
Mannan-BAM, TLR Ligands, Anti-CD40 Antibody (MBTA) Vaccine Immunotherapy: A Review of Current Evidence and Applications in Glioblastoma

 , ,
, , 
Abstract
1. Introduction
2. Current GBM Vaccine Immunotherapy
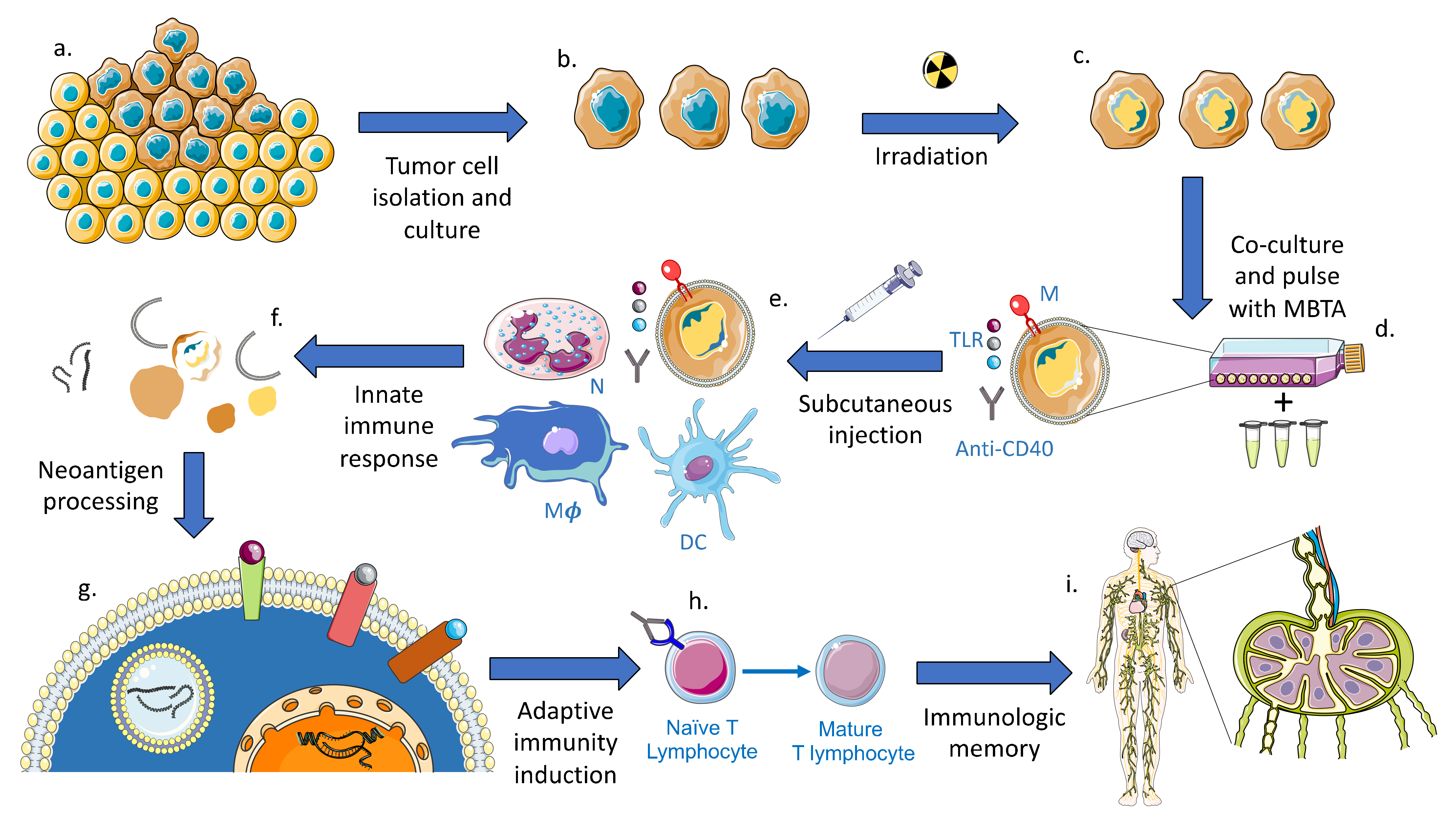
3. Preclinical Investigations of MBTA Immunotherapy in Oncology
4. Future Application of MBTA Vaccine Therapy in GBM
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Caisova, V.; Li, L.; Gupta, G.; Jochmanova, I.; Jha, A.; Uher, O.; Huynh, T.-T.; Miettinen, M.; Pang, Y.; Abunimer, L.; et al. The Significant Reduction or Complete Eradication of Subcutaneous and Metastatic Lesions in a Pheochromocytoma Mouse Model after Immunotherapy Using Mannan-BAM, TLR Ligands, and Anti-CD40. Cancers 2019, 11, 654. [Google Scholar] [CrossRef]
- Caisová, V.; Uher, O.; Nedbalová, P.; Jochmanová, I.; Kvardová, K.; Masáková, K.; Krejčová, G.; Paďouková, L.; Chmelař, J.; Kopecký, J.; et al. Effective cancer immunotherapy based on combination of TLR agonists with stimulation of phagocytosis. Int. Immunopharmacol. 2018, 59, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Itoh, C.; Yasukouchi, T.; Nagamune, T. Rapid Protein Anchoring into the Membranes of Mammalian Cells Using Oleyl Chain and Poly(ethylene glycol) Derivatives. Biotechnol. Prog. 2004, 20, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Janotová, T.; Jalovecká, M.; Auerová, M.; Švecová, I.; Bruzlová, P.; Maierová, V.; Kumžáková, Z.; Čunátová, Š.; Vlčková, Z.; Caisova, V.; et al. The Use of Anchored Agonists of Phagocytic Receptors for Cancer Immunotherapy: B16-F10 Murine Melanoma Model. PLoS ONE 2014, 9, e85222. [Google Scholar] [CrossRef]
- Figueiredo, R.T.; Carneiro, L.A.M.; Bozza, M.T. Fungal Surface and Innate Immune Recognition of Filamentous Fungi. Front. Microbiol. 2011, 2, 248. [Google Scholar] [CrossRef] [PubMed]
- Hassan, S.B.; Sørensen, J.F.; Olsen, B.N.; Pedersen, A.E. Anti-CD40-mediated cancer immunotherapy: An update of recent and ongoing clinical trials. Immunopharmacol. Immunotoxicol. 2014, 36, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Caisová, V.; Vieru, A.M.; Kumžáková, Z.; Glaserová, S.; Husníková, H.; Vácová, N.; Krejčová, G.; Paďouková, L.; Jochmanová, I.; Wolf, K.I.; et al. Innate immunity based cancer immunotherapy: B16-F10 murine melanoma model. BMC Cancer 2016, 16, 940. [Google Scholar] [CrossRef] [PubMed]
- Urban-Wojciuk, Z.; Khan, M.M.; Oyler, B.L.; Fåhraeus, R.; Marek-Trzonkowska, N.; Nita-Lazar, A.; Hupp, T.R.; Goodlett, D.R. The Role of TLRs in Anti-cancer Immunity and Tumor Rejection. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Seo, H.S.; Michalek, S.M.; Nahm, M.H. Lipoteichoic Acid Is Important in Innate Immune Responses to Gram-Positive Bacteria. Infect. Immun. 2007, 76, 206–213. [Google Scholar] [CrossRef]
- Steinhagen, F.; Kinjo, T.; Bode, C.; Klinman, D.M. TLR-based immune adjuvants. Vaccine 2011, 29, 3341–3355. [Google Scholar] [CrossRef]
- Bianchi, F.; Pretto, S.; Tagliabue, E.; Balsari, A.; Sfondrini, L. Exploiting poly(I:C) to induce cancer cell apoptosis. Cancer Biol. Ther. 2017, 18, 747–756. [Google Scholar] [CrossRef] [PubMed]
- Rook, A.H.; Gelfand, J.M.; Wysocka, M.; Troxel, A.B.; Benoit, B.M.; Surber, C.; Elenitsas, R.; Buchanan, M.A.; Leahy, D.S.; Watanabe, R.; et al. Topical resiquimod can induce disease regression and enhance T-cell effector functions in cutaneous T-cell lymphoma. Blood 2015, 126, 1452–1461. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.J.; Huang, D.B.; Tyring, S.K. Resiquimod: A new immune response modifier with potential as a vaccine adjuvant for Th1 immune responses. Antivir. Res. 2004, 64, 79–83. [Google Scholar] [CrossRef]
- Vonderheide, R.H.; Glennie, M.J. Agonistic CD40 Antibodies and Cancer Therapy. Clin. Cancer Res. 2013, 19, 1035–1043. [Google Scholar] [CrossRef]
- Moreau, M.; Yasmin-Karim, S.; Kunjachan, S.; Sinha, N.; Gremse, F.; Kumar, R.; Chow, K.F.; Ngwa, W. Priming the Abscopal Effect Using Multifunctional Smart Radiotherapy Biomaterials Loaded with Immunoadjuvants. Front. Oncol. 2018, 8, 56. [Google Scholar] [CrossRef] [PubMed]
- Fransen, M.F.; Sluijter, M.; Morreau, H.; Arens, R.; Melief, C.J. Local Activation of CD8 T Cells and Systemic Tumor Eradication without Toxicity via Slow Release and Local Delivery of Agonistic CD40 Antibody. Clin. Cancer Res. 2011, 17, 2270–2280. [Google Scholar] [CrossRef] [PubMed]
- Hunn, M.K.; Farrand, K.J.; Broadley, K.W.; Weinkove, R.; Ferguson, P.; Miller, R.J.; Field, C.S.; Petersen, T.; McConnell, M.J.; Hermans, I.F. Vaccination with Irradiated Tumor Cells Pulsed with an Adjuvant That Stimulates NKT Cells Is an Effective Treatment for Glioma. Clin. Cancer Res. 2012, 18, 6446–6459. [Google Scholar] [CrossRef] [PubMed]
- Curry, W.T.; Gorrepati, R.; Piesche, M.; Sasada, T.; Agarwalla, P.; Jones, P.S.; Gerstner, E.R.; Golby, A.J.; Batchelor, T.T.; Wen, P.Y.; et al. Vaccination with Irradiated Autologous Tumor Cells Mixed with Irradiated GM-K562 Cells Stimulates Antitumor Immunity and T Lymphocyte Activation in Patients with Recurrent Malignant Glioma. Clin. Cancer Res. 2016, 22, 2885–2896. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Shi, G.; Yang, G.; Zhang, J.; Li, Y.; Du, T.; Wang, J.; Xu, F.; Cheng, L.; Zhang, X.; et al. Cellular immunotherapy using irradiated lung cancer cell vaccine co-expressing GM-CSF and IL-18 can induce significant antitumor effects. BMC Cancer 2014, 14, 48. [Google Scholar] [CrossRef] [PubMed]
- Koster, B.D.; Santegoets, S.J.A.M.; Harting, J.; Baars, A.; Van Ham, S.M.; Scheper, R.J.; Hooijberg, E.; De Gruijl, T.D.; Eertwegh, A.J.M.V.D. Autologous tumor cell vaccination combined with systemic CpG-B and IFN-α promotes immune activation and induces clinical responses in patients with metastatic renal cell carcinoma: A phase II trial. Cancer Immunol. Immunother. 2019, 68, 1025–1035. [Google Scholar] [CrossRef]
- Uyldegroot, C.; Vermorken, J.; Hannajr, M.; Verboom, P.; Groot, M.; Bonsel, G.; Meijer, C.; Pinedo, H. Immunotherapy with autologous tumor cell-BCG vaccine in patients with colon cancer: A prospective study of medical and economic benefits. Vaccine 2005, 23, 2379–2387. [Google Scholar] [CrossRef] [PubMed]
- Steiner, H.H.; Bonsanto, M.M.; Beckhove, P.; Brysch, M.; Geletneky, K.; Ahmadi, R.; Schuele-Freyer, R.; Kremer, P.; Ranaie, G.; Matejic, D.; et al. Antitumor Vaccination of Patients With Glioblastoma Multiforme: A Pilot Study to Assess Feasibility, Safety, and Clinical Benefit. J. Clin. Oncol. 2004, 22, 4272–4281. [Google Scholar] [CrossRef]
- Medina, R.; Wang, H.; Caisová, V.; Cui, J.; Indig, I.H.; Uher, O.; Ye, J.; Nwankwo, A.; Sanchez, V.; Wu, T.; et al. Induction of Immune Response against Metastatic Tumors via Vaccination of Mannan-BAM, TLR Ligands, and Anti-CD40 Antibody (MBTA). Adv. Ther. 2020, 3, 2000044. [Google Scholar] [CrossRef]
- Belka, C. The fate of irradiated tumor cells. Oncogene 2005, 25, 969–971. [Google Scholar] [CrossRef][Green Version]
- Ostrom, Q.T.; Patil, N.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2013–2017. Neuro-oncology 2020, 22, iv1–iv96. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Chen, J.; Xu, H.; Qin, Z. Geographic variations in the incidence of glioblastoma and prognostic factors predictive of overall survival in US adults from 2004-2013. Front. Aging Neurosci. 2017, 9, 352. [Google Scholar] [CrossRef]
- Weenink, B.; French, P.J.; Smitt, P.A.S.; Debets, R.; Geurts, M. Immunotherapy in Glioblastoma: Current Shortcomings and Future Perspectives. Cancers 2020, 12, 751. [Google Scholar] [CrossRef] [PubMed]
- Ampie, L.; Choy, W.; Lamano, J.B.; Fakurnejad, S.; Bloch, O.; Parsa, A.T. Heat shock protein vaccines against glioblastoma: From bench to bedside. J. Neuro-Oncol. 2015, 123, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Liau, L.M.; Prins, R.M.; Kiertscher, S.M.; Odesa, S.K.; Kremen, T.J.; Giovannone, A.J.; Lin, J.-W.; Chute, D.J.; Mischel, P.S.; Cloughesy, T.F.; et al. Dendritic Cell Vaccination in Glioblastoma Patients Induces Systemic and Intracranial T-cell Responses Modulated by the Local Central Nervous System Tumor Microenvironment. Clin. Cancer Res. 2005, 11, 5515–5525. [Google Scholar] [CrossRef] [PubMed]
- Akgül, S.; Patch, A.-M.; D’Souza, R.C.; Mukhopadhyay, P.; Nones, K.; Kempe, S.; Kazakoff, S.H.; Jeffree, R.L.; Stringer, B.W.; Pearson, J.V.; et al. Intratumoural Heterogeneity Underlies Distinct Therapy Responses and Treatment Resistance in Glioblastoma. Cancers 2019, 11, 190. [Google Scholar] [CrossRef]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.-X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Dudley, M.E.; Yang, J.C.; Sherry, R.; Hughes, M.S.; Royal, R.; Kammula, U.; Robbins, P.F.; Huang, J.; Citrin, D.E.; Leitman, S.F.; et al. Adoptive Cell Therapy for Patients With Metastatic Melanoma: Evaluation of Intensive Myeloablative Chemoradiation Preparative Regimens. J. Clin. Oncol. 2008, 26, 5233–5239. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Medina, R.; Caisova, V.; Uher, O.; Zenka, J.; Pacak, K.; Zhuang, Z. Immu-23. Targeting Metastatic And CNS Tumors Via Mannan-Bam, TLR Ligands and Anti-Cd40 Antibody. Neuro-oncology 2019, 21, vi123–vi124. [Google Scholar] [CrossRef]
- Gromeier, M.; Brown, M.C.; Zhang, G.; Lin, X.; Chen, Y.; Wei, Z.; Beaubier, N.; Yan, H.; Herndon, J.E.; Desjardins, A.; et al. Very low mutation burden is a feature of inflamed recurrent glioblastomas responsive to cancer immunotherapy. Nat. Commun. 2021, 12, 1–7. [Google Scholar] [CrossRef]
- Wank, M.; Schilling, D.; Schmid, T.E.; Meyer, B.; Gempt, J.; Barz, M.; Schlegel, J.; Liesche, F.; Kessel, K.A.; Wiestler, B.; et al. Human Glioma Migration and Infiltration Properties as a Target for Personalized Radiation Medicine. Cancers 2018, 10, 456. [Google Scholar] [CrossRef] [PubMed]
- Santomasso, B.; Bachier, C.; Westin, J.; Rezvani, K.; Shpall, E.J. The Other Side of CAR T-Cell Therapy: Cytokine Release Syndrome, Neurologic Toxicity, and Financial Burden. Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Gust, J.; Hay, K.A.; Hanafi, L.-A.; Li, D.; Myerson, D.; Gonzalez-Cuyar, L.F.; Yeung, C.; Liles, W.C.; Wurfel, M.; Lopez, J.A.; et al. Endothelial Activation and Blood–Brain Barrier Disruption in Neurotoxicity after Adoptive Immunotherapy with CD19 CAR-T Cells. Cancer Discov. 2017, 7, 1404–1419. [Google Scholar] [CrossRef]


{kind=link}
| Clinical Trial | Phase | Target | Number of Patients | Endpoint | Outcome |
|---|---|---|---|---|---|
| ACTIVATe NCT00643097 20921459 | II | EGFRvIII | 18 | PFS at 6 months | 94% v. 59% |
| ACT-II 21149254 | II | EGFRvIII | 22 | OS | 23.6 v. 15.0 mon |
| ACT-III 25586468 | II | EGFRvIII | 65 | PFS at 5.5 months | 66% v. 45% |
| ACT-IV NCT01480479 28844499 | III | EGFRvIII | 371 | OS | No significant difference |
| ReACT NCT01498328 | II | EGFRvIII | 33 | PFS at 6 months | 27% v. 11% |
| HSPPC-96 NCT00905060 II | II | Peptides - Auto. | 46 | OS | 24.0 months |
| HSPPC-96 NCT00293423 24335700 | II | Peptides - Auto. | 41 | OS at 6 months | 90.2% |
| HSPPC-96 NCT01814813 | II | Peptides - Auto. | 30 | OS | No significant difference |
| ITK-1 UMIN000006970 30500939 | III | Tumor Associated Antigens | 58 | OS | No significant difference |
| SL-701 NCT02078648 | II | Tumor Associated Antigens | 74 | OS at 12 months | 43% |
| IMA-950 NCT01920191 30753611 | II | Tumor Associated Antigens | 16 | OS | 19.0 months |
| Clinical Trial | Phase | Antigenic Target | Number of Patients | Endpoint | Outcome |
|---|---|---|---|---|---|
| DENDR1 29632727 | I/II | TL | 24 | PFS at 12 months | 41% |
| DENDR2 NCT02820584 - 1 | I/II | TL | 12 | OS | 7.4 months |
| DENDR2 NCT02820584 - 2 | I/II | TL | 8 | OS | 9.3 months |
| DEND/GM NCT01006044 28499389 | II | TL | 31 | PFS | 12.7 months |
| DCVax-L NCT00045968 29843811 | III | TL | 331 | OS | 23.1 months |
| GBM-Vax NCT01213407 30301187 | II | TL | 34 | PFS at 12 months | No significant difference |
| NCT00576537 18632651 | I/II | TL | 34 | OS | Responders: 21.0 months Non-responders: 14.0 months |
| NCT00323115 21499132 | II | TL | 10 | OS | 28.0 months |
| NCT03879512 30054667 | II | TL | 11 | OS at 6 months | 100% |
| ICT-107 NCT01280552 31320597 | II | Tumor Associated Antigens | 81 | OS | 17.0 months |
| NCT02772094 21715171 | I/II | Irradiated Tumor Cells | 16 | OS at 12 months | 17.0 months |
| NCT01567202 30159779 | I/II | Glioma Stem Cell Associated Antigens | 22 | OS | 7.7 months |
| DC-CAST-GBM NCT00846456 23817721 | I/II | Tumor mRNA | 7 | 25.0 months |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lookian, P.P.; Zhao, D.; Medina, R.; Wang, H.; Zenka, J.; Gilbert, M.R.; Pacak, K.; Zhuang, Z. Mannan-BAM, TLR Ligands, Anti-CD40 Antibody (MBTA) Vaccine Immunotherapy: A Review of Current Evidence and Applications in Glioblastoma. Int. J. Mol. Sci. 2021, 22, 3455. https://doi.org/10.3390/ijms22073455
Lookian PP, Zhao D, Medina R, Wang H, Zenka J, Gilbert MR, Pacak K, Zhuang Z. Mannan-BAM, TLR Ligands, Anti-CD40 Antibody (MBTA) Vaccine Immunotherapy: A Review of Current Evidence and Applications in Glioblastoma. International Journal of Molecular Sciences. 2021; 22(7):3455. https://doi.org/10.3390/ijms22073455
Chicago/Turabian StyleLookian, Pashayar P., David Zhao, Rogelio Medina, Herui Wang, Jan Zenka, Mark R. Gilbert, Karel Pacak, and Zhengping Zhuang. 2021. "Mannan-BAM, TLR Ligands, Anti-CD40 Antibody (MBTA) Vaccine Immunotherapy: A Review of Current Evidence and Applications in Glioblastoma" International Journal of Molecular Sciences 22, no. 7: 3455. https://doi.org/10.3390/ijms22073455
APA StyleLookian, P. P., Zhao, D., Medina, R., Wang, H., Zenka, J., Gilbert, M. R., Pacak, K., & Zhuang, Z. (2021). Mannan-BAM, TLR Ligands, Anti-CD40 Antibody (MBTA) Vaccine Immunotherapy: A Review of Current Evidence and Applications in Glioblastoma. International Journal of Molecular Sciences, 22(7), 3455. https://doi.org/10.3390/ijms22073455