
The Links between ALS and NF-κB


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
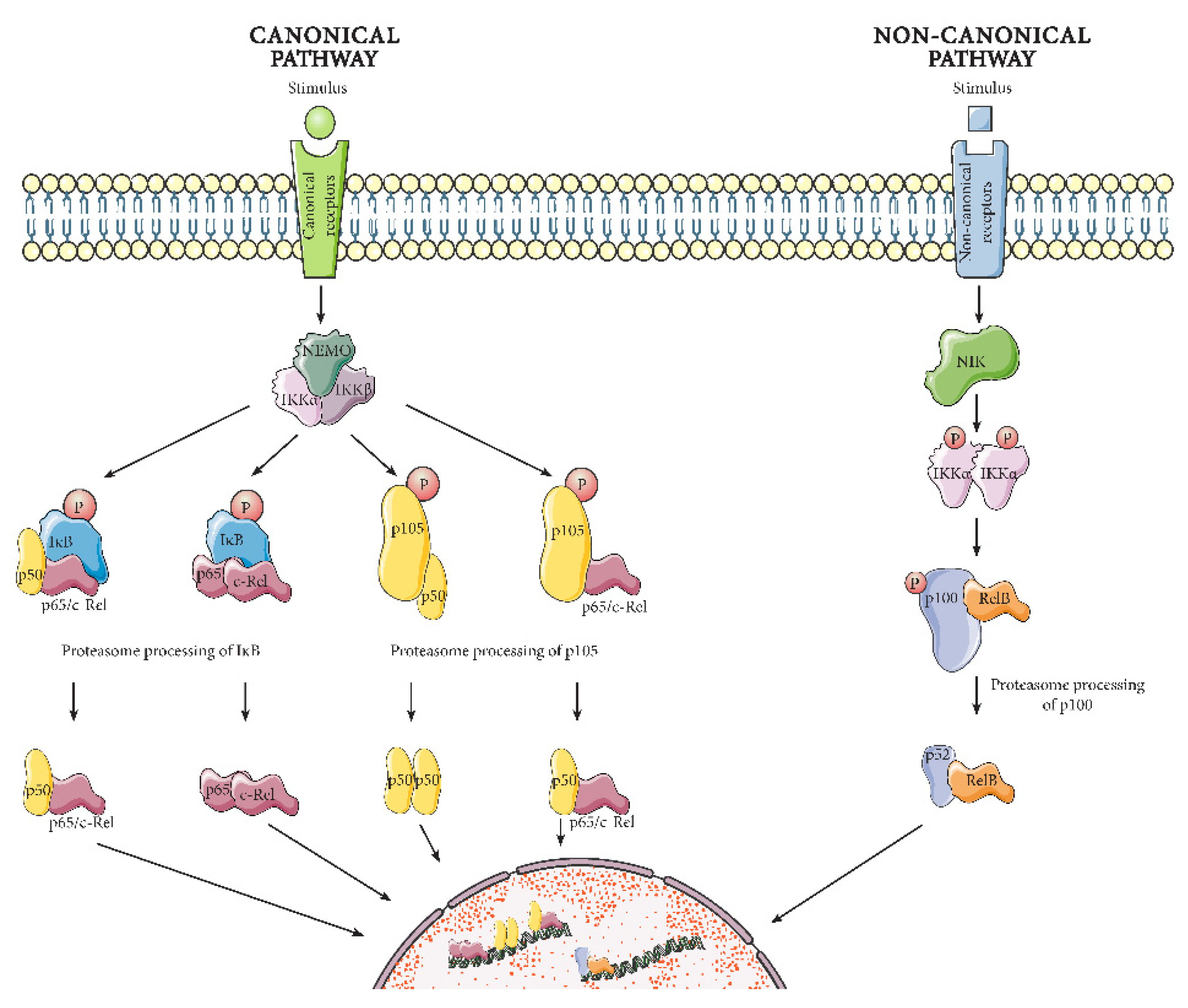
2. NF-κB Signaling
2.1. NF-κB Mechanism of Action
2.1.1. Canonical Pathway
2.1.2. Non-Canonical Pathway
2.2. Functions of NF-κB
3. ALS Genetics
3.1. C9orf72
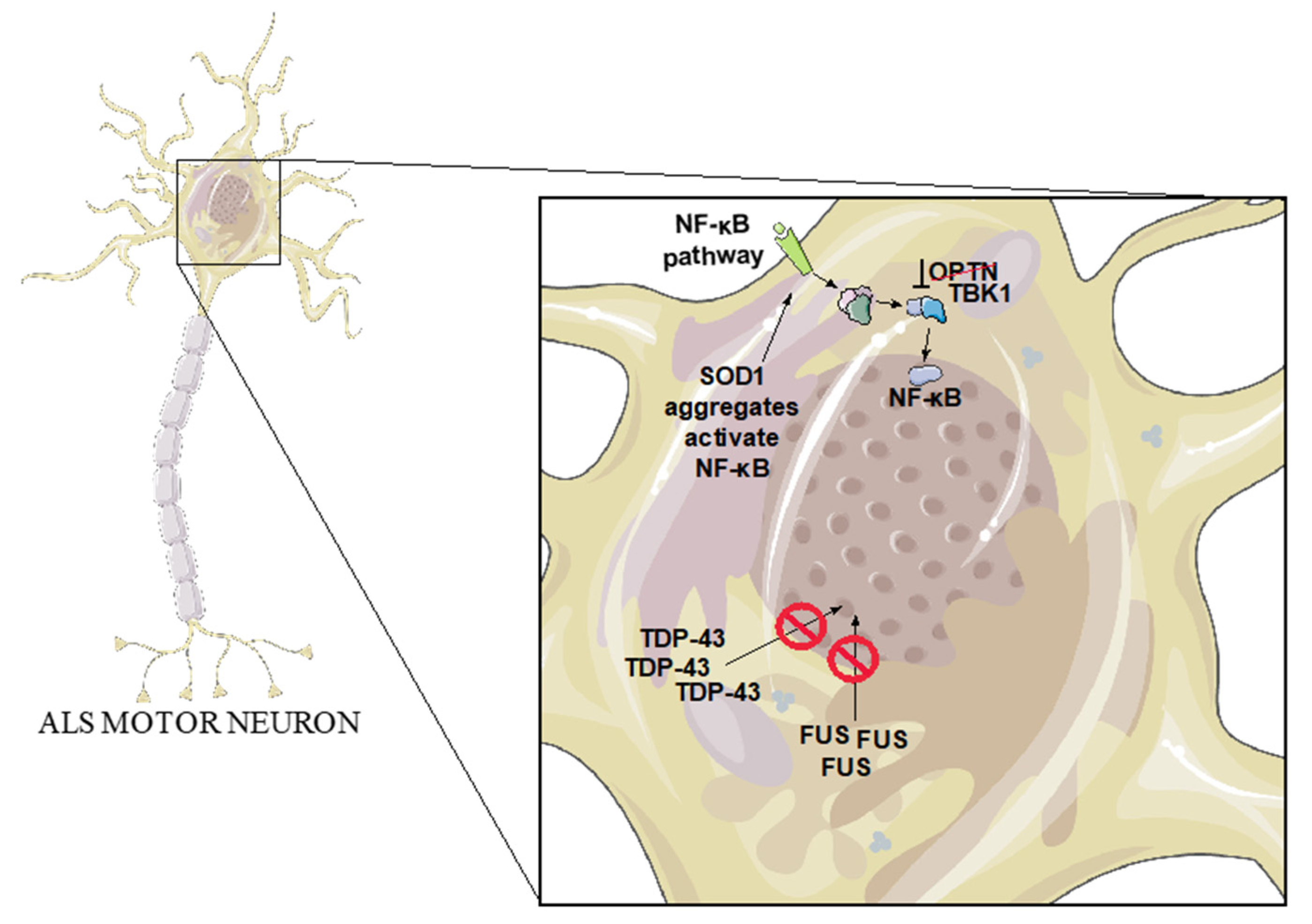
3.2. SOD1
3.3. TDP-43
3.4. FUS
3.5. OPTN
3.6. TBK1
4. ALS Environmental Factors
Bacterial Infections in ALS
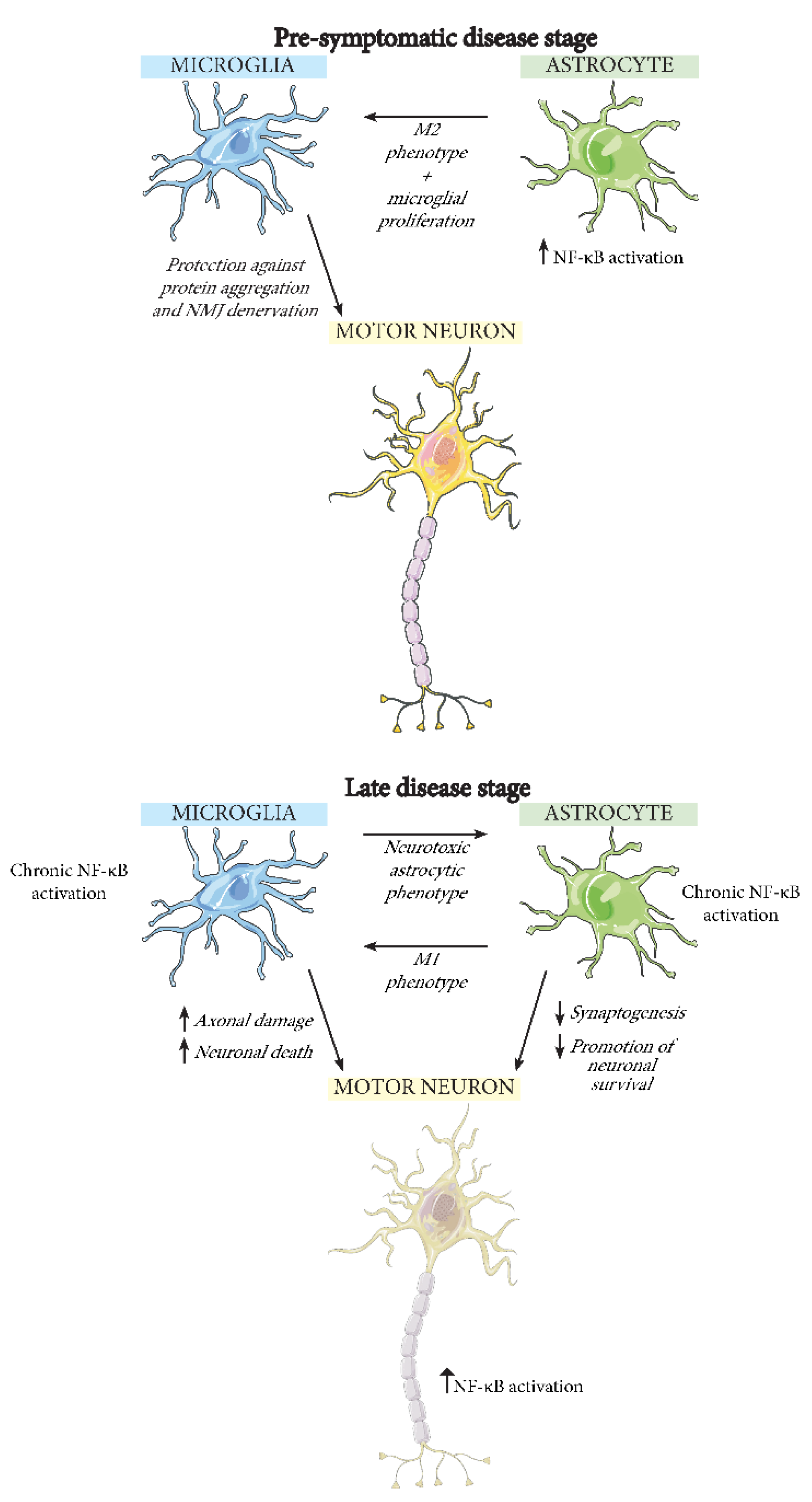
5. The Effect of NF-κB Activation in Different CNS Cell Types during ALS
5.1. Neurons
5.2. Microglia
5.3. Astrocytes
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kumar, D.R.; Aslinia, F.; Yale, S.H.; Mazza, J.J. Jean-Martin Charcot: The Father of Neurology. Clin. Med. Res. 2011, 9, 46. [Google Scholar] [CrossRef]
- Rowland, L.; Shneider, N. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2001, 344, 1688–1700. [Google Scholar] [CrossRef] [PubMed]
- Wijesekera, L.C.; Leigh, P.N. Amyotrophic Lateral Sclerosis. Orphanet J. Rare Dis. 2009, 4, 3. [Google Scholar] [CrossRef]
- Logroscino, G.; Traynor, B.J.; Hardiman, O.; Chiò, A.; Mitchell, D.; Swingler, R.J.; Millul, A.; Benn, E.; Beghi, E. Eurals Incidence of Amyotrophic Lateral Sclerosis in Europe. J. Neurol. Neurosurg. Psychiatry 2010, 81, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Arthur, K.C.; Calvo, A.; Price, T.R.; Geiger, J.T.; Chiò, A.; Traynor, B.J. Projected Increase in Amyotrophic Lateral Sclerosis from 2015 to 2040. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef]
- Renton, A.E.; Chiò, A.; Traynor, B.J. State of Play in Amyotrophic Lateral Sclerosis Genetics. Nat. Neurosci. 2014, 17, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Suescun, J.; Chandra, S.; Schiess, M.C. Chapter 13—The Role of Neuroinflammation in Neurodegenerative Disorders. In Translational Inflammation; Perspectives in Translational Cell Biology; Actor, J.K., Smith, K.C., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 241–267. ISBN 978-0-12-813832-8. [Google Scholar]
- Fakhoury, M. Microglia and Astrocytes in Alzheimer’s Disease: Implications for Therapy. Curr. Neuropharmacol. 2018, 16, 508–518. [Google Scholar] [CrossRef] [PubMed]
- Ponath, G.; Park, C.; Pitt, D. The Role of Astrocytes in Multiple Sclerosis. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Kuhle, J.; Lindberg, R.L.P.; Regeniter, A.; Mehling, M.; Steck, A.J.; Kappos, L.; Czaplinski, A. Increased Levels of Inflammatory Chemokines in Amyotrophic Lateral Sclerosis. Eur. J. Neurol. 2009, 16, 771–774. [Google Scholar] [CrossRef]
- Henkel, J.S.; Beers, D.R.; Siklós, L.; Appel, S.H. The Chemokine MCP-1 and the Dendritic and Myeloid Cells It Attracts Are Increased in the MSOD1 Mouse Model of ALS. Mol. Cell. Neurosci. 2006, 31, 427–437. [Google Scholar] [CrossRef]
- Mishra, P.-S.; Dhull, D.K.; Nalini, A.; Vijayalakshmi, K.; Sathyaprabha, T.N.; Alladi, P.A.; Raju, T.R. Astroglia Acquires a Toxic Neuroinflammatory Role in Response to the Cerebrospinal Fluid from Amyotrophic Lateral Sclerosis Patients. J. Neuroinflamm. 2016, 13, 212. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Günther, R.; Akgün, K.; Hermann, A.; Ziemssen, T. Peripheral Proinflammatory Th1/Th17 Immune Cell Shift Is Linked to Disease Severity in Amyotrophic Lateral Sclerosis. Sci. Rep. 2020, 10, 5941. [Google Scholar] [CrossRef] [PubMed]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X. Motor Neuron Degeneration in Mice That Express a Human Cu, Zn Superoxide Dismutase Mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef]
- Hall, E.D.; Oostveen, J.A.; Gurney, M.E. Relationship of Microglial and Astrocytic Activation to Disease Onset and Progression in a Transgenic Model of Familial ALS. Glia 1998, 23, 249–256. [Google Scholar] [CrossRef]
- Alexianu, M.E.; Kozovska, M.; Appel, S.H. Immune Reactivity in a Mouse Model of Familial ALS Correlates with Disease Progression. Neurology 2001, 57, 1282–1289. [Google Scholar] [CrossRef]
- Frakes, A.E.; Ferraiuolo, L.; Haidet-Phillips, A.M.; Schmelzer, L.; Braun, L.; Miranda, C.J.; Ladner, K.J.; Bevan, A.K.; Foust, K.D.; Godbout, J.P.; et al. Microglia Induce Motor Neuron Death via the Classical NF-ΚB Pathway in Amyotrophic Lateral Sclerosis. Neuron 2014, 81, 1009–1023. [Google Scholar] [CrossRef] [PubMed]
- Crosio, C.; Valle, C.; Casciati, A.; Iaccarino, C.; Carrì, M.T. Astroglial Inhibition of NF-ΚB Does Not Ameliorate Disease Onset and Progression in a Mouse Model for Amyotrophic Lateral Sclerosis (ALS). PLoS ONE 2011, 6, e17187. [Google Scholar] [CrossRef]
- Sen, R.; Baltimore, D. Multiple Nuclear Factors Interact with the Immunoglobulin Enhancer Sequences. Cell 1986, 46, 705–716. [Google Scholar] [CrossRef]
- Steward, R. Dorsal, an Embryonic Polarity Gene in Drosophila, Is Homologous to the Vertebrate Proto-Oncogene, c-Rel. Science 1987, 238, 692–694. [Google Scholar] [CrossRef]
- Gilmore, T.D.; Wolenski, F.S. NF-ΚB: Where Did It Come from and Why? Immunol. Rev. 2012, 246, 14–35. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Ghosh, S. The NF-ΚB Family of Transcription Factors and Its Regulation. Cold Spring Harb. Perspect. Biol. 2009, 1. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.-C. The Non-Canonical NF-ΚB Pathway in Immunity and Inflammation. Nat. Rev. Immunol. 2017, 17, 545. [Google Scholar] [CrossRef]
- Shih, V.F.-S.; Tsui, R.; Caldwell, A.; Hoffmann, A. A Single NFκB System for Both Canonical and Non-Canonical Signaling. Cell Res. 2011, 21, 86–102. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.-C. Non-Canonical NF-ΚB Signaling Pathway. Cell Res. 2011, 21, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-ΚB Signaling in Inflammation. Signal Transduct. Target. Ther. 2017, 2, 1–9. [Google Scholar] [CrossRef]
- Zarnegar, B.J.; Wang, Y.; Mahoney, D.J.; Dempsey, P.W.; Cheung, H.H.; He, J.; Shiba, T.; Yang, X.; Yeh, W.; Mak, T.W.; et al. Activation of Noncanonical NF-ΚB Requires Coordinated Assembly of a Regulatory Complex of the Adaptors CIAP1, CIAP2, TRAF2, TRAF3 and the Kinase NIK. Nat. Immunol. 2008, 9, 1371–1378. [Google Scholar] [CrossRef]
- Rinkenbaugh, A.L.; Baldwin, A.S. The NF-ΚB Pathway and Cancer Stem Cells. Cells 2016, 5, 16. [Google Scholar] [CrossRef]
- Zhang, Q.; Lenardo, M.J.; Baltimore, D. 30 Years of NF-ΚB: A Blossoming of Relevance to Human Pathobiology. Cell 2017, 168, 37–57. [Google Scholar] [CrossRef]
- Cai, D.; Frantz, J.D.; Tawa, N.E.; Melendez, P.A.; Oh, B.-C.; Lidov, H.G.W.; Hasselgren, P.-O.; Frontera, W.R.; Lee, J.; Glass, D.J.; et al. IKKbeta/NF-KappaB Activation Causes Severe Muscle Wasting in Mice. Cell 2004, 119, 285–298. [Google Scholar] [CrossRef]
- Wang, J.; Fu, X.-Q.; Lei, W.-L.; Wang, T.; Sheng, A.-L.; Luo, Z.-G. Nuclear Factor ΚB Controls Acetylcholine Receptor Clustering at the Neuromuscular Junction. J. Neurosci. 2010, 30, 11104–11113. [Google Scholar] [CrossRef]
- Chiot, A.; Zaïdi, S.; Iltis, C.; Ribon, M.; Berriat, F.; Schiaffino, L.; Jolly, A.; de la Grange, P.; Mallat, M.; Bohl, D.; et al. Modifying Macrophages at the Periphery Has the Capacity to Change Microglial Reactivity and to Extend ALS Survival. Nat. Neurosci. 2020, 23, 1339–1351. [Google Scholar] [CrossRef]
- Liu, Z.; Cheng, X.; Zhong, S.; Zhang, X.; Liu, C.; Liu, F.; Zhao, C. Peripheral and Central Nervous System Immune Response Crosstalk in Amyotrophic Lateral Sclerosis. Front. Neurosci. 2020, 14. [Google Scholar] [CrossRef] [PubMed]
- Meffert, M.K.; Chang, J.M.; Wiltgen, B.J.; Fanselow, M.S.; Baltimore, D. NF-Kappa B Functions in Synaptic Signaling and Behavior. Nat. Neurosci. 2003, 6, 1072–1078. [Google Scholar] [CrossRef] [PubMed]
- Meffert, M.K.; Baltimore, D. Physiological Functions for Brain NF-KappaB. Trends Neurosci. 2005, 28, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Albensi, B.C.; Mattson, M.P. Evidence for the Involvement of TNF and NF-KappaB in Hippocampal Synaptic Plasticity. Synapse 2000, 35, 151–159. [Google Scholar] [CrossRef]
- Schuman, E.M.; Madison, D.V. Nitric Oxide and Synaptic Function. Annu. Rev. Neurosci. 1994, 17, 153–183. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.P.; Van Broeckhoven, C.; van der Zee, J. ALS Genes in the Genomic Era and Their Implications for FTD. Trends Genet. 2018, 34, 404–423. [Google Scholar] [CrossRef]
- Picher-Martel, V.; Dutta, K.; Phaneuf, D.; Sobue, G.; Julien, J.-P. Ubiquilin-2 Drives NF-ΚB Activity and Cytosolic TDP-43 Aggregation in Neuronal Cells. Mol. Brain 2015, 8, 71. [Google Scholar] [CrossRef]
- Rolland, P.; Madjd, Z.; Durrant, L.; Ellis, I.O.; Layfield, R.; Spendlove, I. The Ubiquitin-Binding Protein P62 Is Expressed in Breast Cancers Showing Features of Aggressive Disease. Endocr. Relat. Cancer 2007, 14, 73–80. [Google Scholar] [CrossRef]
- Morgan, S.; Orrell, R.W. Pathogenesis of Amyotrophic Lateral Sclerosis. Br. Med. Bull. 2016, 119, 87–98. [Google Scholar] [CrossRef]
- Vanneste, J.; Vercruysse, T.; Boeynaems, S.; Sicart, A.; Van Damme, P.; Daelemans, D.; Van Den Bosch, L. C9orf72-Generated Poly-GR and Poly-PR Do Not Directly Interfere with Nucleocytoplasmic Transport. Sci. Rep. 2019, 9, 15728. [Google Scholar] [CrossRef] [PubMed]
- Bennion Callister, J.; Pickering-Brown, S.M. Pathogenesis/Genetics of Frontotemporal Dementia and How It Relates to ALS. Exp. Neurol. 2014, 262 Pt B, 84–90. [Google Scholar] [CrossRef]
- Balendra, R.; Isaacs, A.M. C9orf72-Mediated ALS and FTD: Multiple Pathways to Disease. Nat. Rev. Neurol. 2018, 14, 544–558. [Google Scholar] [CrossRef]
- Green, K.M.; Linsalata, A.E.; Todd, P.K. RAN Translation—What Makes It Run? Brain Res. 2016, 1647, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Burberry, A.; Suzuki, N.; Wang, J.-Y.; Moccia, R.; Mordes, D.A.; Stewart, M.H.; Suzuki-Uematsu, S.; Ghosh, S.; Singh, A.; Merkle, F.T.; et al. Loss-of-Function Mutations in the C9ORF72 Mouse Ortholog Cause Fatal Autoimmune Disease. Sci. Transl. Med. 2016, 8, 347ra93. [Google Scholar] [CrossRef]
- Atanasio, A.; Decman, V.; White, D.; Ramos, M.; Ikiz, B.; Lee, H.-C.; Siao, C.-J.; Brydges, S.; LaRosa, E.; Bai, Y.; et al. C9orf72 Ablation Causes Immune Dysregulation Characterized by Leukocyte Expansion, Autoantibody Production, and Glomerulonephropathy in Mice. Sci. Rep. 2016, 6, 23204. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, J.G.; Bogdanik, L.; Yáñez, A.; Lall, D.; Wolf, A.J.; Muhammad, A.K.M.G.; Ho, R.; Carmona, S.; Vit, J.P.; Zarrow, J.; et al. C9orf72 Is Required for Proper Macrophage and Microglial Function in Mice. Science 2016, 351, 1324–1329. [Google Scholar] [CrossRef]
- McCauley, M.E.; O’Rourke, J.G.; Yáñez, A.; Markman, J.L.; Ho, R.; Wang, X.; Chen, S.; Lall, D.; Jin, M.; Muhammad, A.K.M.G.; et al. C9orf72 in Myeloid Cells Suppresses STING-Induced Inflammation. Nature 2020, 585, 96–101. [Google Scholar] [CrossRef]
- Burberry, A.; Wells, M.F.; Limone, F.; Couto, A.; Smith, K.S.; Keaney, J.; Gillet, G.; van Gastel, N.; Wang, J.-Y.; Pietilainen, O.; et al. C9orf72 Suppresses Systemic and Neural Inflammation Induced by Gut Bacteria. Nature 2020, 582, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Fomin, V.; Richard, P.; Hoque, M.; Li, C.; Gu, Z.; Fissore-O’Leary, M.; Tian, B.; Prives, C.; Manley, J.L. The C9ORF72 Gene, Implicated in ALS/FTD, Encodes a Protein That Functions in Control of Endothelin and Glutamate Signaling. Mol. Cell. Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.J.; McKeown, S.R.; Rashid, S. Mutant SOD1 Mediated Pathogenesis of Amyotrophic Lateral Sclerosis. Gene 2016, 577, 109–118. [Google Scholar] [CrossRef]
- Chattopadhyay, M.; Valentine, J.S. Aggregation of Copper-Zinc Superoxide Dismutase in Familial and Sporadic ALS. Antioxid. Redox Signal. 2009, 11, 1603–1614. [Google Scholar] [CrossRef]
- Rochat, C.; Bernard-Marissal, N.; Pradervand, S.; Perrin, F.E.; Raoul, C.; Aebischer, P.; Schneider, B.L. Expression of a MiRNA Targeting Mutated SOD1 in Astrocytes Induces Motoneuron Plasticity and Improves Neuromuscular Function in ALS Mice. bioRxiv 2021. [Google Scholar] [CrossRef]
- Boillée, S.; Yamanaka, K.; Lobsiger, C.S.; Copeland, N.G.; Jenkins, N.A.; Kassiotis, G.; Kollias, G.; Cleveland, D.W. Onset and Progression in Inherited ALS Determined by Motor Neurons and Microglia. Science 2006, 312, 1389–1392. [Google Scholar] [CrossRef]
- Beers, D.R.; Henkel, J.S.; Xiao, Q.; Zhao, W.; Wang, J.; Yen, A.A.; Siklos, L.; McKercher, S.R.; Appel, S.H. Wild-Type Microglia Extend Survival in PU.1 Knockout Mice with Familial Amyotrophic Lateral Sclerosis. Proc. Natl. Acad. Sci. USA 2006, 103, 16021–16026. [Google Scholar] [CrossRef]
- Guttenplan, K.A.; Weigel, M.K.; Adler, D.I.; Couthouis, J.; Liddelow, S.A.; Gitler, A.D.; Barres, B.A. Knockout of Reactive Astrocyte Activating Factors Slows Disease Progression in an ALS Mouse Model. Nat. Commun. 2020, 11, 3753. [Google Scholar] [CrossRef] [PubMed]
- Nivon, M.; Fort, L.; Muller, P.; Richet, E.; Simon, S.; Guey, B.; Fournier, M.; Arrigo, A.-P.; Hetz, C.; Atkin, J.D.; et al. NFκB Is a Central Regulator of Protein Quality Control in Response to Protein Aggregation Stresses via Autophagy Modulation. Mol. Biol. Cell 2016, 27, 1712–1727. [Google Scholar] [CrossRef] [PubMed]
- Prell, T.; Lautenschläger, J.; Weidemann, L.; Ruhmer, J.; Witte, O.W.; Grosskreutz, J. Endoplasmic Reticulum Stress Is Accompanied by Activation of NF-ΚB in Amyotrophic Lateral Sclerosis. J. Neuroimmunol. 2014, 270, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Ederle, H.; Funk, C.; Abou-Ajram, C.; Hutten, S.; Funk, E.B.E.; Kehlenbach, R.H.; Bailer, S.M.; Dormann, D. Nuclear Egress of TDP-43 and FUS Occurs Independently of Exportin-1/CRM1. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef]
- Khalfallah, Y.; Kuta, R.; Grasmuck, C.; Prat, A.; Durham, H.D.; Vande Velde, C. TDP-43 Regulation of Stress Granule Dynamics in Neurodegenerative Disease-Relevant Cell Types. Sci. Rep. 2018, 8, 7551. [Google Scholar] [CrossRef] [PubMed]
- Swarup, V.; Phaneuf, D.; Dupré, N.; Petri, S.; Strong, M.; Kriz, J.; Julien, J.-P. Deregulation of TDP-43 in Amyotrophic Lateral Sclerosis Triggers Nuclear Factor ΚB–Mediated Pathogenic Pathways. J. Exp. Med. 2011, 208, 2429–2447. [Google Scholar] [CrossRef]
- Yu, C.-H.; Davidson, S.; Harapas, C.R.; Hilton, J.B.; Mlodzianoski, M.J.; Laohamonthonkul, P.; Louis, C.; Low, R.R.J.; Moecking, J.; De Nardo, D.; et al. TDP-43 Triggers Mitochondrial DNA Release via MPTP to Activate CGAS/STING in ALS. Cell 2020, 183, 636–649.e18. [Google Scholar] [CrossRef] [PubMed]
- Jara, J.H.; Gautam, M.; Kocak, N.; Xie, E.F.; Mao, Q.; Bigio, E.H.; Özdinler, P.H. MCP1-CCR2 and Neuroinflammation in the ALS Motor Cortex with TDP-43 Pathology. J. Neuroinflamm. 2019, 16, 196. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kim, S.; Kang, H.-Y.; Lim, H.R.; Kwon, Y.; Jo, M.; Jeon, Y.-M.; Kim, S.R.; Kim, K.; Ha, C.M.; et al. The Overexpression of TDP-43 in Astrocytes Causes Neurodegeneration via a PTP1B-Mediated Inflammatory Response. J. Neuroinflamm. 2020, 17. [Google Scholar] [CrossRef] [PubMed]
- Zamudio, F.; Loon, A.R.; Smeltzer, S.; Benyamine, K.; Navalpur Shanmugam, N.K.; Stewart, N.J.F.; Lee, D.C.; Nash, K.; Selenica, M.-L.B. TDP-43 Mediated Blood-Brain Barrier Permeability and Leukocyte Infiltration Promote Neurodegeneration in a Low-Grade Systemic Inflammation Mouse Model. J. Neuroinflamm. 2020, 17, 283. [Google Scholar] [CrossRef]
- Correia, A.S.; Patel, P.; Dutta, K.; Julien, J.-P. Inflammation Induces TDP-43 Mislocalization and Aggregation. PLoS ONE 2015, 10. [Google Scholar] [CrossRef]
- Vance, C.; Scotter, E.L.; Nishimura, A.L.; Troakes, C.; Mitchell, J.C.; Kathe, C.; Urwin, H.; Manser, C.; Miller, C.C.; Hortobágyi, T.; et al. ALS Mutant FUS Disrupts Nuclear Localization and Sequesters Wild-Type FUS within Cytoplasmic Stress Granules. Hum. Mol. Genet. 2013, 22, 2676–2688. [Google Scholar] [CrossRef]
- Nakaya, T.; Maragkakis, M. Amyotrophic Lateral Sclerosis Associated FUS Mutation Shortens Mitochondria and Induces Neurotoxicity. Sci. Rep. 2018, 8, 15575. [Google Scholar] [CrossRef]
- López-Erauskin, J.; Tadokoro, T.; Baughn, M.W.; Myers, B.; McAlonis-Downes, M.; Chillon-Marinas, C.; Asiaban, J.N.; Artates, J.; Bui, A.T.; Vetto, A.P.; et al. ALS/FTD-Linked Mutation in FUS Suppresses Intra-Axonal Protein Synthesis and Drives Disease Without Nuclear Loss-of-Function of FUS. Neuron 2018, 100, 816–830.e7. [Google Scholar] [CrossRef]
- Wang, J.-W.; Brent, J.R.; Tomlinson, A.; Shneider, N.A.; McCabe, B.D. The ALS-Associated Proteins FUS and TDP-43 Function Together to Affect Drosophila Locomotion and Life Span. J. Clin. Investig. 2011, 121, 4118–4126. [Google Scholar] [CrossRef]
- Uranishi, H.; Tetsuka, T.; Yamashita, M.; Asamitsu, K.; Shimizu, M.; Itoh, M.; Okamoto, T. Involvement of the Pro-Oncoprotein TLS (Translocated in Liposarcoma) in Nuclear Factor-ΚB P65-Mediated Transcription as a Coactivator. J. Biol. Chem. 2001, 276, 13395–13401. [Google Scholar] [CrossRef]
- Shelkovnikova, T.A.; An, H.; Skelt, L.; Tregoning, J.S.; Humphreys, I.R.; Buchman, V.L. Antiviral Immune Response as a Trigger of FUS Proteinopathy in Amyotrophic Lateral Sclerosis. Cell Rep. 2019, 29, 4496–4508.e4. [Google Scholar] [CrossRef]
- Ajmone-Cat, M.A.; Onori, A.; Toselli, C.; Stronati, E.; Morlando, M.; Bozzoni, I.; Monni, E.; Kokaia, Z.; Lupo, G.; Minghetti, L.; et al. Increased FUS Levels in Astrocytes Leads to Astrocyte and Microglia Activation and Neuronal Death. Sci. Rep. 2019, 9, 4572. [Google Scholar] [CrossRef] [PubMed]
- Markovinovic, A.; Cimbro, R.; Ljutic, T.; Kriz, J.; Rogelj, B.; Munitic, I. Optineurin in Amyotrophic Lateral Sclerosis: Multifunctional Adaptor Protein at the Crossroads of Different Neuroprotective Mechanisms. Prog. Neurobiol. 2017, 154, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Ryan, T.A.; Tumbarello, D.A. Optineurin: A Coordinator of Membrane-Associated Cargo Trafficking and Autophagy. Front. Immunol. 2018, 9, 1024. [Google Scholar] [CrossRef] [PubMed]
- Slowicka, K.; van Loo, G. Optineurin Functions for Optimal Immunity. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Akizuki, M.; Yamashita, H.; Uemura, K.; Maruyama, H.; Kawakami, H.; Ito, H.; Takahashi, R. Optineurin Suppression Causes Neuronal Cell Death via NF-ΚB Pathway. J. Neurochem. 2013, 126, 699–704. [Google Scholar] [CrossRef] [PubMed]
- Kamada, M.; Izumi, Y.; Ayaki, T.; Nakamura, M.; Kagawa, S.; Kudo, E.; Sako, W.; Maruyama, H.; Nishida, Y.; Kawakami, H.; et al. Clinicopathologic Features of Autosomal Recessive Amyotrophic Lateral Sclerosis Associated with Optineurin Mutation. Neuropathology 2014, 34, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Sako, W.; Ito, H.; Yoshida, M.; Koizumi, H.; Kamada, M.; Fujita, K.; Hashizume, Y.; Izumi, Y.; Kaji, R. Nuclear Factor κ B Expression in Patients with Sporadic Amyotrophic Lateral Sclerosis and Hereditary Amyotrophic Lateral Sclerosis with Optineurin Mutations. Clin. Neuropathol. 2012, 31, 418–423. [Google Scholar] [CrossRef]
- Ito, Y.; Ofengeim, D.; Najafov, A.; Das, S.; Saberi, S.; Li, Y.; Hitomi, J.; Zhu, H.; Chen, H.; Mayo, L.; et al. RIPK1 Mediates Axonal Degeneration by Promoting Inflammation and Necroptosis in ALS. Science 2016, 353, 603–608. [Google Scholar] [CrossRef]
- De Diego, R.P.; Rodríguez-Gallego, C. Chapter 34—Other TLR Pathway Defects. In Stiehm’s Immune Deficiencies; Sullivan, K.E., Stiehm, E.R., Eds.; Academic Press: Amsterdam, The Netherlands, 2014; pp. 687–710. ISBN 978-0-12-405546-9. [Google Scholar]
- Sweeney, S.E.; Mo, L.; Firestein, G.S. Antiviral Gene Expression in Rheumatoid Arthritis: Role of IKKϵ and Interferon Regulatory Factor 3. Arthritis Rheum. 2007, 56, 743–752. [Google Scholar] [CrossRef]
- Pomerantz, J.L.; Baltimore, D. NF-KappaB Activation by a Signaling Complex Containing TRAF2, TANK and TBK1, a Novel IKK-Related Kinase. EMBO J. 1999, 18, 6694–6704. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Yue, B.Y.J.T. Chapter five—Cellular and Molecular Biology of Optineurin. In International Review of Cell and Molecular Biology; Jeon, K.W., Ed.; Academic Press: Cambridge, MA, USA, 2012; Volume 294, pp. 223–258. [Google Scholar]
- De Majo, M.; Topp, S.D.; Smith, B.N.; Nishimura, A.L.; Chen, H.-J.; Gkazi, A.S.; Miller, J.; Wong, C.H.; Vance, C.; Baas, F.; et al. ALS-Associated Missense and Nonsense TBK1 Mutations Can Both Cause Loss of Kinase Function. Neurobiol. Aging 2018, 71, 266.e1–266.e10. [Google Scholar] [CrossRef] [PubMed]
- Gerbino, V.; Kaunga, E.; Ye, J.; Canzio, D.; O’Keeffe, S.; Rudnick, N.D.; Guarnieri, P.; Lutz, C.M.; Maniatis, T. The Loss of TBK1 Kinase Activity in Motor Neurons or in All Cell Types Differentially Impacts ALS Disease Progression in SOD1 Mice. Neuron 2020, 106, 789–805.e5. [Google Scholar] [CrossRef]
- Brenner, D.; Sieverding, K.; Bruno, C.; Lüningschrör, P.; Buck, E.; Mungwa, S.; Fischer, L.; Brockmann, S.J.; Ulmer, J.; Bliederhäuser, C.; et al. Heterozygous Tbk1 Loss Has Opposing Effects in Early and Late Stages of ALS in Mice. J. Exp. Med. 2019, 216, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.; Logroscino, G.; Hernán, M.A. Smoking and the Risk of Amyotrophic Lateral Sclerosis: A Systematic Review and Meta-Analysis. J. Neurol. Neurosurg. Psychiatry 2010, 81, 1249–1252. [Google Scholar] [CrossRef]
- Wang, H.; O’Reilly, E.; Weisskopf, M.; Logroscino, G.; McCullough, M.; Thun, M.; Schatzkin, A.; Kolonel, L.; Ascherio, A. Smoking and Risk of Amyotrophic Lateral Sclerosis: A Pooled Analysis of 5 Prospective Cohorts. Arch. Neurol. 2011, 68, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Seelen, M.; van Doormaal, P.T.C.; Visser, A.E.; Huisman, M.H.B.; Roozekrans, M.H.J.; de Jong, S.W.; van der Kooi, A.J.; de Visser, M.; Voermans, N.C.; Veldink, J.H.; et al. Prior Medical Conditions and the Risk of Amyotrophic Lateral Sclerosis. J. Neurol. 2014, 261, 1949–1956. [Google Scholar] [CrossRef] [PubMed]
- McCambridge, M.; Stinson, M.J. Advances in Chronic Traumatic Encephalopathy. JAAPA Off. J. Am. Acad. Physician Assist. 2020, 33, 39–42. [Google Scholar] [CrossRef]
- Oskarsson, B.; Horton, D.K.; Mitsumoto, H. Potential Environmental Factors in Amyotrophic Lateral Sclerosis. Neurol. Clin. 2015, 33, 877–888. [Google Scholar] [CrossRef]
- Huisman, M.H.B.; Seelen, M.; de Jong, S.W.; Dorresteijn, K.R.I.S.; van Doormaal, P.T.C.; van der Kooi, A.J.; de Visser, M.; Schelhaas, H.J.; van den Berg, L.H.; Veldink, J.H. Lifetime Physical Activity and the Risk of Amyotrophic Lateral Sclerosis. J. Neurol. Neurosurg. Psychiatry 2013, 84, 976–981. [Google Scholar] [CrossRef]
- Fang, F.; Kwee, L.C.; Allen, K.D.; Umbach, D.M.; Ye, W.; Watson, M.; Keller, J.; Oddone, E.Z.; Sandler, D.P.; Schmidt, S.; et al. Association between Blood Lead and the Risk of Amyotrophic Lateral Sclerosis. Am. J. Epidemiol. 2010, 171, 1126–1133. [Google Scholar] [CrossRef]
- Liu, Y.-Z.; Wang, Y.-X.; Jiang, C.-L. Inflammation: The Common Pathway of Stress-Related Diseases. Front. Hum. Neurosci. 2017, 11. [Google Scholar] [CrossRef]
- Bozzoni, V.; Pansarasa, O.; Diamanti, L.; Nosari, G.; Cereda, C.; Ceroni, M. Amyotrophic Lateral Sclerosis and Environmental Factors. Funct. Neurol. 2016, 31, 7–19. [Google Scholar] [CrossRef]
- Banks, C.N.; Lein, P.J. A Review of Experimental Evidence Linking Neurotoxic Organophosphorus Compounds and Inflammation. Neurotoxicology 2012, 33, 575–584. [Google Scholar] [CrossRef]
- Ascherio, A.; Weisskopf, M.G.; O’reilly, E.J.; Jacobs, E.J.; McCullough, M.L.; Calle, E.E.; Cudkowicz, M.; Thun, M.J. Vitamin E Intake and Risk of Amyotrophic Lateral Sclerosis. Ann. Neurol. 2005, 57, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Zhang, X.; Li, J.J. The Role of NF-ΚB in the Regulation of Cell Stress Responses. Int. Immunopharmacol. 2002, 2, 1509–1520. [Google Scholar] [CrossRef]
- Martorana, F.; Foti, M.; Virtuoso, A.; Gaglio, D.; Aprea, F.; Latronico, T.; Rossano, R.; Riccio, P.; Papa, M.; Alberghina, L.; et al. Differential Modulation of NF-ΚB in Neurons and Astrocytes Underlies Neuroprotection and Antigliosis Activity of Natural Antioxidant Molecules. Available online: https://www.hindawi.com/journals/omcl/2019/8056904/ (accessed on 3 February 2021).
- Kratsovnik, E.; Bromberg, Y.; Sperling, O.; Zoref-Shani, E. Oxidative Stress Activates Transcription Factor NF-KB-Mediated Protective Signaling in Primary Rat Neuronal Cultures. J. Mol. Neurosci. 2005, 26, 27–32. [Google Scholar] [CrossRef]
- Delzor, A.; Couratier, P.; Boumédiène, F.; Nicol, M.; Druet-Cabanac, M.; Paraf, F.; Méjean, A.; Ploux, O.; Leleu, J.-P.; Brient, L.; et al. Searching for a Link between the L-BMAA Neurotoxin and Amyotrophic Lateral Sclerosis: A Study Protocol of the French BMAALS Programme. BMJ Open 2014, 4, e005528. [Google Scholar] [CrossRef]
- Silva, D.F.; Candeias, E.; Esteves, A.R.; Magalhães, J.D.; Ferreira, I.L.; Nunes-Costa, D.; Rego, A.C.; Empadinhas, N.; Cardoso, S.M. Microbial BMAA Elicits Mitochondrial Dysfunction, Innate Immunity Activation, and Alzheimer’s Disease Features in Cortical Neurons. J. Neuroinflamm. 2020, 17, 332. [Google Scholar] [CrossRef]
- Alonso, R.; Pisa, D.; Carrasco, L. Searching for Bacteria in Neural Tissue From Amyotrophic Lateral Sclerosis. Front. Neurosci. 2019, 13. [Google Scholar] [CrossRef]
- Nicolson, G.L.; Nasralla, M.Y.; Haier, J.; Pomfret, J. High Frequency of Systemic Mycoplasmal Infections in Gulf War Veterans and Civilians with Amyotrophic Lateral Sclerosis (ALS). J. Clin. Neurosci. 2002, 9, 525–529. [Google Scholar] [CrossRef]
- Hänsel, Y.; Ackerl, M.; Stanek, G. ALS-like sequelae in chronic neuroborreliosis. Wien. Med. Wochenschr. 1995, 145, 186–188. [Google Scholar]
- Halperin, J.J.; Kaplan, G.P.; Brazinsky, S.; Tsai, T.F.; Cheng, T.; Ironside, A.; Wu, P.; Delfiner, J.; Golightly, M.; Brown, R.H. Immunologic Reactivity against Borrelia Burgdorferi in Patients with Motor Neuron Disease. Arch. Neurol. 1990, 47, 586–594. [Google Scholar] [CrossRef]
- Allen, H.B. Alzheimer’s Disease: Assessing the Role of Spirochetes, Biofilms, the Immune System, and Amyloid-β with Regard to Potential Treatment and Prevention. J. Alzheimers Dis. 2016, 53, 1271–1276. [Google Scholar] [CrossRef]
- Nicolson, G.L. Chronic Bacterial and Viral Infections in Neurodegenerative and Neurobehavioral Diseases. Lab. Med. 2008, 39, 291–299. [Google Scholar] [CrossRef][Green Version]
- Kashyap, S.; Sarkar, M. Mycoplasma Pneumonia: Clinical Features and Management. Lung India Off. Organ Indian Chest Soc. 2010, 27, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Ebnet, K.; Brown, K.D.; Siebenlist, U.K.; Simon, M.M.; Shaw, S. Borrelia Burgdorferi Activates Nuclear Factor-Kappa B and Is a Potent Inducer of Chemokine and Adhesion Molecule Gene Expression in Endothelial Cells and Fibroblasts. J. Immunol. 1997, 158, 3285–3292. [Google Scholar]
- Brown, G.C. The Endotoxin Hypothesis of Neurodegeneration. J. Neuroinflamm. 2019, 16, 180. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Miller, R.G.; Gascon, R.; Champion, S.; Katz, J.; Lancero, M.; Narvaez, A.; Honrada, R.; Ruvalcaba, D.; McGrath, M.S. Circulating Endotoxin and Systemic Immune Activation in Sporadic Amyotrophic Lateral Sclerosis (SALS). J. Neuroimmunol. 2009, 206, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Wu, X.; Block, M.L.; Liu, Y.; Breese, G.R.; Hong, J.-S.; Knapp, D.J.; Crews, F.T. Systemic LPS Causes Chronic Neuroinflammation and Progressive Neurodegeneration. Glia 2007, 55, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Zakaria, R.; Wan Yaacob, W.M.; Othman, Z.; Long, I.; Ahmad, A.H.; Al-Rahbi, B. Lipopolysaccharide-Induced Memory Impairment in Rats: A Model of Alzheimer’s Disease. Physiol. Res. 2017, 66, 553–565. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.C.; Gilliam, E.A.; Li, L. Innate Immune Programing by Endotoxin and Its Pathological Consequences. Front. Immunol. 2015, 5. [Google Scholar] [CrossRef]
- Coque, E.; Salsac, C.; Espinosa-Carrasco, G.; Varga, B.; Degauque, N.; Cadoux, M.; Crabé, R.; Virenque, A.; Soulard, C.; Fierle, J.K.; et al. Cytotoxic CD8+ T Lymphocytes Expressing ALS-Causing SOD1 Mutant Selectively Trigger Death of Spinal Motoneurons. Proc. Natl. Acad. Sci. USA 2019, 116, 2312–2317. [Google Scholar] [CrossRef]
- Morimoto, K.; Nakajima, K. Role of the Immune System in the Development of the Central Nervous System. Front. Neurosci. 2019, 13. [Google Scholar] [CrossRef] [PubMed]
- Kassed, C.A.; Herkenham, M. NF-ΚB P50-Deficient Mice Show Reduced Anxiety-like Behaviors in Tests of Exploratory Drive and Anxiety. Behav. Brain Res. 2004, 154, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, K.; Odero, G.L.; Platt, E.; Neuendorff, M.; Hatherell, A.; Bernstein, M.J.; Albensi, B.C. NF-ΚB P50 Subunit Knockout Impairs Late LTP and Alters Long Term Memory in the Mouse Hippocampus. BMC Neurosci. 2012, 13, 45. [Google Scholar] [CrossRef]
- Ahn, H.J.; Hernandez, C.M.; Levenson, J.M.; Lubin, F.D.; Liou, H.-C.; Sweatt, J.D. C-Rel, an NF-ΚB Family Transcription Factor, Is Required for Hippocampal Long-Term Synaptic Plasticity and Memory Formation. Learn. Mem. 2008, 15, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Boersma, M.C.H.; Dresselhaus, E.C.; Biase, L.M.D.; Mihalas, A.B.; Bergles, D.E.; Meffert, M.K. A Requirement for Nuclear Factor-ΚB in Developmental and Plasticity-Associated Synaptogenesis. J. Neurosci. 2011, 31, 5414–5425. [Google Scholar] [CrossRef]
- Guerrini, L.; Molteni, A.; Wirth, T.; Kistler, B.; Blasi, F. Glutamate-Dependent Activation of NF-ΚB During Mouse Cerebellum Development. J. Neurosci. 1997, 17, 6057–6063. [Google Scholar] [CrossRef]
- Dresselhaus, E.C.; Meffert, M.K. Cellular Specificity of NF-ΚB Function in the Nervous System. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Dutta, K.; Thammisetty, S.S.; Boutej, H.; Bareil, C.; Julien, J.-P. Mitigation of ALS Pathology by Neuron-Specific Inhibition of Nuclear Factor Kappa B Signaling. J. Neurosci. 2020, 40, 5137–5154. [Google Scholar] [CrossRef]
- Kyrargyri, V.; Vega-Flores, G.; Gruart, A.; Delgado-García, J.M.; Probert, L. Differential Contributions of Microglial and Neuronal IKKβ to Synaptic Plasticity and Associative Learning in Alert Behaving Mice. Glia 2015, 63, 549–566. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.R.; Cagnin, A.; Turkheimer, F.E.; Miller, C.C.J.; Shaw, C.E.; Brooks, D.J.; Leigh, P.N.; Banati, R.B. Evidence of Widespread Cerebral Microglial Activation in Amyotrophic Lateral Sclerosis: An [11C](R)-PK11195 Positron Emission Tomography Study. Neurobiol. Dis. 2004, 15, 601–609. [Google Scholar] [CrossRef]
- Geloso, M.C.; Corvino, V.; Marchese, E.; Serrano, A.; Michetti, F.; D’Ambrosi, N. The Dual Role of Microglia in ALS: Mechanisms and Therapeutic Approaches. Front. Aging Neurosci. 2017, 9. [Google Scholar] [CrossRef]
- Crain, J.M.; Nikodemova, M.; Watters, J.J. Microglia Express Distinct M1 and M2 Phenotypic Markers in the Postnatal and Adult Central Nervous System in Male and Female Mice. J. Neurosci. Res. 2013, 91, 1143–1151. [Google Scholar] [CrossRef] [PubMed]
- Appel, S.H.; Zhao, W.; Beers, D.R.; Henkel, J.S. The Microglial-Motoneuron Dialogue in ALS. Acta Myol. 2011, 30, 4. [Google Scholar] [PubMed]
- Henkel, J.S.; Beers, D.R.; Zhao, W.; Appel, S.H. Microglia in ALS: The Good, The Bad, and The Resting. J. Neuroimmune Pharmacol. 2009, 4, 389–398. [Google Scholar] [CrossRef]
- Zhang, Y.; Reichel, J.M.; Han, C.; Zuniga-Hertz, J.P.; Cai, D. Astrocytic Process Plasticity and IKKβ/NF-ΚB in Central Control of Blood Glucose, Blood Pressure, and Body Weight. Cell Metab. 2017, 25, 1091–1102.e4. [Google Scholar] [CrossRef]
- Ghosh, M.; Yang, Y.; Rothstein, J.D.; Robinson, M.B. Nuclear Factor-ΚB Contributes to Neuron-Dependent Induction of Glutamate Transporter-1 Expression in Astrocytes. J. Neurosci. 2011, 31, 9159–9169. [Google Scholar] [CrossRef]
- Giovannoni, F.; Quintana, F.J. The Role of Astrocytes in CNS Inflammation. Trends Immunol. 2020, 41, 805–819. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic Reactive Astrocytes Are Induced by Activated Microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Ouali Alami, N.; Schurr, C.; Olde Heuvel, F.; Tang, L.; Li, Q.; Tasdogan, A.; Kimbara, A.; Nettekoven, M.; Ottaviani, G.; Raposo, C.; et al. NF-κB Activation in Astrocytes Drives a Stage-specific Beneficial Neuroimmunological Response in ALS. EMBO J. 2018, 37. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, K.; Chun, S.J.; Boillee, S.; Fujimori-Tonou, N.; Yamashita, H.; Gutmann, D.H.; Takahashi, R.; Misawa, H.; Cleveland, D.W. Astrocytes as Determinants of Disease Progression in Inherited ALS. Nat. Neurosci. 2008, 11, 251–253. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Källstig, E.; McCabe, B.D.; Schneider, B.L. The Links between ALS and NF-κB. Int. J. Mol. Sci. 2021, 22, 3875. https://doi.org/10.3390/ijms22083875
Källstig E, McCabe BD, Schneider BL. The Links between ALS and NF-κB. International Journal of Molecular Sciences. 2021; 22(8):3875. https://doi.org/10.3390/ijms22083875
Chicago/Turabian StyleKällstig, Emma, Brian D. McCabe, and Bernard L. Schneider. 2021. "The Links between ALS and NF-κB" International Journal of Molecular Sciences 22, no. 8: 3875. https://doi.org/10.3390/ijms22083875
APA StyleKällstig, E., McCabe, B. D., & Schneider, B. L. (2021). The Links between ALS and NF-κB. International Journal of Molecular Sciences, 22(8), 3875. https://doi.org/10.3390/ijms22083875