic, S.; Franckenberg, S.; Nagel, H.W.; Frick, L.; Schawkat, K.; Eberhard, M.; Blüthgen, C.; Volbracht, J.; et al. Brown fat does not cause cachexia in cancer patients: A large retrospective longitudinal FDG-PET/CT cohort study. PLoS One 2020, 15, e0239990. [Google Scholar] [CrossRef]
- Miller, J.; Dreczkowski, G.; Ramage, M.I.; Wigmore, S.J.; Gallagher, I.J.; Skipworth, R.J.E. Adipose depot gene expression and intelectin-1 in the metabolic response to cancer and cachexia. J. Cachexia Sarcopenia Muscle 2020, 11, 1141–1153. [Google Scholar] [CrossRef]
- Scott, H.R.; McMillan, D.C.; Crilly, A.; McArdle, C.S.; Milroy, R. The relationship between weight loss and interleukin 6 in non-small-cell lung cancer. Br. J. Cancer 1996, 73, 1560–1562. [Google Scholar] [CrossRef]
- Onesti, J.K.; Guttridge, D.C. Inflammation Based Regulation of Cancer Cachexia. Biomed Res. Int. 2014, 2014, 168407. [Google Scholar] [CrossRef] [PubMed]
- Saltiel, A.R.; Olefsky, J.M. Inflammatory mechanisms linking obesity and metabolic disease. J. Clin. Investig. 2017, 127, 1–4. [Google Scholar] [CrossRef]
- Haddad, F.; Zaldivar, F.; Cooper, D.M.; Adams, G.R. IL-6-induced skeletal muscle atrophy. J. Appl. Physiol. 2005, 98, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Goodman, M.N. Tumor necrosis factor induces skeletal muscle protein breakdown in rats. Am. J. Physiol.-Endocrinol. Metab. 1991, 260, E727–E730. [Google Scholar] [CrossRef] [PubMed]
- Strassmann, G.; Fong, M.; Kenney, J.S.; Jacob, C.O. Evidence for the involvement of interleukin 6 in experimental cancer cachexia. J. Clin. Investig. 1992, 89, 1681–1684. [Google Scholar] [CrossRef] [PubMed]
- White, J.P.; Baynes, J.W.; Welle, S.L.; Kostek, M.C.; Matesic, L.E.; Sato, S.; Carson, J.A. The Regulation of Skeletal Muscle Protein Turnover during the Progression of Cancer Cachexia in the ApcMin/+ Mouse. PLoS One 2011, 6, e24650. [Google Scholar] [CrossRef] [PubMed]
- Baltgalvis, K.A.; Berger, F.G.; Pena, M.M.O.; Davis, J.M.; Muga, S.J.; Carson, J.A. Interleukin-6 and cachexia in ApcMin/+ mice. Am. J. Physiol. 2008, 294, R393–R401. [Google Scholar] [CrossRef]
- Prado, B.L.; Qian, Y. Anti-cytokines in the treatment of cancer cachexia. Ann. Palliat. Med. 2019, 8, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Fearon, K.; Strasser, F.; Anker, S.D.; Bosaeus, I.; Bruera, E.; Fainsinger, R.L.; Jatoi, A.; Loprinzi, C.; MacDonald, N.; Mantovani, G.; et al. Definition and classification of cancer cachexia: An international consensus. Lancet Oncol. 2011, 12, 489–495. [Google Scholar] [CrossRef]
- Christensen, J.F.; Jones, L.W.; Andersen, J.L.; Daugaard, G.; Rorth, M.; Hojman, P. Muscle dysfunction in cancer patients. Ann. Oncol. 2014, 25, 947–958. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, B.T.; Lee, K.Y.; Klaus, K.; Softic, S.; Krumpoch, M.T.; Fentz, J.; Stanford, K.I.; Robinson, M.M.; Cai, W.; Kleinridders, A.; et al. Insulin and IGF-1 receptors regulate FoxO-mediated signaling in muscle proteostasis. J. Clin. Investig. 2016, 126, 3433–3446. [Google Scholar] [CrossRef]
- Ebner, N.; Anker, S.D.; von Haehling, S. Recent developments in the field of cachexia, sarcopenia, and muscle wasting: Highlights from the 12th Cachexia Conference. J. Cachexia Sarcopenia Muscle 2020, 11, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Dolly, A.; Dumas, J.F.; Servais, S. Cancer cachexia and skeletal muscle atrophy in clinical studies: What do we really know? J. Cachexia Sarcopenia Muscle 2020, 11, 1413–1428. [Google Scholar] [CrossRef] [PubMed]
- Argilés, J.M.; Stemmler, B.; López-Soriano, F.J.; Busquets, S. Inter-tissue communication in cancer cachexia. Nat. Rev. Endocrinol. 2019, 15, 9–20. [Google Scholar] [CrossRef]
- Biswas, A.K.; Acharyya, S. Understanding cachexia in the context of metastatic progression. Nat. Rev. Cancer 2020, 20, 274–284. [Google Scholar] [CrossRef]
- Cohen, S.; Nathan, J.A.; Goldberg, A.L. Muscle wasting in disease: Molecular mechanisms and promising therapies. Nat. Rev. Drug Discov. 2015, 14, 58–74. [Google Scholar] [CrossRef]
- Sandri, M. Protein breakdown in cancer cachexia. Semin. Cell Dev. Biol. 2016, 54, 11–19. [Google Scholar] [CrossRef]
- Emery, P.W.; Edwards, R.H.; Rennie, M.J.; Souhami, R.L.; Halliday, D. Protein synthesis in muscle measured in vivo in cachectic patients with cancer. BMJ 1984, 289, 584–586. [Google Scholar] [CrossRef]
- Brown, J.L.; Lee, D.E.; Rosa-Caldwell, M.E.; Brown, L.A.; Perry, R.A.; Haynie, W.S.; Huseman, K.; Sataranatarajan, K.; Van Remmen, H.; Washington, T.A.; et al. Protein imbalance in the development of skeletal muscle wasting in tumour-bearing mice. J. Cachexia Sarcopenia Muscle 2018, 9, 987–1002. [Google Scholar] [CrossRef] [PubMed]
- Lima, M.; Sato, S.; Enos, R.T.; Baynes, J.W.; Carson, J.A. Development of an UPLC mass spectrometry method for measurement of myofibrillar protein synthesis: Application to analysis of murine muscles during cancer cachexia. J. Appl. Physiol. 2013, 114, 824–828. [Google Scholar] [CrossRef]
- Smith, K.; Tisdale, M. Increased protein degradation and decreased protein synthesis in skeletal muscle during cancer cachexia. Br. J. Cancer 1993, 67, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Murton, A.J.; Maddocks, M.; Stephens, F.B.; Marimuthu, K.; England, R.; Wilcock, A. Consequences of Late-Stage Non–Small-Cell Lung Cancer Cachexia on Muscle Metabolic Processes. Clin. Lung Cancer 2017, 18, e1–e11. [Google Scholar] [CrossRef] [PubMed]
- Op Den Kamp, C.M.; Langen, R.C.; Snepvangers, F.J.; De Theije, C.C.; Schellekens, J.M.; Laugs, F.; Dingemans, A.M.C.; Schols, A.M. Nuclear transcription factor κB activation and protein turnover adaptations in skeletal muscle of patients with progressive stages of lung cancer cachexia. Am. J. Clin. Nutr. 2013, 98, 738–748. [Google Scholar] [CrossRef]
- Puig-Vilanova, E.; Rodriguez, D.A.; Lloreta, J.; Ausin, P.; Pascual-Guardia, S.; Broquetas, J.; Roca, J.; Gea, J.; Barreiro, E. Oxidative stress, redox signaling pathways, and autophagy in cachectic muscles of male patients with advanced COPD and lung cancer. Free Radic. Biol. Med. 2015, 79, 91–108. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, J.; Wang, X.; Gao, T.; Tian, H.; Zhou, D.; Zhang, L.; Li, G.; Wang, X. The autophagic-lysosomal and ubiquitin proteasome systems are simultaneously activated in the skeletal muscle of gastric cancer patients with cachexia. Am. J. Clin. Nutr. 2020, 111, 570–579. [Google Scholar] [CrossRef] [PubMed]
- Srikanthan, P.; Karlamangla, A.S. Relative muscle mass is inversely associated with insulin resistance and prediabetes. Findings from the third national health and nutrition examination survey. J. Clin. Endocrinol. Metab. 2011, 96, 2898–2903. [Google Scholar] [CrossRef]
- Wall, B.T.; Gorissen, S.H.; Pennings, B.; Koopman, R.; Groen, B.B.L.; Verdijk, L.B.; van Loon, L.J.C. Aging Is Accompanied by a Blunted Muscle Protein Synthetic Response to Protein Ingestion. PLoS One 2015, 10, e0140903. [Google Scholar] [CrossRef]
- Volpi, E.; Mittendorfer, B.; Rasmussen, B.B.; Wolfe, R.R. The Response of Muscle Protein Anabolism to Combined Hyperaminoacidemia and Glucose-Induced Hyperinsulinemia Is Impaired in the Elderly 1. J. Clin. Endocrinol. Metab. 2000, 85, 4481–4490. [Google Scholar] [CrossRef]
- Guillet, C.; Prod’homme, M.; Balage, M.; Gachon, P.; Giraudet, C.; Morin, L.; Grizard, J.; Boirie, Y. Impaired anabolic response of muscle protein synthesis is associated with S6K1 dysregulation in elderly humans. FASEB J. 2004, 18, 1586–1587. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.; Song, W.; Droujinine, I.A.; Hu, Y.; Asara, J.M.; Perrimon, N. Systemic organ wasting induced by localized expression of the secreted Insulin/IGF antagonist ImpL2. Dev. Cell 2015, 33, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Savikj, M.; Zierath, J.R. Train like an athlete: Applying exercise interventions to manage type 2 diabetes. Diabetologia 2020, 63, 1491–1499. [Google Scholar] [CrossRef]
- Sylow, L.; Richter, E.A. Current advances in our understanding of exercise as medicine in metabolic disease. Curr. Opin. Physiol. 2019, 12, 12–19. [Google Scholar] [CrossRef]
- Hargreaves, M.; Spriet, L.L. Skeletal muscle energy metabolism during exercise. Nat. Metab. 2020, 2, 817–828. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, J.W.; Hirshman, M.F.; Gervino, E.V.; Ocel, J.V.; Forse, R.A.; Hoenig, S.J.; Aronson, D.; Goodyear, L.J.; Horton, E.S. Acute exercise induces GLUT4 translocation in skeletal muscle of normal human subjects and subjects with type 2 diabetes. Diabetes 1999, 48, 1192–1197. [Google Scholar] [CrossRef] [PubMed]
- Wojtaszewski, J.F.P.; Higaki, Y.; Hirshman, M.F.; Michael, M.D.; Dufresne, S.D.; Kahn, C.R.; Goodyear, L.J. Exercise modulates postreceptor insulin signaling and glucose transport in muscle-specific insulin receptor knockout mice. J. Clin. Investig. 1999, 104, 1257–1264. [Google Scholar] [CrossRef]
- Sylow, L.; Kleinert, M.; Richter, E.A.; Jensen, T.E. Exercise-stimulated glucose uptake-regulation and implications for glycaemic control. Nat. Rev. Endocrinol. 2017, 13, 133–148. [Google Scholar] [CrossRef]
- O’Neill, H.M.; Maarbjerg, S.J.; Crane, J.D.; Jeppesen, J.; Jørgensen, S.B.; Schertzer, J.D.; Shyroka, O.; Kiens, B.; van Denderen, B.J.; Tarnopolsky, M.A.; et al. AMP-activated protein kinase (AMPK) beta1beta2 muscle null mice reveal an essential role for AMPK in maintaining mitochondrial content and glucose uptake during exercise. Proc. Natl. Acad. Sci. USA 2011, 108, 16092–16097. [Google Scholar] [CrossRef]
- Jørgensen, S.B.; Viollet, B.; Andreelli, F.; Frøsig, C.; Birk, J.B.; Schjerling, P.; Vaulont, S.; Richter, E.A.; Wojtaszewski, J.F.P. Knockout of the α 2 but Not α 1 5′-AMP-activated Protein Kinase Isoform Abolishes 5-Aminoimidazole-4-carboxamide-1-β-4-ribofuranosidebut Not Contraction-induced Glucose Uptake in Skeletal Muscle. J. Biol. Chem. 2004, 279, 1070–1079. [Google Scholar] [CrossRef] [PubMed]
- Lee-young, R.S.; Griffee, S.R.; Lynes, S.E.; Bracy, D.P.; Ayala, J.E.; Mcguinness, O.P.; Wasserman, D.H. Skeletal Muscle AMP-activated Protein Kinase Is Essential for the Metabolic Response to Exercise In Vivo. J. Biol. Chem. 2009, 284, 23825–23934. [Google Scholar] [CrossRef]
- Sylow, L.; Møller, L.L.V.; Kleinert, M.; D’Hulst, G.; De Groote, E.; Schjerling, P.; Steinberg, G.R.; Jensen, T.E.; Richter, E.A. Rac1 and AMPK Account for the Majority of Muscle Glucose Uptake Stimulated by Ex Vivo Contraction but Not In Vivo Exercise. Diabetes 2017, 66, 1548–1559. [Google Scholar] [CrossRef] [PubMed]
- Sylow, L.; Jensen, T.E.; Kleinert, M.; Mouatt, J.R.; Maarbjerg, S.J.; Jeppesen, J.; Prats, C.; Chiu, T.T.; Boguslavsky, S.; Klip, A.; et al. Rac1 is a novel regulator of contraction-stimulated glucose uptake in skeletal muscle. Diabetes 2013, 62, 1139–1151. [Google Scholar] [CrossRef] [PubMed]
- Sylow, L.; Nielsen, I.L.; Kleinert, M.; Møller, L.L.V.; Ploug, T.; Schjerling, P.; Bilan, P.J.; Klip, A.; Jensen, T.E.; Richter, E.A. Rac1 governs exercise-stimulated glucose uptake in skeletal muscle through regulation of GLUT4 translocation in mice. J. Physiol. 2016, 594, 4997–5008. [Google Scholar] [CrossRef]
- Henríquez-Olguin, C.; Knudsen, J.R.; Raun, S.H.; Li, Z.; Dalbram, E.; Treebak, J.T.; Sylow, L.; Holmdahl, R.; Richter, E.A.; Jaimovich, E.; et al. Cytosolic ROS production by NADPH oxidase 2 regulates muscle glucose uptake during exercise. Nat. Commun. 2019, 10, 4623. [Google Scholar] [CrossRef]
- Hoffman, N.J.; Parker, B.L.; Chaudhuri, R.; Fisher-Wellman, K.H.; Kleinert, M.; Humphrey, S.J.; Yang, P.; Holliday, M.; Trefely, S.; Fazakerley, D.J.; et al. Global Phosphoproteomic Analysis of Human Skeletal Muscle Reveals a Network of Exercise-Regulated Kinases and AMPK Substrates. Cell Metab. 2015, 22, 948. [Google Scholar] [CrossRef]
- Deshmukh, A.S.; Steenberg, D.E.; Hostrup, M.; Birk, J.B.; Larsen, J.K.; Santos, A.; Kjøbsted, R.; Hingst, J.R.; Schéele, C.C.; Murgia, M.; et al. Deep muscle-proteomic analysis of freeze-dried human muscle biopsies reveals fiber type-specific adaptations to exercise training. Nat. Commun. 2021, 12. [Google Scholar] [CrossRef]
- Richter, E.A.; Garetto, L.P.; Goodman, M.N.; Ruderman, N.B. Muscle glucose metabolism following exercise in the rat. Increased sensitivity to insulin. J. Clin. Investig. 1982, 69, 785–793. [Google Scholar] [CrossRef] [PubMed]
- Richter, E.A.; Mikines, K.J.; Galbo, H.; Kiens, B. Effect of exercise on insulin action in human skeletal muscle. J Appl Physiol 1989, 66, 876–885. [Google Scholar] [CrossRef] [PubMed]
- Cartee, G.D.; Young, D.A.; Sleeper, M.D.; Zierath, J.; Wallberg-Henriksson, H.; Holloszy, J.O. Prolonged increase in insulin-stimulated glucose transport in muscle after exercise. Am. J. Physiol. Endocrinol. Metab. 1989, 256, E494–E499. [Google Scholar] [CrossRef] [PubMed]
- Wojtaszewski, J.F.P.; Hansen, B.F.; Gade, J.; Kiens, B.; Markuns, J.F.; Goodyear, L.J.; Richter, E.A. Insulin signaling and insulin sensitivity after exercise in human skeletal muscle. Diabetes 2000, 49, 325–331. [Google Scholar] [CrossRef]
- Wojtaszewski, J.F.P.; Hansen, B.F.; Kiens, B.; Richter, E.A. Insulin Signaling in Human Skeletal Muscle: Time Course and Effect of Exercise. Diabetes 1997, 46, 1775–1781. [Google Scholar] [CrossRef] [PubMed]
- Steenberg, D.E.; Hingst, J.R.; Birk, J.B.; Thorup, A.; Kristensen, J.M.; Sjøberg, K.A.; Kiens, B.; Richter, E.A.; Wojtaszewski, J.F.P. A Single Bout of One-Legged Exercise to Local Exhaustion Decreases Insulin Action in Nonexercised Muscle Leading to Decreased Whole-Body Insulin Action. Diabetes 2020, 69, 578–590. [Google Scholar] [CrossRef] [PubMed]
- Mikines, K.J.; Sonne, B.; Farrell, P.A.; Tronier, B.; Galbo, H. Effect of physical exercise on sensitivity and responsiveness to insulin in humans. Am. J. Physiol. Metab. 1988, 254, E248–E259. [Google Scholar] [CrossRef]
- Sjøberg, K.A.; Frøsig, C.; Kjøbsted, R.; Sylow, L.; Kleinert, M.; Betik, A.C.; Shaw, C.S.; Kiens, B.; Wojtaszewski, J.F.P.; Rattigan, S.; et al. Exercise Increases Human Skeletal Muscle Insulin Sensitivity via Coordinated Increases in Microvascular Perfusion and Molecular Signaling. Diabetes 2017, 66, 1501–1510. [Google Scholar] [CrossRef] [PubMed]
- Kjøbsted, R.; Chadt, A.; Jørgensen, N.O.; Kido, K.; Larsen, J.K.; de Wendt, C.; Al-Hasani, H.; Wojtaszewski, J.F.P. TBC1D4 Is Necessary for Enhancing Muscle Insulin Sensitivity in Response to AICAR and Contraction. Diabetes 2019, 68, 1756–1766. [Google Scholar] [CrossRef]
- Kjøbsted, R.; Munk-Hansen, N.; Birk, J.B.; Foretz, M.; Viollet, B.; Björnholm, M.; Zierath, J.R.; Treebak, J.T.; Wojtaszewski, J.F.P. Enhanced Muscle Insulin Sensitivity After Contraction/Exercise Is Mediated by AMPK. Diabetes 2017, 66, 598–612. [Google Scholar] [CrossRef]
- Sylow, L.; Møller, L.L.V.; D’Hulst, G.; Schjerling, P.; Jensen, T.E.; Richter, E.A. Rac1 in Muscle Is Dispensable for Improved Insulin Action After Exercise in Mice. Endocrinology 2016, 157, 3009–3015. [Google Scholar] [CrossRef] [PubMed]
- Mikines, K.J.; Richter, E.A.; Dela, F.; Galbo, H. Seven days of bed rest decrease insulin action on glucose uptake in leg and whole body. J. Appl. Physiol. 1991, 70, 1245–1254. [Google Scholar] [CrossRef]
- Richter, E.A.; Kiens, B.; Mizuno, M.; Strange, S. Insulin action in human thighs after one-legged immobilization. J. Appl. Physiol. 1989, 67, 19–23. [Google Scholar] [CrossRef]
- Mortensen, B.; Friedrichsen, M.; Andersen, N.R.; Alibegovic, A.C.; Højbjerre, L.; Sonne, M.P.; Stallknecht, B.; Dela, F.; Wojtaszewski, J.F.P.; Vaag, A. Physical inactivity affects skeletal muscle insulin signaling in a birth weight-dependent manner. J. Diabetes Complications 2014, 28, 71–78. [Google Scholar] [CrossRef]
- Van Dijk, J.W.; Venema, M.; Van Mechelen, W.; Stehouwer, C.D.A.; Hartgens, F.; Van Loon, L.J.C. Effect of moderate-intensity exercise versus activities of daily living on 24-hour blood glucose homeostasis in male patients with type 2 diabetes. Diabetes Care 2013, 36, 3448–3453. [Google Scholar] [CrossRef]
- Mikines, K.J.; Sonne, B.; Farrell, P.A.; Tronier, B.; Galbo, H. Effect of training on the dose-response relationship for insulin action in men. J. Appl. Physiol. 1989, 66, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Bird, S.R.; Hawley, J.A. Update on the effects of physical activity on insulin sensitivity in humans. BMJ Open Sport Exerc. Med. 2017, 2. [Google Scholar] [CrossRef]
- Steenberg, D.E.; Jørgensen, N.B.; Birk, J.B.; Sjøberg, K.A.; Kiens, B.; Richter, E.A.; Wojtaszewski, J.F.P. Exercise training reduces the insulin-sensitizing effect of a single bout of exercise in human skeletal muscle. J. Physiol. 2019, 597, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Hvid, T.; Winding, K.; Rinnov, A.; Dejgaard, T.; Thomsen, C.; Iversen, P.; Brasso, K.; Mikines, K.J.; Van Hall, G.; Lindegaard, B.; et al. Endurance training improves insulin sensitivity and body composition in prostate cancer patients treated with androgen deprivation therapy. Endocr. Relat. Cancer 2013, 20, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.C.; Rickels, M.R.; Troxel, A.B.; Zemel, B.S.; Damjanov, N.; Ky, B.; Rhim, A.D.; Rustgi, A.K.; Courneya, K.S.; Schmitz, K.H. Dose-response effects of exercise on insulin among colon cancer survivors. Endocr. Relat. Cancer 2018. [Google Scholar] [CrossRef]
- Lee, D.H.; Kim, J.Y.; Lee, M.K.; Lee, C.; Min, J.-H.; Jeong, D.H.; Lee, J.-W.; Chu, S.H.; Meyerhardt, J.A.; Ligibel, J.; et al. Effects of a 12-week home-based exercise program on the level of physical activity, insulin, and cytokines in colorectal cancer survivors: A pilot study. Support. Care Cancer 2013, 21, 2537–2545. [Google Scholar] [CrossRef]
- Dieli-Conwright, C.M.; Courneya, K.S.; Demark-Wahnefried, W.; Sami, N.; Lee, K.; Buchanan, T.A.; Spicer, D.V.; Tripathy, D.; Bernstein, L.; Mortimer, J.E. Effects of aerobic and resistance exercise on metabolic syndrome, sarcopenic obesity, and circulating biomarkers in overweight or obese survivors of breast cancer: A randomized controlled trial. J. Clin. Oncol. 2018, 36, 875–883. [Google Scholar] [CrossRef] [PubMed]
- Viskochil, R.; Blankenship, J.M.; Makari-Judson, G.; Staudenmayer, J.; Freedson, P.S.; Hankinson, S.E.; Braun, B. Metrics of diabetes risk are only minimally improved by exercise training in postmenopausal breast cancer survivors. J. Clin. Endocrinol. Metab. 2020, 105, e1958–e1966. [Google Scholar] [CrossRef] [PubMed]
- Møller, A.B.; Lønbro, S.; Farup, J.; Voss, T.S.; Rittig, N.; Wang, J.; Højris, I.; Mikkelsen, U.R.; Jessen, N. Molecular and cellular adaptations to exercise training in skeletal muscle from cancer patients treated with chemotherapy. J. Cancer Res. Clin. Oncol. 2019, 145, 1449–1460. [Google Scholar] [CrossRef] [PubMed]
- Puppa, M.J.; White, J.P.; Velázquez, K.T.; Baltgalvis, K.A.; Sato, S.; Baynes, J.W.; Carson, J.A. The effect of exercise on IL-6-induced cachexia in the ApcMin/+ mouse. J. Cachexia Sarcopenia Muscle 2012, 3, 117–137. [Google Scholar] [CrossRef] [PubMed]
- Sennott, J.; Morrissey, J.; Standley, P.R.; Broderick, T.L. Treadmill exercise training fails to reverse defects in glucose, insulin and muscle GLUT4 content in the db/db mouse model of diabetes. Pathophysiology 2008, 15, 173–179. [Google Scholar] [CrossRef]
- McKie, G.L.; Medak, K.D.; Knuth, C.M.; Shamshoum, H.; Townsend, L.K.; Peppler, W.T.; Wright, D.C. Housing temperature affects the acute and chronic metabolic adaptations to exercise in mice. J. Physiol. 2019, 597, 4581–4600. [Google Scholar] [CrossRef]
- Sun, Y.; Cui, D.; Zhang, Z.; Zhang, Q.; Ji, L.; Ding, S. Voluntary wheel exercise alters the levels of miR-494 and miR-696 in the skeletal muscle of C57BL/6 mice. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2016, 202, 16–22. [Google Scholar] [CrossRef]
- Ritchie, I.R.W.; Wright, D.C.; Dyck, D.J. Adiponectin is not required for exercise training-induced improvements in glucose and insulin tolerance in mice. Physiol. Rep. 2014, 2, 1–12. [Google Scholar] [CrossRef]
- Peppler, W.T.; Anderson, Z.G.; Sutton, C.D.; Rector, R.S.; Wright, D.C. Voluntary wheel running attenuates lipopolysaccharide-induced liver inflammation in mice. Am. J. Physiol. 2016, 310, R934–R942. [Google Scholar] [CrossRef] [PubMed]
- Raun, S.H.; Henriquez-Olguín, C.; Karavaeva, I.; Ali, M.; Møller, L.L.V.; Kot, W.; Castro-Mejía, J.L.; Nielsen, D.S.; Gerhart-Hines, Z.; Richter, E.A.; et al. Housing temperature influences exercise training adaptations in mice. Nat. Commun. 2020, 11, 1560. [Google Scholar] [CrossRef]
- Stallknecht, B.; Dela, F.; Helge, J.W. Are blood flow and lipolysis in subcutaneous adipose tissue influenced by contractions in adjacent muscles in humans? Am. J. Physiol. Metab. 2007, 292, E394–E399. [Google Scholar] [CrossRef]
- Mulla, N.; Simonsen, L.; Bülow, J. Post-exercise adipose tissue and skeletal muscle lipid metabolism in humans: The effects of exercise intensity. J. Physiol. 2000, 524, 919–928. [Google Scholar] [CrossRef]
- Moro, C.; Crampes, F.; Sengenes, C.; De Glisezinski, I.; Galitzky, J.; Thalamas, C.; Lafontan, M.; Berlan, M. Atrial natriuretic peptide contributes to the physiological control of lipid mobilization in humans. FASEB J. 2004, 18, 908–910. [Google Scholar] [CrossRef]
- Stallknecht, B.; Lorentsen, J.; Enevoldsen, L.H.; Bülow, J.; Biering-Sørensen, F.; Galbo, H.; Kjær, M. Role of the sympathoadrenergic system in adipose tissue metabolism during exercise in humans. J. Physiol. 2001, 536, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, R.R.; Klein, S.; Carraro, F.; Weber, J.M. Role of triglyceride-fatty acid cycle in controlling fat metabolism in humans during and after exercise. Am. J. Physiol. Metab. 1990, 258, E382–E389. [Google Scholar] [CrossRef]
- Lundsgaard, A.M.; Fritzen, A.M.; Kiens, B. The importance of fatty acids as nutrients during post-exercise recovery. Nutrients 2020, 12, 280. [Google Scholar] [CrossRef] [PubMed]
- Honkala, S.M.; Motiani, P.; Kivelä, R.; Hemanthakumar, K.A.; Tolvanen, E.; Motiani, K.K.; Eskelinen, J.-J.; Virtanen, K.A.; Kemppainen, J.; Heiskanen, M.A.; et al. Exercise training improves adipose tissue metabolism and vasculature regardless of baseline glucose tolerance and sex. BMJ Open Diabetes Res. Care 2020, 8, e000830. [Google Scholar] [CrossRef] [PubMed]
- Duncan, R.E.; Ahmadian, M.; Jaworski, K.; Sarkadi-Nagy, E.; Sul, H.S. Regulation of Lipolysis in Adipocytes. Annu. Rev. Nutr. 2007, 27, 79–101. [Google Scholar] [CrossRef]
- Plaisance, E.P.; Fisher, G. Exercise and Dietary-Mediated Reductions in Postprandial Lipemia. J. Nutr. Metab. 2014, 2014, 902065. [Google Scholar] [CrossRef] [PubMed]
- Schlierf, G.; Dinsenbacher, A.; Kather, H.; Kohlmeier, M.; Haberbosch, W. Mitigation of alimentary lipemia by postprandial exercise—Phenomena and mechanisms. Metabolism 1987, 36, 726–730. [Google Scholar] [CrossRef]
- Tsetsonis, N.V.; Hardman, A.E.; Mastana, S.S. Acute effects of exercise on postprandial lipemia: A comparative study in trained and untrained middle-aged women. Am. J. Clin. Nutr. 1997, 65, 525–533. [Google Scholar] [CrossRef]
- ZIOGAS, G.G.; THOMAS, T.R.; HARRIS, W.S. Exercise training, postprandial hypertriglyceridemia, and LDL subfraction distribution. Med. Sci. Sport. Exerc. 1997, 29, 986–991. [Google Scholar] [CrossRef]
- Harley Hartung, G.; Lawrence, S.J.; Reeves, R.S.; Foreyt, J.P. Effect of alcohol and exercise on postprandial lipemia and triglyceride clearance in men. Atherosclerosis 1993, 100, 33–40. [Google Scholar] [CrossRef]
- Merrill, J.R.; Holly, R.G.; Anderson, R.L.; Rifai, N.; King, M.E.; DeMeersman, R. Hyperlipemic response of young trained and untrained men after a high fat meal. Arteriosclerosis 1989, 9, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.C.; Noakes, T.D.; Benade, A.J.S. Postprandial lipemia and chylomicron clearance in athletes and in sedentary men. Am. J. Clin. Nutr. 1989, 49, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Talanian, J.L.; Holloway, G.P.; Snook, L.A.; Heigenhauser, G.J.F.; Bonen, A.; Spriet, L.L. Exercise training increases sarcolemmal and mitochondrial fatty acid transport proteins in human skeletal muscle. Am. J. Physiol. Metab. 2010, 299, E180–E188. [Google Scholar] [CrossRef]
- Louche, K.; Badin, P.-M.; Montastier, E.; Laurens, C.; Bourlier, V.; de Glisezinski, I.; Thalamas, C.; Viguerie, N.; Langin, D.; Moro, C. Endurance Exercise Training Up-Regulates Lipolytic Proteins and Reduces Triglyceride Content in Skeletal Muscle of Obese Subjects. J. Clin. Endocrinol. Metab. 2013, 98, 4863–4871. [Google Scholar] [CrossRef]
- Alsted, T.J.; Nybo, L.; Schweiger, M.; Fledelius, C.; Jacobsen, P.; Zimmermann, R.; Zechner, R.; Kiens, B. Adipose triglyceride lipase in human skeletal muscle is upregulated by exercise training. Am. J. Physiol. Metab. 2009, 296, E445–E453. [Google Scholar] [CrossRef]
- Jong-Yeon, K.; Hickner, R.C.; Dohm, G.L.; Houmard, J.A. Long- and medium-chain fatty acid oxidation is increased in exercise-trained human skeletal muscle. Metabolism 2002, 51, 460–464. [Google Scholar] [CrossRef] [PubMed]
- Fritzen, A.M.; Lundsgaard, A.-M.; Kiens, B. Tuning fatty acid oxidation in skeletal muscle with dietary fat and exercise. Nat. Rev. Endocrinol. 2020, 16, 683–696. [Google Scholar] [CrossRef]
- Bacchi, E.; Negri, C.; Targher, G.; Faccioli, N.; Lanza, M.; Zoppini, G.; Zanolin, E.; Schena, F.; Bonora, E.; Moghetti, P. Both resistance training and aerobic training reduce hepatic fat content in type 2 diabetic subjects with nonalcoholic fatty liver disease (the RAED2 randomized trial). Hepatology 2013, 58, 1287–1295. [Google Scholar] [CrossRef]
- Lee, S.J.; Bacha, F.; Hannon, T.; Kuk, J.L.; Boesch, C.; Arslanian, S. Effects of aerobic versus resistance exercise without caloric restriction on abdominal fat, intrahepatic lipid, and insulin sensitivity in obese adolescent boys a randomized, controlled trial. Diabetes 2012, 61, 2787–2795. [Google Scholar] [CrossRef] [PubMed]
- Thyfault, J.P.; Scott Rector, R. Exercise combats hepatic steatosis: Potential mechanisms and clinical implications. Diabetes 2020, 69, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Hvid, T.; Lindegaard, B.; Winding, K.; Iversen, P.; Brasso, K.; Solomon, T.P.J.; Pedersen, B.K.; Hojman, P. Effect of a 2-year home-based endurance training intervention on physiological function and PSA doubling time in prostate cancer patients. Cancer Causes Control 2016, 27, 165–174. [Google Scholar] [CrossRef]
- Holloszy, J.O. Biochemical adaptations in muscle. Effects of exercise on mitochondrial oxygen uptake and respiratory enzyme activity in skeletal muscle. J. Biol. Chem. 1967, 242, 2278–2282. [Google Scholar] [CrossRef]
- Granata, C.; Jamnick, N.A.; Bishop, D.J. Training-Induced Changes in Mitochondrial Content and Respiratory Function in Human Skeletal Muscle. Sport. Med. 2018, 48, 1809–1828. [Google Scholar] [CrossRef]
- Khadir, A.; Tiss, A.; Abubaker, J.; Abu-Farha, M.; Al-Khairi, I.; Cherian, P.; John, J.; Kavalakatt, S.; Warsame, S.; Al-Madhoun, A.; et al. MAP kinase phosphatase DUSP1 is overexpressed in obese humans and modulated by physical exercise. Am. J. Physiol. Metab. 2015, 308, E71–E83. [Google Scholar] [CrossRef] [PubMed]
- Ruschke, K.; Fishbein, L.; Dietrich, A.; Klöting, N.; Tönjes, A.; Oberbach, A.; Fasshauer, M.; Jenkner, J.; Schön, M.R.; Stumvoll, M.; et al. Gene expression of PPARγ and PGC-1α in human omental and subcutaneous adipose tissues is related to insulin resistance markers and mediates beneficial effects of physical training. Eur. J. Endocrinol. 2010, 162, 515–523. [Google Scholar] [CrossRef]
- Rönn, T.; Volkov, P.; Tornberg, Å.; Elgzyri, T.; Hansson, O.; Eriksson, K.-F.; Groop, L.; Ling, C. Extensive changes in the transcriptional profile of human adipose tissue including genes involved in oxidative phosphorylation after a 6-month exercise intervention. Acta Physiol. 2014, 211, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Riis, S.; Christensen, B.; Nellemann, B.; Møller, A.B.; Husted, A.S.; Pedersen, S.B.; Schwartz, T.W.; Jørgensen, J.O.L.; Jessen, N. Molecular adaptations in human subcutaneous adipose tissue after ten weeks of endurance exercise training in healthy males. J. Appl. Physiol. 2019, 126, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Stallknecht, B.; Vinten, J.; Ploug, T.; Galbo, H. Increased activities of mitochondrial enzymes in white adipose tissue in trained rats. Am. J. Physiol. Metab. 1991, 261, E410–E414. [Google Scholar] [CrossRef]
- Stanford, K.I.; Middelbeek, R.J.W.; Townsend, K.L.; Lee, M.-Y.; Takahashi, H.; So, K.; Hitchcox, K.M.; Markan, K.R.; Hellbach, K.; Hirshman, M.F.; et al. A novel role for subcutaneous adipose tissue in exercise-induced improvements in glucose homeostasis. Diabetes 2015, 64, 2002–2014. [Google Scholar] [CrossRef]
- Lehnig, A.C.; Stanford, K.I. Exercise-induced adaptations to white and brown adipose tissue. J. Exp. Biol. 2018, 221, jeb161570. [Google Scholar] [CrossRef]
- Stinkens, R.; Brouwers, B.; Jocken, J.W.; Blaak, E.E.; Teunissen-Beekman, K.F.; Hesselink, M.K.; van Baak, M.A.; Schrauwen, P.; Goossens, G.H. Exercise training-induced effects on the abdominal subcutaneous adipose tissue phenotype in humans with obesity. J. Appl. Physiol. 2018, 125, 1585–1593. [Google Scholar] [CrossRef]
- Camera, D.M.; Anderson, M.J.; Hawley, J.A.; Carey, A.L. Short-term endurance training does not alter the oxidative capacity of human subcutaneous adipose tissue. Eur. J. Appl. Physiol. 2010, 109, 307–316. [Google Scholar] [CrossRef]
- Fletcher, J.A.; Meers, G.M.; Linden, M.A.; Kearney, M.L.; Morris, E.M.; Thyfault, J.P.; Rector, R.S. Impact of Various Exercise Modalities on Hepatic Mitochondrial Function. Med. Sci. Sport. Exerc. 2014, 46, 1089–1097. [Google Scholar] [CrossRef] [PubMed]
- Laye, M.J.; Rector, R.S.; Borengasser, S.J.; Naples, S.P.; Uptergrove, G.M.; Ibdah, J.A.; Booth, F.W.; Thyfault, J.P. Cessation of daily wheel running differentially alters fat oxidation capacity in liver, muscle, and adipose tissue. J. Appl. Physiol. 2009, 106, 161–168. [Google Scholar] [CrossRef]
- Hock, M.B.; Kralli, A. Transcriptional Control of Mitochondrial Biogenesis and Function. Annu. Rev. Physiol. 2009, 71, 177–203. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Handschin, C.; Spiegelman, B.M. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005, 1, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Patti, M.E.; Corvera, S. The role of mitochondria in the pathogenesis of type 2 diabetes. Endocr. Rev. 2010, 31, 364–395. [Google Scholar] [CrossRef] [PubMed]
- Ballarò, R.; Beltrà, M.; De Lucia, S.; Pin, F.; Ranjbar, K.; Hulmi, J.J.; Costelli, P.; Penna, F. Moderate exercise in mice improves cancer plus chemotherapy-induced muscle wasting and mitochondrial alterations. FASEB J. 2019, 33, 5482–5494. [Google Scholar] [CrossRef]
- RANJBAR, K.; BALLARÒ, R.; BOVER, Q.; PIN, F.; BELTRÀ, M.; PENNA, F.; COSTELLI, P. Combined Exercise Training Positively Affects Muscle Wasting in Tumor-Bearing Mice. Med. Sci. Sport. Exerc. 2019, 51, 1387–1395. [Google Scholar] [CrossRef] [PubMed]
- Pin, F.; Busquets, S.; Toledo, M.; Camperi, A.; Lopez-Soriano, F.J.; Costelli, P.; Argilés, J.M.; Penna, F. Combination of exercise training and erythropoietin prevents cancer-induced muscle alterations. Oncotarget 2015, 6, 43202–43215. [Google Scholar] [CrossRef] [PubMed]
- Padrão, A.I.; Figueira, A.C.C.; Faustino-Rocha, A.I.; Gama, A.; Loureiro, M.M.; Neuparth, M.J.; Moreira-Gonçalves, D.; Vitorino, R.; Amado, F.; Santos, L.L.; et al. Long-term exercise training prevents mammary tumorigenesis-induced muscle wasting in rats through the regulation of TWEAK signalling. Acta Physiol. 2017, 219, 803–813. [Google Scholar] [CrossRef] [PubMed]
- Mijwel, S.; Cardinale, D.A.; Norrbom, J.; Chapman, M.; Ivarsson, N.; Wengström, Y.; Sundberg, C.J.; Rundqvist, H. Exercise training during chemotherapy preserves skeletal muscle fiber area, capillarization, and mitochondrial content in patients with breast cancer. FASEB J. 2018, 32, 5495–5505. [Google Scholar] [CrossRef] [PubMed]
- Martins, R.A.; Neves, A.P.; Coelho-Silva, M.J.; Veríssimo, M.T.; Teixeira, A.M. The effect of aerobic versus strength-based training on high-sensitivity C-reactive protein in older adults. Eur. J. Appl. Physiol. 2010, 110, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Balducci, S.; Zanuso, S.; Cardelli, P.; Salerno, G.; Fallucca, S.; Nicolucci, A.; Pugliese, G. Supervised exercise training counterbalances the adverse effects of insulin therapy in overweight/obese subjects with type 2 diabetes. Diabetes Care 2012, 35, 39–41. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Merino, D.; Drogou, C.; Guezennec, C.Y.; Chennaoui, M. Effects of chronic exercise on cytokine production in white adipose tissue and skeletal muscle of rats. Cytokine 2007, 40, 23–29. [Google Scholar] [CrossRef]
- Vieira, V.J.; Valentine, R.J.; Wilund, K.R.; Antao, N.; Baynard, T.; Woods, J.A. Effects of exercise and low-fat diet on adipose tissue inflammation and metabolic complications in obese mice. Am. J. Physiol. Metab. 2009, 296, E1164–E1171. [Google Scholar] [CrossRef]
- Vieira, V.J.; Valentine, R.J.; Wilund, K.R.; Woods, J.A. Effects of diet and exercise on metabolic disturbances in high-fat diet-fed mice. Cytokine 2009, 46, 339–345. [Google Scholar] [CrossRef]
- Bruun, J.M.; Helge, J.W.; Richelsen, B.; Stallknecht, B. Diet and exercise reduce low-grade inflammation and macrophage infiltration in adipose tissue but not in skeletal muscle in severely obese subjects. Am. J. Physiol. Endocrinol. Metab. 2006, 290, E961–E967. [Google Scholar] [CrossRef] [PubMed]
- Fabre, O.; Ingerslev, L.R.; Garde, C.; Donkin, I.; Simar, D.; Barrès, R. Exercise training alters the genomic response to acute exercise in human adipose tissue. Epigenomics 2018, 10, 1033–1050. [Google Scholar] [CrossRef]
- Pedersen, B.K. Muscles and their myokines. J. Exp. Biol. 2011, 214, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, L.; Idorn, M.; Olofsson, G.H.; Lauenborg, B.; Nookaew, I.; Hansen, R.H.; Johannesen, H.H.; Becker, J.C.; Pedersen, K.S.; Dethlefsen, C.; et al. Voluntary running suppresses tumor growth through epinephrine- and IL-6-dependent NK cell mobilization and redistribution. Cell Metab. 2016, 23, 554–562. [Google Scholar] [CrossRef]
- Khosravi, N.; Stoner, L.; Farajivafa, V.; Hanson, E.D. Exercise training, circulating cytokine levels and immune function in cancer survivors: A meta-analysis. Brain. Behav. Immun. 2019, 81, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Hiensch, A.E.; Mijwel, S.; Bargiela, D.; Wengström, Y.; May, A.M.; Rundqvist, H. Inflammation Mediates Exercise Effects on Fatigue in Patients with Breast Cancer. Med. Sci. Sport. Exerc. 2020, 53, 496. [Google Scholar] [CrossRef]
- Brown, J.C.; Zhang, S.; Ligibel, J.A.; Irwin, M.L.; Jones, L.W.; Campbell, N.; Pollak, M.N.; Sorrentino, A.; Cartmel, B.; Harrigan, M.; et al. Effect of Exercise or Metformin on Biomarkers of Inflammation in Breast and Colorectal Cancer: A Randomized Trial. Cancer Prev. Res. 2020, 13, 1055–1062. [Google Scholar] [CrossRef]
- Jee, H.; Chang, J.-E.; Yang, E.J. Positive Prehabilitative Effect of Intense Treadmill Exercise for Ameliorating Cancer Cachexia Symptoms in a Mouse Model. J. Cancer 2016, 7, 2378–2387. [Google Scholar] [CrossRef]
- Joanisse, S.; Lim, C.; McKendry, J.; Mcleod, J.C.; Stokes, T.; Phillips, S.M. Recent advances in understanding resistance exercise training-induced skeletal muscle hypertrophy in humans. F1000Research 2020, 9, 141. [Google Scholar] [CrossRef]
- Sigal, R.J.; Kenny, G.P.; Boulé, N.G.; Wells, G.A.; Prud’homme, D.; Fortier, M.; Reid, R.D.; Tulloch, H.; Coyle, D.; Phillips, P.; et al. Effects of Aerobic Training, Resistance Training, or Both on Glycemic Control in Type 2 Diabetes. Ann. Intern. Med. 2007, 147, 357. [Google Scholar] [CrossRef]
- Church, T.S.; Blair, S.N.; Cocreham, S.; Johannsen, N.; Johnson, W.; Kramer, K.; Mikus, C.R.; Myers, V.; Nauta, M.; Rodarte, R.Q.; et al. Effects of Aerobic and Resistance Training on Hemoglobin A 1c Levels in Patients With Type 2 Diabetes. JAMA 2010, 304, 2253. [Google Scholar] [CrossRef] [PubMed]
- Schiaffino, S.; Reggiani, C.; Akimoto, T.; Blaauw, B. Molecular Mechanisms of Skeletal Muscle Hypertrophy. J. Neuromuscul. Dis. 2020, 1–15. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Tipton, K.D.; Hamilton, D.L.; Gallagher, I.J. Assessing the Role of Muscle Protein Breakdown in Response to Nutrition and Exercise in Humans. Sport. Med. 2018, 48, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Solheim, T.S.; Laird, B.J.A.; Balstad, T.R.; Stene, G.B.; Bye, A.; Johns, N.; Pettersen, C.H.; Fallon, M.; Fayers, P.; Fearon, K.; et al. A randomized phase II feasibility trial of a multimodal intervention for the management of cachexia in lung and pancreatic cancer. J. Cachexia Sarcopenia Muscle 2017, 8, 778–788. [Google Scholar] [CrossRef] [PubMed]
- Lønbro, S.; Dalgas, U.; Primdahl, H.; Johansen, J.; Nielsen, J.L.; Aagaard, P.; Hermann, A.P.; Overgaard, J.; Overgaard, K. Progressive resistance training rebuilds lean body mass in head and neck cancer patients after radiotherapy – Results from the randomized DAHANCA 25B trial. Radiother. Oncol. 2013, 108, 314–319. [Google Scholar] [CrossRef]
- Schmitt, J.; Lindner, N.; Reuss-Borst, M.; Holmberg, H.-C.; Sperlich, B. A 3-week multimodal intervention involving high-intensity interval training in female cancer survivors: A randomized controlled trial. Physiol. Rep. 2016, 4, e12693. [Google Scholar] [CrossRef]
- Mijwel, S.; Backman, M.; Bolam, K.A.; Olofsson, E.; Norrbom, J.; Bergh, J.; Sundberg, C.J.; Wengström, Y.; Rundqvist, H. Highly favorable physiological responses to concurrent resistance and high-intensity interval training during chemotherapy: The OptiTrain breast cancer trial. Breast Cancer Res. Treat. 2018, 169, 93–103. [Google Scholar] [CrossRef]
- Adamsen, L.; Quist, M.; Andersen, C.; Møller, T.; Herrstedt, J.; Kronborg, D.; Baadsgaard, M.T.; Vistisen, K.; Midtgaard, J.; Christiansen, B.; et al. Effect of a multimodal high intensity exercise intervention in cancer patients undergoing chemotherapy: Randomised controlled trial. BMJ 2009, 339, 895–898. [Google Scholar] [CrossRef]
- Lønbro, S.; Farup, J.; Bentsen, S.; Voss, T.; Rittig, N.; Wang, J.; Ørskov, M.; Højris, I.; Mikkelsen, U.R. Lean body mass, muscle fibre size and muscle function in cancer patients during chemotherapy and 10 weeks exercise. JCSM Clin. Reports 2017, 2, 1–15. [Google Scholar] [CrossRef][Green Version]
- Stene, G.B.; Helbostad, J.L.; Balstad, T.R.; Riphagen, I.I.; Kaasa, S.; Oldervoll, L.M. Effect of physical exercise on muscle mass and strength in cancer patients during treatment—A systematic review. Crit. Rev. Oncol. Hematol. 2013, 88, 573–593. [Google Scholar] [CrossRef]
- Lee, J. The effects of resistance training on muscular strength and hypertrophy in elderly cancer patients: A systematic review and meta-analysis. J. Sport Heal. Sci. 2021. [Google Scholar] [CrossRef]
- Tanaka, M.; Sugimoto, K.; Fujimoto, T.; Xie, K.; Takahashi, T.; Akasaka, H.; Kurinami, H.; Yasunobe, Y.; Matsumoto, T.; Fujino, H.; et al. Preventive effects of low-intensity exercise on cancer cachexia–induced muscle atrophy. FASEB J. 2019, 33, 7852–7862. [Google Scholar] [CrossRef]
- Salomão, E.M.; Toneto, A.T.; Silva, G.O.; Gomes-Marcondes, M.C.C. Physical Exercise and a Leucine-Rich Diet Modulate the Muscle Protein Metabolism in Walker Tumor-Bearing Rats. Nutr. Cancer 2010, 62, 1095–1104. [Google Scholar] [CrossRef] [PubMed]
- Khamoui, A.V.; Park, B.-S.; Kim, D.-H.; Yeh, M.-C.; Oh, S.-L.; Elam, M.L.; Jo, E.; Arjmandi, B.H.; Salazar, G.; Grant, S.C.; et al. Aerobic and resistance training dependent skeletal muscle plasticity in the colon-26 murine model of cancer cachexia. Metabolism 2016, 65, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Sugimoto, K.; Fujimoto, T.; Xie, K.; Takahashi, T.; Akasaka, H.; Yasunobe, Y.; Takeya, Y.; Yamamoto, K.; Hirabayashi, T.; et al. Differential effects of pre-exercise on cancer cachexia-induced muscle atrophy in fast- and slow-twitch muscles. FASEB J. 2020, 34, 14389–14406. [Google Scholar] [CrossRef] [PubMed]
- Hardee, J.P.; Fix, D.K.; Koh, H.-J.; Wang, X.; Goldsmith, E.C.; Carson, J.A. Repeated eccentric contractions positively regulate muscle oxidative metabolism and protein synthesis during cancer cachexia in mice. J. Appl. Physiol. 2020, 128, 1666–1676. [Google Scholar] [CrossRef]
- Staples, A.W.; Burd, N.A.; West, D.W.D.; Currie, K.D.; Atherton, P.J.; Moore, D.R.; Rennie, M.; Macdonald, M.J.; Baker, S.K.; Phillips, S.M. Carbohydrate Does Not Augment Exercise-Induced Protein Accretion versus Protein Alone. Med. Sci. Sport. Exerc. 2011, 43, 1154–1161. [Google Scholar] [CrossRef]
- Pennings, B.; Koopman, R.; Beelen, M.; Senden, J.M.; Saris, W.H.; van Loon, L.J. Exercising before protein intake allows for greater use of dietary protein–derived amino acids for de novo muscle protein synthesis in both young and elderly men. Am. J. Clin. Nutr. 2011, 93, 322–331. [Google Scholar] [CrossRef]
- Turner, R.R.; Steed, L.; Quirk, H.; Greasley, R.U.; Saxton, J.M.; Taylor, S.J.; Rosario, D.J.; Thaha, M.A.; Bourke, L. Interventions for promoting habitual exercise in people living with and beyond cancer. Cochrane database Syst. Rev. 2018, 9, CD010192. [Google Scholar] [CrossRef]
- Schmitz, K.H.; Courneya, K.S.; Matthews, C.; Demark-Wahnefried, W.; GALVÃO, D.A.; Pinto, B.M.; IRWIN, M.L.; WOLIN, K.Y.; SEGAL, R.J.; LUCIA, A.; et al. American College of Sports Medicine Roundtable on Exercise Guidelines for Cancer Survivors. Med. Sci. Sport. Exerc. 2010, 42, 1409–1426. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raun, S.H.; Buch-Larsen, K.; Schwarz, P.; Sylow, L. Exercise—A Panacea of Metabolic Dysregulation in Cancer: Physiological and Molecular Insights. Int. J. Mol. Sci. 2021, 22, 3469. https://doi.org/10.3390/ijms22073469
Raun SH, Buch-Larsen K, Schwarz P, Sylow L. Exercise—A Panacea of Metabolic Dysregulation in Cancer: Physiological and Molecular Insights. International Journal of Molecular Sciences. 2021; 22(7):3469. https://doi.org/10.3390/ijms22073469
Chicago/Turabian StyleRaun, Steffen H., Kristian Buch-Larsen, Peter Schwarz, and Lykke Sylow. 2021. "Exercise—A Panacea of Metabolic Dysregulation in Cancer: Physiological and Molecular Insights" International Journal of Molecular Sciences 22, no. 7: 3469. https://doi.org/10.3390/ijms22073469
APA StyleRaun, S. H., Buch-Larsen, K., Schwarz, P., & Sylow, L. (2021). Exercise—A Panacea of Metabolic Dysregulation in Cancer: Physiological and Molecular Insights. International Journal of Molecular Sciences, 22(7), 3469. https://doi.org/10.3390/ijms22073469