
Gastric Cancer: Advances in Carcinogenesis Research and New Therapeutic Strategies

 , ,
, ,
Abstract
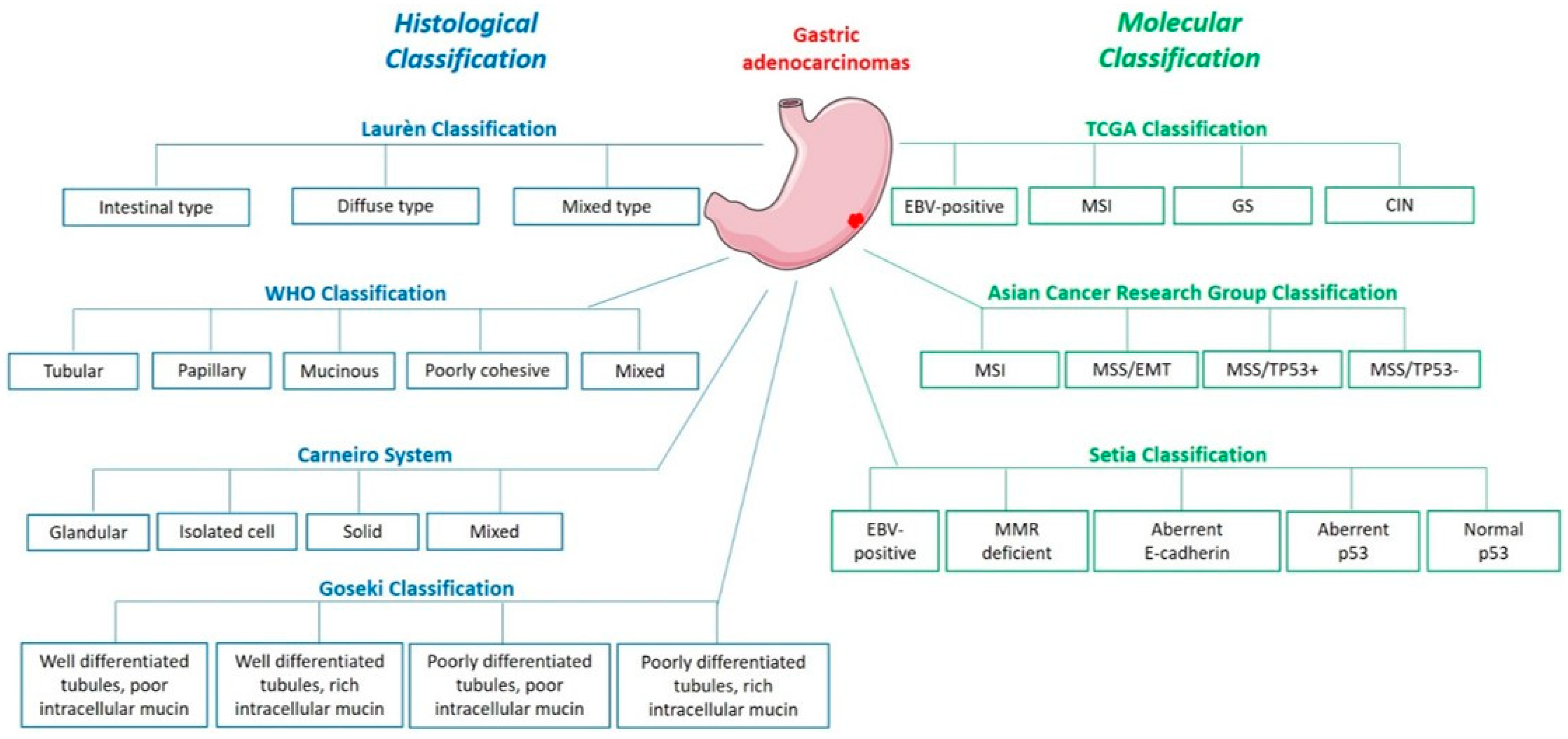
1. Histological and Molecular Classifications
2. Epidemiology and Risk Factors
3. Gastric Carcinogenesis
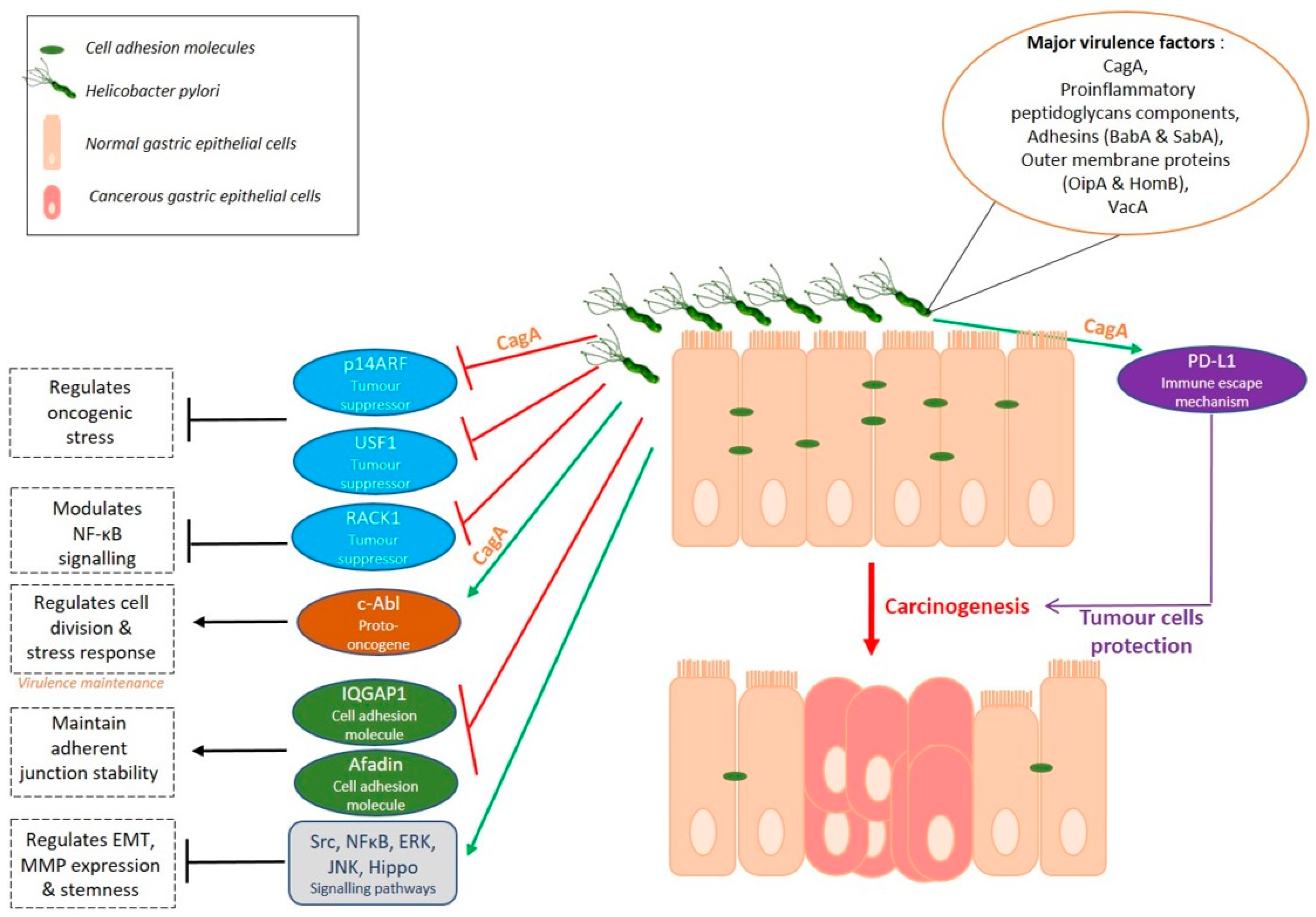
3.1. A Helicobacter Disease
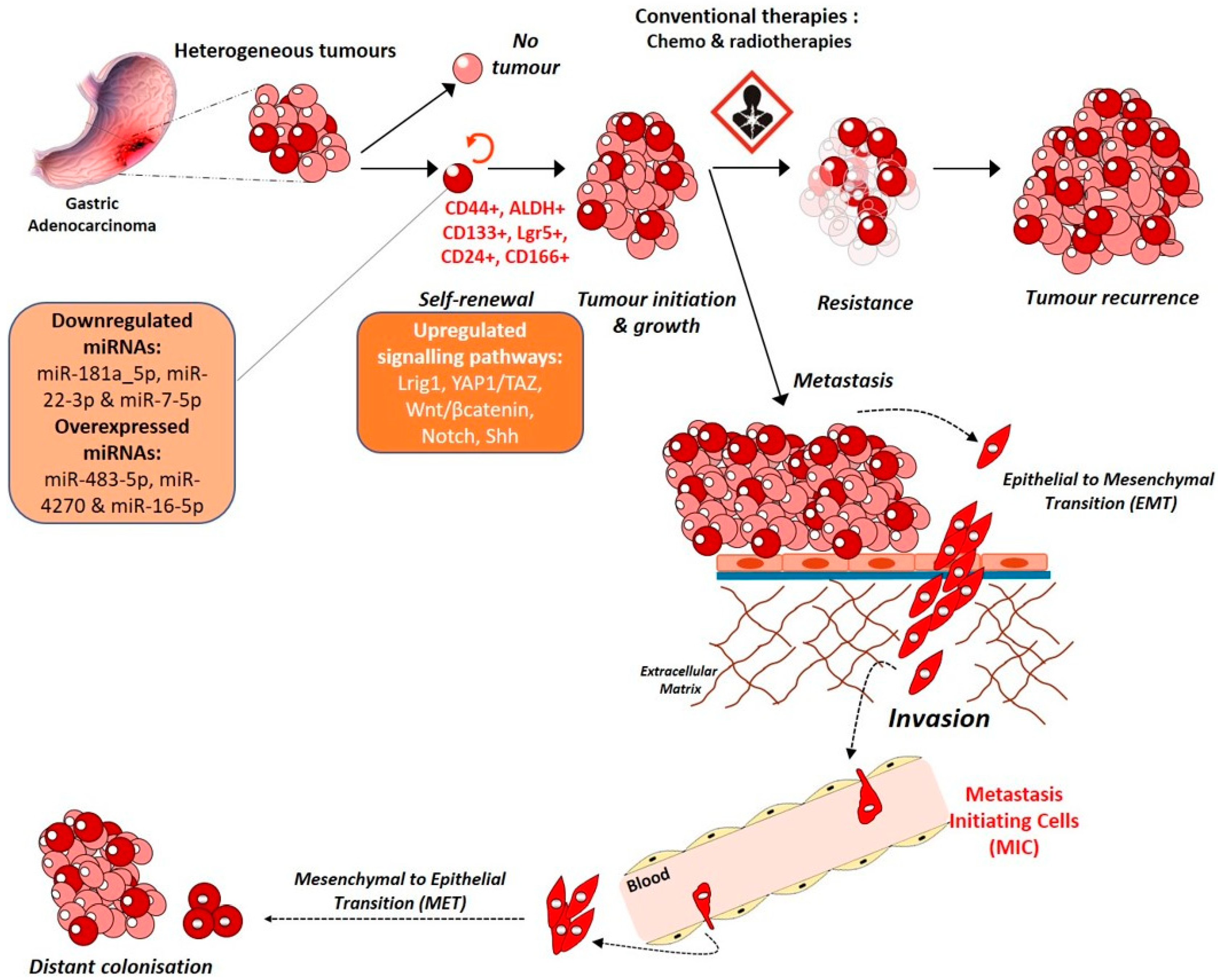
3.2. A Stem Cell Disease
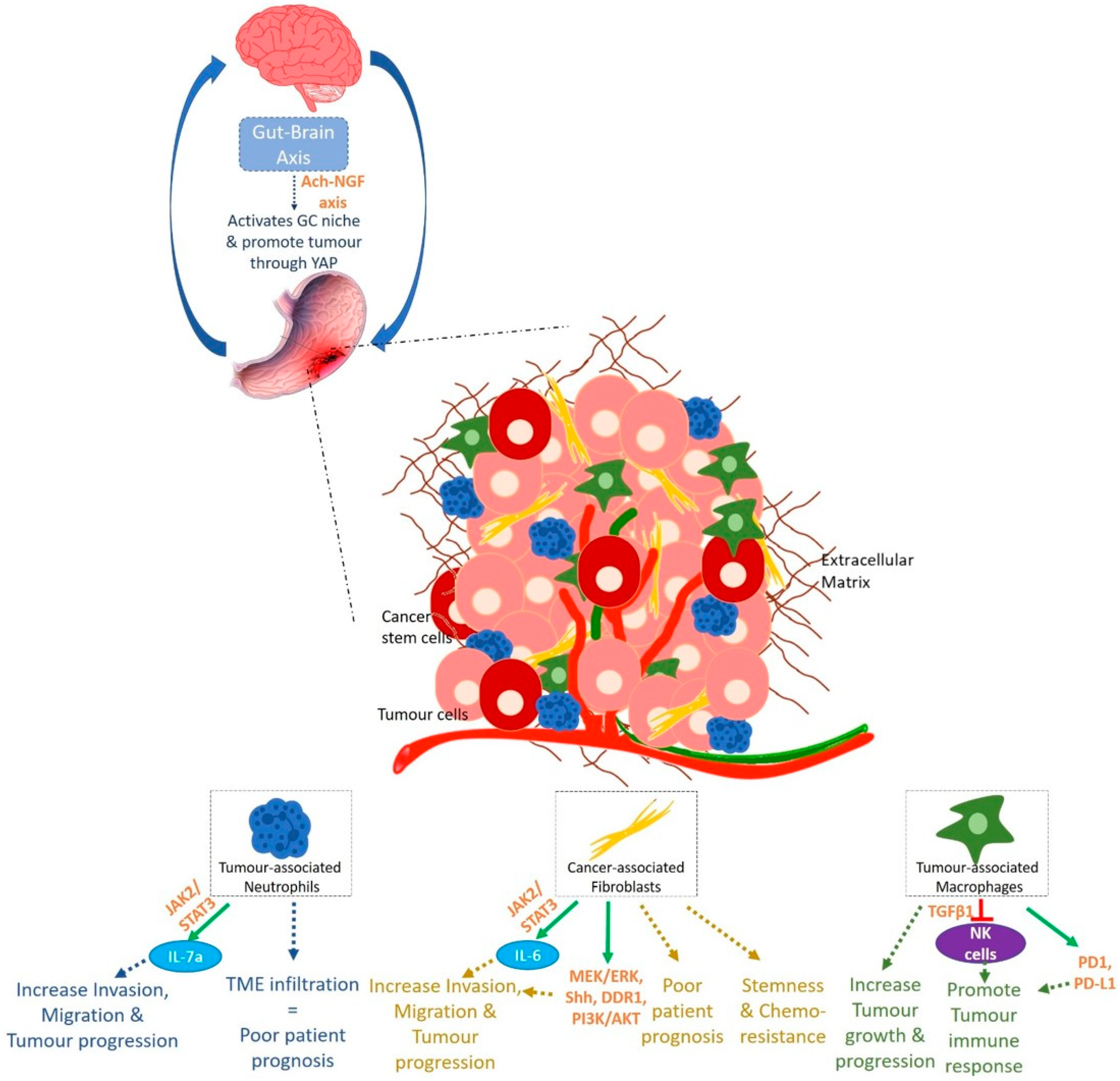
3.3. A Microenvironment Disease
3.4. A Microbiome Disease
4. Biomarkers and Therapeutic Strategies
4.1. Biomarkers and Targeting
4.2. GCSC Targeting
4.3. Liquid Biopsies as Biomarkers
4.4. Immunotherapy
5. New Experimental Models
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| MDPI | Multidisciplinary Digital Publishing Institute |
| DOAJ | Directory of open access journals |
| TLA | Three letter acronym |
| LD | linear dichroism |
References
- Knight, W.R.; Allum, W.H. Gastric Tumours. Medicine 2019, 47, 309–313. [Google Scholar] [CrossRef]
- Cisło, M.; Filip, A.A.; Arnold Offerhaus, G.J.; Ciseł, B.; Rawicz-Pruszyński, K.; Skierucha, M.; Polkowski, W.P. Distinct Molecular Subtypes of Gastric Cancer: From Laurén to Molecular Pathology. Oncotarget 2018, 9, 19427–19442. [Google Scholar] [CrossRef]
- WHO Classification of Tumours of the Digestive System|Bosman FT, Carneiro F, Hruban RH, Theise ND|Download. Available online: https://b-ok.cc/book/3418007/99908b (accessed on 28 July 2020).
- Guo, J.; Yu, W.; Su, H.; Pang, X. Genomic Landscape of Gastric Cancer: Molecular Classification and Potential Targets. Sci. China Life Sci. 2017, 60, 126–137. [Google Scholar] [CrossRef][Green Version]
- Yoda, Y.; Takeshima, H.; Niwa, T.; Kim, J.G.; Ando, T.; Kushima, R.; Sugiyama, T.; Katai, H.; Noshiro, H.; Ushijima, T. Integrated Analysis of Cancer-Related Pathways Affected by Genetic and Epigenetic Alterations in Gastric Cancer. Gastric Cancer 2015, 18, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Fewings, E.; Larionov, A.; Redman, J.; Goldgraben, M.A.; Scarth, J.; Richardson, S.; Brewer, C.; Davidson, R.; Ellis, I.; Evans, D.G.; et al. Germline Pathogenic Variants in PALB2 and Other Cancer-Predisposing Genes in Families with Hereditary Diffuse Gastric Cancer without CDH1 Mutation: A Whole-Exome Sequencing Study. Lancet Gastroenterol. Hepatol. 2018, 3, 489–498. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network Comprehensive Molecular Characterization of Gastric Adenocarcinoma. Nature 2014, 513, 202–209. [CrossRef]
- Cristescu, R.; Lee, J.; Nebozhyn, M.; Kim, K.-M.; Ting, J.C.; Wong, S.S.; Liu, J.; Yue, Y.G.; Wang, J.; Yu, K.; et al. Molecular Analysis of Gastric Cancer Identifies Subtypes Associated with Distinct Clinical Outcomes. Nat. Med. 2015, 21, 449–456. [Google Scholar] [CrossRef]
- Setia, N.; Agoston, A.T.; Han, H.S.; Mullen, J.T.; Duda, D.G.; Clark, J.W.; Deshpande, V.; Mino-Kenudson, M.; Srivastava, A.; Lennerz, J.K.; et al. A Protein and MRNA Expression-Based Classification of Gastric Cancer. Mod. Pathol. 2016, 29, 772–784. [Google Scholar] [CrossRef]
- Li, Y.; Kang, K.; Krahn, J.M.; Croutwater, N.; Lee, K.; Umbach, D.M.; Li, L. A Comprehensive Genomic Pan-Cancer Classification Using the Cancer Genome Atlas Gene Expression Data. BMC Genom. 2017, 18, 508. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Hu, S.; Min, M.; Ni, Y.; Lu, Z.; Sun, X.; Wu, J.; Liu, B.; Ying, X.; Liu, Y. Dissecting Transcriptional Heterogeneity in Primary Gastric Adenocarcinoma by Single Cell RNA Sequencing. Gut 2021, 70, 464–475. [Google Scholar] [CrossRef]
- Cancer Today. Available online: http://gco.iarc.fr/today/home (accessed on 9 March 2020).
- Balakrishnan, M.; George, R.; Sharma, A.; Graham, D.Y. Changing Trends in Stomach Cancer Throughout the World. Curr Gastroenterol. Rep. 2017, 19, 36. [Google Scholar] [CrossRef] [PubMed]
- Correa, P.; Piazuelo, M.B. The Gastric Precancerous Cascade. J. Dig. Dis. 2012, 13, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Camargo, M.C.; Figueiredo, C.; Machado, J.C. Review: Gastric Malignancies: Basic Aspects. Helicobacter 2019, 24 (Suppl. 1), e12642. [Google Scholar] [CrossRef]
- Huang, S.-C.; Ng, K.-F.; Yeh, T.-S.; Cheng, C.-T.; Lin, J.-S.; Liu, Y.-J.; Chuang, H.-C.; Chen, T.-C. Subtraction of Epstein–Barr Virus and Microsatellite Instability Genotypes from the Lauren Histotypes: Combined Molecular and Histologic Subtyping with Clinicopathological and Prognostic Significance Validated in a Cohort of 1,248 Cases. Int. J. Cancer 2019, 145, 3218–3230. [Google Scholar] [CrossRef]
- Yusefi, A.R.; Bagheri Lankarani, K.; Bastani, P.; Radinmanesh, M.; Kavosi, Z. Risk Factors for Gastric Cancer: A Systematic Review. Asian Pac. J. Cancer Prev. 2018, 19, 591–603. [Google Scholar] [CrossRef]
- Rokkas, T.; Rokka, A.; Portincasa, P. A Systematic Review and Meta-Analysis of the Role of Helicobacter pylori Eradication in Preventing Gastric Cancer. Ann. Gastroenterol. 2017, 30, 414–423. [Google Scholar] [CrossRef]
- Choi, I.J.; Kook, M.-C.; Kim, Y.-I.; Cho, S.-J.; Lee, J.Y.; Kim, C.G.; Park, B.; Nam, B.-H. Helicobacter pylori Therapy for the Prevention of Metachronous Gastric Cancer. N. Engl. J. Med. 2018, 378, 1085–1095. [Google Scholar] [CrossRef] [PubMed]
- Ramos, A.; Camargo, F.D. The Hippo Signaling Pathway and Stem Cell Biology. Trends Cell Biol. 2012, 22, 339–346. [Google Scholar] [CrossRef]
- Wroblewski, L.E.; Peek, R.M.; Wilson, K.T. Helicobacter pylori and Gastric Cancer: Factors That Modulate Disease Risk. Clin. Microbiol. Rev. 2010, 23, 713–739. [Google Scholar] [CrossRef]
- Varon, C.; Mégraud, F. Stomach Cancer. In World Cancer Report: Cancer Research for Cancer Prevention; Wild, C.P., Weiderpass, E., Stewart, B.W., Eds.; International Agency for Research on Cancer: Lyon, France, 2020; pp. 333–343. ISBN 978-92-832-0447-3. [Google Scholar]
- Agudo, A.; Cayssials, V.; Bonet, C.; Tjønneland, A.; Overvad, K.; Boutron-Ruault, M.-C.; Affret, A.; Fagherazzi, G.; Katzke, V.; Schübel, R.; et al. Inflammatory Potential of the Diet and Risk of Gastric Cancer in the European Prospective Investigation into Cancer and Nutrition (EPIC) Study. Am. J. Clin. Nutr. 2018, 107, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Bouvard, V.; Loomis, D.; Guyton, K.Z.; Grosse, Y.; Ghissassi, F.E.; Benbrahim-Tallaa, L.; Guha, N.; Mattock, H.; Straif, K. International Agency for Research on Cancer Monograph Working Group Carcinogenicity of Consumption of Red and Processed Meat. Lancet Oncol. 2015, 16, 1599–1600. [Google Scholar] [CrossRef]
- Praud, D.; Rota, M.; Pelucchi, C.; Bertuccio, P.; Rosso, T.; Galeone, C.; Zhang, Z.-F.; Matsuo, K.; Ito, H.; Hu, J.; et al. Cigarette Smoking and Gastric Cancer in the Stomach Cancer Pooling (StoP) Project. Eur. J. Cancer Prev. 2018, 27, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, C.; Seruca, R.; Carneiro, F. Genetics, Pathology, and Clinics of Familial Gastric Cancer. Int. J. Surg. Pathol. 2016. [Google Scholar] [CrossRef]
- Horvat, A.; Noto, J.M.; Ramatchandirin, B.; Zaika, E.; Palrasu, M.; Wei, J.; Schneider, B.G.; El-Rifai, W.; Peek, R.M.; Zaika, A.I. Helicobacter pylori Pathogen Regulates P14ARF Tumor Suppressor and Autophagy in Gastric Epithelial Cells. Oncogene 2018, 37, 5054–5065. [Google Scholar] [CrossRef]
- Costa, L.; Corre, S.; Michel, V.; Le Luel, K.; Fernandes, J.; Ziveri, J.; Jouvion, G.; Danckaert, A.; Mouchet, N.; Da Silva Barreira, D.; et al. USF1 Defect Drives P53 Degradation during Helicobacter pylori Infection and Accelerates Gastric Carcinogenesis. Gut 2020, 69, 1582–1591. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Liu, J.-P.; Li, X.-Y.; Cai, Y.; He, C.; Li, N.-S.; Xie, C.; Xiong, Z.-J.; Ge, Z.-M.; Lu, N.-H.; et al. Downregulation of Tumor Suppressor RACK1 by Helicobacter pylori Infection Promotes Gastric Carcinogenesis through the Integrin β-1/NF-ΚB Signaling Pathway. Cancer Lett. 2019, 450, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Mueller, D.; Tegtmeyer, N.; Brandt, S.; Yamaoka, Y.; De Poire, E.; Sgouras, D.; Wessler, S.; Torres, J.; Smolka, A.; Backert, S. C-Src and c-Abl Kinases Control Hierarchic Phosphorylation and Function of the CagA Effector Protein in Western and East Asian Helicobacter pylori Strains. J. Clin. Investig. 2012, 122, 1553–1566. [Google Scholar] [CrossRef]
- Posselt, G.; Wiesauer, M.; Chichirau, B.E.; Engler, D.; Krisch, L.M.; Gadermaier, G.; Briza, P.; Schneider, S.; Boccellato, F.; Meyer, T.F.; et al. Helicobacter pylori-Controlled c-Abl Localization Promotes Cell Migration and Limits Apoptosis. Cell Commun. Signal. 2019, 17, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Caron, T.J.; Scott, K.E.; Fox, J.G.; Hagen, S.J. Tight Junction Disruption: Helicobacter pylori and Dysregulation of the Gastric Mucosal Barrier. World J. Gastroenterol. 2015, 21, 11411–11427. [Google Scholar] [CrossRef]
- Hagen, S.J.; Ang, L.-H.; Zheng, Y.; Karahan, S.N.; Wu, J.; Wang, Y.E.; Caron, T.J.; Gad, A.P.; Muthupalani, S.; Fox, J.G. Loss of Tight Junction Protein Claudin 18 Promotes Progressive Neoplasia Development in Mouse Stomach. Gastroenterology 2018, 155, 1852–1867. [Google Scholar] [CrossRef]
- Baud, J.; Varon, C.; Chabas, S.; Chambonnier, L.; Darfeuille, F.; Staedel, C. Helicobacter pylori Initiates a Mesenchymal Transition through ZEB1 in Gastric Epithelial Cells. PLoS ONE 2013, 8, e60315. [Google Scholar] [CrossRef] [PubMed]
- Bessède, E.; Staedel, C.; Acuña Amador, L.A.; Nguyen, P.H.; Chambonnier, L.; Hatakeyama, M.; Belleannée, G.; Mégraud, F.; Varon, C. Helicobacter pylori Generates Cells with Cancer Stem Cell Properties via Epithelial-Mesenchymal Transition-like Changes. Oncogene 2014, 33, 4123–4131. [Google Scholar] [CrossRef]
- Bessède, E.; Molina, S.; Amador, L.A.; Dubus, P.; Staedel, C.; Chambonnier, L.; Buissonnière, A.; Sifré, E.; Giese, A.; Bénéjat, L.; et al. Deletion of IQGAP1 Promotes Helicobacter pylori-Induced Gastric Dysplasia in Mice and Acquisition of Cancer Stem Cell Properties in Vitro. Oncotarget 2016, 7, 80688–80699. [Google Scholar] [CrossRef] [PubMed]
- Marques, M.S.; Melo, J.; Cavadas, B.; Mendes, N.; Pereira, L.; Carneiro, F.; Figueiredo, C.; Leite, M. Afadin Downregulation by Helicobacter pylori Induces Epithelial to Mesenchymal Transition in Gastric Cells. Front. Microbiol. 2018, 9, 2712. [Google Scholar] [CrossRef]
- Costa, A.M.; Ferreira, R.M.; Pinto-Ribeiro, I.; Sougleri, I.S.; Oliveira, M.J.; Carreto, L.; Santos, M.A.; Sgouras, D.N.; Carneiro, F.; Leite, M.; et al. Helicobacter pylori Activates Matrix Metalloproteinase 10 in Gastric Epithelial Cells via EGFR and ERK-Mediated Pathways. J. Infect. Dis. 2016, 213, 1767–1776. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Feng, Y.; Hu, Y.; He, C.; Xie, C.; Ouyang, Y.; Artim, S.C.; Huang, D.; Zhu, Y.; Luo, Z.; et al. Helicobacter pylori CagA Promotes Epithelial Mesenchymal Transition in Gastric Carcinogenesis via Triggering Oncogenic YAP Pathway. J. Exp. Clin. Cancer Res. 2018, 37, 280. [Google Scholar] [CrossRef]
- Molina Castro, S.E.; Tiffon, C.; Giraud, J.; Boeuf, H.; Sifre, E.; Giese, A.; Belleannée, G.; Lehours, P.; Bessède, E.; Mégraud, F.; et al. The Hippo Kinase LATS2 Controls Helicobacter pylori-Induced Epithelial-Mesenchymaltransition and Intestinal Metaplasia in Gastric Mucosa. Cell. Mol. Gastroenterol. Hepatol. 2020, 9, 257–276. [Google Scholar] [CrossRef] [PubMed]
- Tiffon, C.; Giraud, J.; Molina-Castro, S.E.; Peru, S.; Seeneevassen, L.; Sifré, E.; Staedel, C.; Bessède, E.; Dubus, P.; Mégraud, F.; et al. TAZ Controls Helicobacter pylori-Induced Epithelial-Mesenchymal Transition and Cancer Stem Cell-Like Invasive and Tumorigenic Properties. Cells 2020, 9, 1462. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Lu, N.; Xie, C. The Hippo and Wnt Signalling Pathways: Crosstalk during Neoplastic Progression in Gastrointestinal Tissue. FEBS J. 2019, 286, 3745–3756. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xu, C.-X.; Gong, R.-J.; Chi, J.-S.; Liu, P.; Liu, X.-M. How Does Helicobacter pylori Cause Gastric Cancer through Connexins: An Opinion Review. World J. Gastroenterol. 2019, 25, 5220–5232. [Google Scholar] [CrossRef] [PubMed]
- Holokai, L.; Chakrabarti, J.; Broda, T.; Chang, J.; Hawkins, J.A.; Sundaram, N.; Wroblewski, L.E.; Peek, R.M.; Wang, J.; Helmrath, M.; et al. Increased Programmed Death-Ligand 1 Is an Early Epithelial Cell Response to Helicobacter pylori Infection. PLoS Pathog. 2019, 15, e1007468. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, A.; Abe, H.; Kunita, A.; Saito, R.; Kanda, T.; Yamashita, H.; Seto, Y.; Ishikawa, S.; Fukayama, M. Viral Loads Correlate with Upregulation of PD-L1 and Worse Patient Prognosis in Epstein-Barr Virus-Associated Gastric Carcinoma. PLoS ONE 2019, 14, e0211358. [Google Scholar] [CrossRef]
- Wang, S.; Chen, Z.; Zhu, S.; Lu, H.; Peng, D.; Soutto, M.; Naz, H.; Peek, R.; Xu, H.; Zaika, A.; et al. PRDX2 Protects against Oxidative Stress Induced by H. pylori and Promotes Resistance to Cisplatin in Gastric Cancer. Redox Biol. 2019, 28, 101319. [Google Scholar] [CrossRef]
- Noto, J.M.; Chopra, A.; Loh, J.T.; Romero-Gallo, J.; Piazuelo, M.B.; Watson, M.; Leary, S.; Beckett, A.C.; Wilson, K.T.; Cover, T.L.; et al. Pan-Genomic Analyses Identify Key Helicobacter pylori Pathogenic Loci Modified by Carcinogenic Host Microenvironments. Gut 2018, 67, 1793–1804. [Google Scholar] [CrossRef]
- Berthenet, E.; Yahara, K.; Thorell, K.; Pascoe, B.; Meric, G.; Mikhail, J.M.; Engstrand, L.; Enroth, H.; Burette, A.; Megraud, F.; et al. A GWAS on Helicobacter pylori Strains Points to Genetic Variants Associated with Gastric Cancer Risk. BMC Biol. 2018, 16, 84. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, D.; Dick, J.E. Human Acute Myeloid Leukemia Is Organized as a Hierarchy That Originates from a Primitive Hematopoietic Cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective Identification of Tumorigenic Breast Cancer Cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a Cancer Stem Cell in Human Brain Tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar] [PubMed]
- Ricci-Vitiani, L.; Lombardi, D.G.; Pilozzi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; De Maria, R. Identification and Expansion of Human Colon-Cancer-Initiating Cells. Nature 2007, 445, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Takaishi, S.; Okumura, T.; Tu, S.; Wang, S.S.W.; Shibata, W.; Vigneshwaran, R.; Gordon, S.A.K.; Shimada, Y.; Wang, T.C. Identification of Gastric Cancer Stem Cells Using the Cell Surface Marker CD44. Stem Cells 2009, 27, 1006–1020. [Google Scholar] [CrossRef]
- Fukuda, K.; Saikawa, Y.; Ohashi, M.; Kumagai, K.; Kitajima, M.; Okano, H.; Matsuzaki, Y.; Kitagawa, Y. Tumor Initiating Potential of Side Population Cells in Human Gastric Cancer. Int. J. Oncol. 2009, 34, 1201–1207. [Google Scholar] [PubMed]
- Nguyen, P.H.; Giraud, J.; Chambonnier, L.; Dubus, P.; Wittkop, L.; Belleannée, G.; Collet, D.; Soubeyran, I.; Evrard, S.; Rousseau, B.; et al. Characterization of Biomarkers of Tumorigenic and Chemoresistant Cancer Stem Cells in Human Gastric Carcinoma. Clin. Cancer Res. 2017, 23, 1586–1597. [Google Scholar] [CrossRef] [PubMed]
- Kodama, H.; Murata, S.; Ishida, M.; Yamamoto, H.; Yamaguchi, T.; Kaida, S.; Miyake, T.; Takebayashi, K.; Kushima, R.; Tani, M. Prognostic Impact of CD44-Positive Cancer Stem-like Cells at the Invasive Front of Gastric Cancer. Br. J. Cancer 2017, 116, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Bessède, E.; Dubus, P.; Mégraud, F.; Varon, C. Helicobacter pylori Infection and Stem Cells at the Origin of Gastric Cancer. Oncogene 2015, 34, 2547–2555. [Google Scholar] [CrossRef]
- Wroblewski, L.E.; Choi, E.; Petersen, C.; Delgado, A.G.; Piazuelo, M.B.; Romero-Gallo, J.; Lantz, T.L.; Zavros, Y.; Coffey, R.J.; Goldenring, J.R.; et al. Targeted Mobilization of Lrig1+ Gastric Epithelial Stem Cell Populations by a Carcinogenic Helicobacter pylori Type IV Secretion System. Proc. Natl. Acad. Sci. USA 2019, 116, 19652–19658. [Google Scholar] [CrossRef]
- Giraud, J.; Molina-Castro, S.; Seeneevassen, L.; Sifré, E.; Izotte, J.; Tiffon, C.; Staedel, C.; Boeuf, H.; Fernandez, S.; Barthelemy, P.; et al. Verteporfin Targeting YAP1/TAZ-TEAD Transcriptional Activity Inhibits the Tumorigenic Properties of Gastric Cancer Stem Cells. Int. J. Cancer 2019, 146, 2255–2267. [Google Scholar] [CrossRef]
- Seeneevassen, L.; Giraud, J.; Molina-Castro, S.; Sifré, E.; Tiffon, C.; Beauvoit, C.; Staedel, C.; Mégraud, F.; Lehours, P.; Martin, O.C.B.; et al. Leukaemia Inhibitory Factor (LIF) Inhibits Cancer Stem Cells Tumorigenic Properties through Hippo Kinases Activation in Gastric Cancer. Cancers 2020, 12, 2011. [Google Scholar] [CrossRef]
- Bertaux-Skeirik, N.; Feng, R.; Schumacher, M.A.; Li, J.; Mahe, M.M.; Engevik, A.C.; Javier, J.E.; Peek, R.M.; Ottemann, K.; Orian-Rousseau, V.; et al. CD44 Plays a Functional Role in Helicobacter pylori-Induced Epithelial Cell Proliferation. PLoS Pathog. 2015, 11, e1004663. [Google Scholar] [CrossRef] [PubMed]
- Lau, W.M.; Teng, E.; Chong, H.S.; Lopez, K.A.P.; Tay, A.Y.L.; Salto-Tellez, M.; Shabbir, A.; So, J.B.Y.; Chan, S.L. CD44v8-10 Is a Cancer-Specific Marker for Gastric Cancer Stem Cells. Cancer Res. 2014, 74, 2630–2641. [Google Scholar] [CrossRef] [PubMed]
- Yamakawa, Y.; Kusuhara, M.; Terashima, M.; Kinugasa, Y.; Sugino, T.; Abe, M.; Mochizuki, T.; Hatakeyama, K.; Kami, K.; Yamaguchi, K. CD44 Variant 9 Expression as a Predictor for Gastric Cancer Recurrence: Immunohistochemical and Metabolomic Analysis of Surgically Resected Tissues. Biomed. Res. 2017, 38, 41–52. [Google Scholar] [CrossRef]
- Tsugawa, H.; Kato, C.; Mori, H.; Matsuzaki, J.; Kameyama, K.; Saya, H.; Hatakeyama, M.; Suematsu, M.; Suzuki, H. Cancer Stem-Cell Marker CD44v9-Positive Cells Arise from Helicobacter pylori-Infected CAPZA1-Overexpressing Cells. Cell Mol. Gastroenterol. Hepatol. 2019, 8, 319–334. [Google Scholar] [CrossRef]
- Pereira, C.; Ferreira, D.; Mendes, N.; Granja, P.L.; Almeida, G.M.; Oliveira, C. Expression of CD44v6-Containing Isoforms Influences Cisplatin Response in Gastric Cancer Cells. Cancers 2020, 12, 858. [Google Scholar] [CrossRef]
- Jung, D.H.; Bae, Y.J.; Kim, J.-H.; Shin, Y.K.; Jeung, H.-C. HER2 Regulates Cancer Stem Cell Activities via the Wnt Signaling Pathway in Gastric Cancer Cells. Oncology 2019, 1–8. [Google Scholar] [CrossRef]
- Xin, L.; Liu, L.; Liu, C.; Zhou, L.-Q.; Zhou, Q.; Yuan, Y.-W.; Li, S.-H.; Zhang, H.-T. DNA-Methylation-Mediated Silencing of MiR-7-5p Promotes Gastric Cancer Stem Cell Invasion via Increasing Smo and Hes1. J. Cell. Physiol. 2019, 235, 2643–2654. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.-C.; Long, D.; Liao, C.-C.; Zhang, S. Association between Density of Tumor-Infiltrating Lymphocytes and Prognoses of Patients with Gastric Cancer. Medicine 2018, 97, e11387. [Google Scholar] [CrossRef]
- Pernot, S.; Terme, M.; Radosevic-Robin, N.; Castan, F.; Badoual, C.; Marcheteau, E.; Penault-Llorca, F.; Bouche, O.; Bennouna, J.; Francois, E.; et al. Infiltrating and Peripheral Immune Cell Analysis in Advanced Gastric Cancer According to the Lauren Classification and Its Prognostic Significance. Gastric Cancer 2020, 23, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Baj, J.; Brzozowska, K.; Forma, A.; Maani, A.; Sitarz, E.; Portincasa, P. Immunological Aspects of the Tumor Microenvironment and Epithelial-Mesenchymal Transition in Gastric Carcinogenesis. Int. J. Mol. Sci. 2020, 21, 2544. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Saxena, S.; Singh, R.K. Neutrophils in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1224, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Szczerba, B.M.; Castro-Giner, F.; Vetter, M.; Krol, I.; Gkountela, S.; Landin, J.; Scheidmann, M.C.; Donato, C.; Scherrer, R.; Singer, J.; et al. Neutrophils Escort Circulating Tumour Cells to Enable Cell Cycle Progression. Nature 2019, 566, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Cong, X.; Gao, H.; Lan, X.; Li, Z.; Wang, W.; Song, S.; Wang, Y.; Li, C.; Zhang, H.; et al. Tumor-Associated Neutrophils Induce EMT by IL-17a to Promote Migration and Invasion in Gastric Cancer Cells. J. Exp. Clin. Cancer Res. 2019, 38, 6. [Google Scholar] [CrossRef]
- Ma, Y.; Zhu, J.; Chen, S.; Ma, J.; Zhang, X.; Huang, S.; Hu, J.; Yue, T.; Zhang, J.; Wang, P.; et al. Low Expression of SPARC in Gastric Cancer-Associated Fibroblasts Leads to Stemness Transformation and 5-Fluorouracil Resistance in Gastric Cancer. Cancer Cell Int. 2019, 19, 137. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Tao, P.; Zhou, Q.; Li, J.; Yu, Z.; Wang, X.; Li, J.; Li, C.; Yan, M.; Zhu, Z.; et al. IL-6 Secreted by Cancer-Associated Fibroblasts Promotes Epithelial-Mesenchymal Transition and Metastasis of Gastric Cancer via JAK2/STAT3 Signaling Pathway. Oncotarget 2017, 8, 20741–20750. [Google Scholar] [CrossRef] [PubMed]
- Karakasheva, T.A.; Lin, E.W.; Tang, Q.; Qiao, E.; Waldron, T.J.; Soni, M.; Klein-Szanto, A.J.; Sahu, V.; Basu, D.; Ohashi, S.; et al. IL-6 Mediates Cross-Talk between Tumor Cells and Activated Fibroblasts in the Tumor Microenvironment. Cancer Res. 2018, 78, 4957–4970. [Google Scholar] [CrossRef] [PubMed]
- Soutto, M.; Chen, Z.; Bhat, A.A.; Wang, L.; Zhu, S.; Gomaa, A.; Bates, A.; Bhat, N.S.; Peng, D.; Belkhiri, A.; et al. Activation of STAT3 Signaling Is Mediated by TFF1 Silencing in Gastric Neoplasia. Nat. Commun. 2019, 10, 3039. [Google Scholar] [CrossRef] [PubMed]
- Chong, Y.; Tang, D.; Xiong, Q.; Jiang, X.; Xu, C.; Huang, Y.; Wang, J.; Zhou, H.; Shi, Y.; Wu, X.; et al. Galectin-1 from Cancer-Associated Fibroblasts Induces Epithelial-Mesenchymal Transition through Β1 Integrin-Mediated Upregulation of Gli1 in Gastric Cancer. J. Exp. Clin. Cancer Res. 2016, 35, 175. [Google Scholar] [CrossRef]
- Jin, H.; Ham, I.-H.; Oh, H.J.; Bae, C.A.; Lee, D.; Kim, Y.-B.; Son, S.-Y.; Chwae, Y.-J.; Han, S.-U.; Brekken, R.A.; et al. Inhibition of Discoidin Domain Receptor 1 Prevents Stroma-Induced Peritoneal Metastasis in Gastric Carcinoma. Mol. Cancer Res. 2018, 16, 1590–1600. [Google Scholar] [CrossRef]
- Ning, X.; Zhang, H.; Wang, C.; Song, X. Exosomes Released by Gastric Cancer Cells Induce Transition of Pericytes Into Cancer-Associated Fibroblasts. Med. Sci. Monit. 2018, 24, 2350–2359. [Google Scholar] [CrossRef]
- Krzysiek-Maczka, G.; Wrobel, T.; Targosz, A.; Szczyrk, U.; Strzalka, M.; Ptak-Belowska, A.; Czyz, J.; Brzozowski, T. Helicobacter pylori-Activated Gastric Fibroblasts Induce Epithelial-Mesenchymal Transition of Gastric Epithelial Cells in Vitro in a TGF-β-Dependent Manner. Helicobacter 2019, 24, e12653. [Google Scholar] [CrossRef]
- Sathe, A.; Grimes, S.M.; Lau, B.T.; Chen, J.; Suarez, C.; Huang, R.J.; Poultsides, G.; Ji, H.P. Single-Cell Genomic Characterization Reveals the Cellular Reprogramming of the Gastric Tumor Microenvironment. Clin. Cancer Res. 2020, 26, 2640–2653. [Google Scholar] [CrossRef]
- Peng, L.-S.; Zhang, J.-Y.; Teng, Y.-S.; Zhao, Y.-L.; Wang, T.-T.; Mao, F.-Y.; Lv, Y.-P.; Cheng, P.; Li, W.-H.; Chen, N.; et al. Tumor-Associated Monocytes/Macrophages Impair NK-Cell Function via TGFβ1 in Human Gastric Cancer. Cancer Immunol. Res. 2017, 5, 248–256. [Google Scholar] [CrossRef]
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 Expression by Tumour-Associated Macrophages Inhibits Phagocytosis and Tumour Immunity. Nature 2017, 545, 495–499. [Google Scholar] [CrossRef]
- Wang, L.; Zhou, D.; Ren, H.; Chen, Y. Effects of Modified FOLFOX-6 Chemotherapy on Cellular Immune Function in Patients with Gastric Cancer. Oncol. Lett. 2018, 15, 8635–8640. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhao, J.; Li, Q.; Wang, Q.; Zhou, Y.; Tong, Z. Gastric Cancer Patients Have Elevated Plasmacytoid and CD1c+ Dendritic Cells in the Peripheral Blood. Oncol. Lett. 2018, 15, 5087–5092. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Sun, Y.; Huang, J.; Xu, W.; Liu, J.; Yuan, Z. CD4/CD8 + T Cells, DC Subsets, Foxp3, and IDO Expression Are Predictive Indictors of Gastric Cancer Prognosis. Cancer Med. 2019, 8, 7330–7344. [Google Scholar] [CrossRef] [PubMed]
- Sigal, M.; Logan, C.Y.; Kapalczynska, M.; Mollenkopf, H.-J.; Berger, H.; Wiedenmann, B.; Nusse, R.; Amieva, M.R.; Meyer, T.F. Stromal R-Spondin Orchestrates Gastric Epithelial Stem Cells and Gland Homeostasis. Nature 2017, 548, 451–455. [Google Scholar] [CrossRef]
- Sigal, M.; Reinés, M.D.M.; Müllerke, S.; Fischer, C.; Kapalczynska, M.; Berger, H.; Bakker, E.R.M.; Mollenkopf, H.-J.; Rothenberg, M.E.; Wiedenmann, B.; et al. R-Spondin-3 Induces Secretory, Antimicrobial Lgr5+ Cells in the Stomach. Nat. Cell Biol. 2019, 21, 812–823. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, Y.; Sakitani, K.; Konishi, M.; Asfaha, S.; Niikura, R.; Tomita, H.; Renz, B.W.; Tailor, Y.; Macchini, M.; Middelhoff, M.; et al. Nerve Growth Factor Promotes Gastric Tumorigenesis through Aberrant Cholinergic Signaling. Cancer Cell 2017, 31, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Sohn, S.-H.; Kim, N.; Jo, H.J.; Kim, J.; Park, J.H.; Nam, R.H.; Seok, Y.-J.; Kim, Y.-R.; Lee, D.H. Analysis of Gastric Body Microbiota by Pyrosequencing: Possible Role of Bacteria Other Than Helicobacter pylori in the Gastric Carcinogenesis. J. Cancer Prev. 2017, 22, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Fritsch, J.; Abreu, M.T. The Microbiota and the Immune Response: What Is the Chicken and What Is the Egg? Gastrointest. Endosc. Clin. N. Am. 2019, 29, 381–393. [Google Scholar] [CrossRef]
- Lofgren, J.L.; Whary, M.T.; Ge, Z.; Muthupalani, S.; Taylor, N.S.; Mobley, M.; Potter, A.; Varro, A.; Eibach, D.; Suerbaum, S.; et al. Lack of Commensal Flora in Helicobacter pylori-Infected INS-GAS Mice Reduces Gastritis and Delays Intraepithelial Neoplasia. Gastroenterology 2011, 140, 210–220. [Google Scholar] [CrossRef]
- Lertpiriyapong, K.; Whary, M.T.; Muthupalani, S.; Lofgren, J.L.; Gamazon, E.R.; Feng, Y.; Ge, Z.; Wang, T.C.; Fox, J.G. Gastric Colonisation with a Restricted Commensal Microbiota Replicates the Promotion of Neoplastic Lesions by Diverse Intestinal Microbiota in the Helicobacter pylori INS-GAS Mouse Model of Gastric Carcinogenesis. Gut 2014, 63, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.M.; Pereira-Marques, J.; Pinto-Ribeiro, I.; Costa, J.L.; Carneiro, F.; Machado, J.C.; Figueiredo, C. Gastric Microbial Community Profiling Reveals a Dysbiotic Cancer-Associated Microbiota. Gut 2018, 67, 226–236. [Google Scholar] [CrossRef]
- Hsieh, Y.-Y.; Tung, S.-Y.; Pan, H.-Y.; Yen, C.-W.; Xu, H.-W.; Lin, Y.-J.; Deng, Y.-F.; Hsu, W.-T.; Wu, C.-S.; Li, C. Increased Abundance of Clostridium and Fusobacterium in Gastric Microbiota of Patients with Gastric Cancer in Taiwan. Sci. Rep. 2018, 8, 158. [Google Scholar] [CrossRef] [PubMed]
- Castaño-Rodríguez, N.; Goh, K.-L.; Fock, K.M.; Mitchell, H.M.; Kaakoush, N.O. Dysbiosis of the Microbiome in Gastric Carcinogenesis. Sci. Rep. 2017, 7, 15957. [Google Scholar] [CrossRef] [PubMed]
- Carrasco-Garcia, E.; García-Puga, M.; Arevalo, S.; Matheu, A. Towards Precision Medicine: Linking Genetic and Cellular Heterogeneity in Gastric Cancer. Ther. Adv. Med. Oncol. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, T.; Yashiro, M. Biomarkers of Gastric Cancer: Current Topics and Future Perspective. World J. Gastroenterol. 2018, 24, 2818–2832. [Google Scholar] [CrossRef]
- Apicella, M.; Corso, S.; Giordano, S. Targeted Therapies for Gastric Cancer: Failures and Hopes from Clinical Trials. Oncotarget 2017, 8, 57654–57669. [Google Scholar] [CrossRef]
- Chu, T.-H.; Huang, S.-T.; Yang, S.-F.; Li, C.-J.; Lin, H.-W.; Weng, B.-C.; Yang, S.-M.; Huang, S.-C.; Wu, J.-C.; Chang, Y.-C.; et al. Hepatoma-Derived Growth Factor Participates in Helicobacter pylori-Induced Neutrophils Recruitment, Gastritis and Gastric Carcinogenesis. Oncogene 2019, 38, 6461–6477. [Google Scholar] [CrossRef]
- Andres, F.; Iamele, L.; Meyer, T.; Stüber, J.C.; Kast, F.; Gherardi, E.; Niemann, H.H.; Plückthun, A. Inhibition of the MET Kinase Activity and Cell Growth in MET-Addicted Cancer Cells by Bi-Paratopic Linking. J. Mol. Biol. 2019, 431, 2020–2039. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, H.; Ning, T.; Liu, D.; Deng, T.; Liu, R.; Bai, M.; Zhu, K.; Li, J.; Fan, Q.; et al. Exosome-Delivered c-Met SiRNA Could Reverse Chemoresistance to Cisplatin in Gastric Cancer. Int. J. Nanomed. 2020, 15, 2323–2335. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Cao, Y.; Guan, Y.; Zheng, C. BST2 Promotes Cell Proliferation, Migration and Induces NF-ΚB Activation in Gastric Cancer. Biotechnol. Lett. 2018, 40, 1015–1027. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.K.; Hoppe, M.M.; Srivastava, S.; Samanta, A.; Sharma, N.; Tan, K.T.; Yang, H.; Voon, D.C.; Pang, B.; Teh, M.; et al. CEACAM6 Is Upregulated by Helicobacter pylori CagA and Is a Biomarker for Early Gastric Cancer. Oncotarget 2016, 7, 55290–55301. [Google Scholar] [CrossRef]
- Bekaii-Saab, T.; El-Rayes, B. Identifying and Targeting Cancer Stem Cells in the Treatment of Gastric Cancer. Cancer 2017, 123, 1303–1312. [Google Scholar] [CrossRef]
- Nguyen, P.H.; Giraud, J.; Staedel, C.; Chambonnier, L.; Dubus, P.; Chevret, E.; Bœuf, H.; Gauthereau, X.; Rousseau, B.; Fevre, M.; et al. All-Trans Retinoic Acid Targets Gastric Cancer Stem Cells and Inhibits Patient-Derived Gastric Carcinoma Tumor Growth. Oncogene 2016, 35, 5619–5628. [Google Scholar] [CrossRef] [PubMed]
- Courtois, S.; Durán, R.V.; Giraud, J.; Sifré, E.; Izotte, J.; Mégraud, F.; Lehours, P.; Varon, C.; Bessède, E. Metformin Targets Gastric Cancer Stem Cells. Eur. J. Cancer 2017, 84, 193–201. [Google Scholar] [CrossRef]
- Giraud, J.; Bouriez, D.; Seeneevassen, L.; Rousseau, B.; Sifré, E.; Giese, A.; Mégraud, F.; Lehours, P.; Dubus, P.; Gronnier, C.; et al. Orthotopic Patient-Derived Xenografts of Gastric Cancer to Decipher Drugs Effects on Cancer Stem Cells and Metastatic Dissemination. Cancers 2019, 11, 560. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, S.; Tsugawa, H.; Matsuzaki, J.; Hirata, K.; Mori, H.; Saya, H.; Kanai, T.; Suzuki, H. Inhibiting XCT Improves 5-Fluorouracil Resistance of Gastric Cancer Induced by CD44 Variant 9 Expression. Anticancer Res. 2018, 38, 6163–6170. [Google Scholar] [CrossRef]
- Staedel, C.; Varon, C.; Nguyen, P.H.; Vialet, B.; Chambonnier, L.; Rousseau, B.; Soubeyran, I.; Evrard, S.; Couillaud, F.; Darfeuille, F. Inhibition of Gastric Tumor Cell Growth Using Seed-Targeting LNA as Specific, Long-Lasting MicroRNA Inhibitors. Mol. Ther Nucleic Acids 2015, 4, e246. [Google Scholar] [CrossRef] [PubMed]
- Bonelli, P.; Borrelli, A.; Tuccillo, F.M.; Silvestro, L.; Palaia, R.; Buonaguro, F.M. Precision Medicine in Gastric Cancer. World J. Gastrointest. Oncol. 2019, 11, 804–829. [Google Scholar] [CrossRef]
- Tavakolian, S.; Goudarzi, H.; Faghihloo, E. Evaluating the Expression Level of MiR-9-5p and MiR-192-5p in Gastrointestinal Cancer: Introducing Novel Screening Biomarkers for Patients. BMC Res. Notes 2020, 13, 226. [Google Scholar] [CrossRef]
- Jin, L.; Zhang, N.; Zhang, Q.; Ding, G.; Yang, Z.; Zhang, Z. Serum MicroRNAs as Potential New Biomarkers for Cisplatin Resistance in Gastric Cancer Patients. PeerJ 2020, 8, e8943. [Google Scholar] [CrossRef] [PubMed]
- Ohzawa, H.; Saito, A.; Kumagai, Y.; Kimura, Y.; Yamaguchi, H.; Hosoya, Y.; Lefor, A.K.; Sata, N.; Kitayama, J. Reduced Expression of Exosomal MiR-29s in Peritoneal Fluid Is a Useful Predictor of Peritoneal Recurrence after Curative Resection of Gastric Cancer with Serosal Involvement. Oncol. Rep. 2020, 43, 1081–1088. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, R.; Kanda, T.; Yokosuka, O.; Kato, N.; Matsuoka, S.; Moriyama, M. Exosomes and Hepatocellular Carcinoma: From Bench to Bedside. Int. J. Mol. Sci. 2019, 20, 1406. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zhou, J.; Tang, J.; Min, X.; Yi, T.; Zhao, J.; Ren, Y. Identification of Serum Exosomal LncRNA MIAT as a Novel Diagnostic and Prognostic Biomarker for Gastric Cancer. J. Clin. Lab. Anal. 2020, 34, e23323. [Google Scholar] [CrossRef] [PubMed]
- Akin Telli, T.; Bregni, G.; Camera, S.; Deleporte, A.; Hendlisz, A.; Sclafani, F. PD-1 and PD-L1 Inhibitors in Oesophago-Gastric Cancers. Cancer Lett. 2020, 469, 142–150. [Google Scholar] [CrossRef]
- Han, Y.; Liu, C.; Li, G.; Li, J.; Lv, X.; Shi, H.; Liu, J.; Liu, S.; Yan, P.; Wang, S.; et al. Antitumor Effects and Persistence of a Novel HER2 CAR T Cells Directed to Gastric Cancer in Preclinical Models. Am. J. Cancer Res. 2018, 8, 106–119. [Google Scholar] [PubMed]
- Kim, M.; Pyo, S.; Kang, C.H.; Lee, C.O.; Lee, H.K.; Choi, S.U.; Park, C.H. Folate Receptor 1 (FOLR1) Targeted Chimeric Antigen Receptor (CAR) T Cells for the Treatment of Gastric Cancer. PLoS ONE 2018, 13, e0198347. [Google Scholar] [CrossRef] [PubMed]
- Long, B.; Qin, L.; Zhang, B.; Li, Q.; Wang, L.; Jiang, X.; Ye, H.; Zhang, G.; Yu, Z.; Jiao, Z. CAR T-cell Therapy for Gastric Cancer: Potential and Perspective (Review). Int. J. Oncol. 2020. [Google Scholar] [CrossRef]
- Barker, N.; Huch, M.; Kujala, P.; van de Wetering, M.; Snippert, H.J.; van Es, J.H.; Sato, T.; Stange, D.E.; Begthel, H.; van den Born, M.; et al. Lgr5+ve Stem Cells Drive Self-Renewal in the Stomach and Build Long-Lived Gastric Units In Vitro. Cell Stem Cell 2010, 6, 25–36. [Google Scholar] [CrossRef]
- Schlaermann, P.; Toelle, B.; Berger, H.; Schmidt, S.C.; Glanemann, M.; Ordemann, J.; Bartfeld, S.; Mollenkopf, H.J.; Meyer, T.F. A Novel Human Gastric Primary Cell Culture System for Modelling Helicobacter pylori Infection in Vitro. Gut 2016, 65, 202–213. [Google Scholar] [CrossRef]
- Boccellato, F.; Woelffling, S.; Imai-Matsushima, A.; Sanchez, G.; Goosmann, C.; Schmid, M.; Berger, H.; Morey, P.; Denecke, C.; Ordemann, J.; et al. Polarised Epithelial Monolayers of the Gastric Mucosa Reveal Insights into Mucosal Homeostasis and Defence against Infection. Gut 2019, 68, 400–413. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule/Strategy | Targets | Known/Tested Use in GC | References |
|---|---|---|---|
| Anti-CA19-9 antibodies | CA19-9 | CA19-9-positive GC biomarker for diagnosis | Matsuoka et al. 2018 [99] |
| Cetuximab | EGFR | Potential targeted therapy against tyrosine kinase receptors | Carrasco-Garcia et al. 2018 [98], Apicella et al. 2017 [100] |
| Rilotumumab | HGF | Potential targeted therapy against tyrosine kinase receptors | Carrasco-Garcia et al. 2018 [98], Apicella et al. 2017 [100] |
| Dovitinib | VEGFR-1/2/3; PDGFR-β; FGFR1/2/3 | Potential targeted therapy against tyrosine kinase receptors | Carrasco-Garcia et al. 2018 [98], Apicella et al. 2017 [100] |
| Anti-FGFR2 antibodies | FGFR2 | Under clinical trial for FGFR2 overexpressing GC | Carrasco-Garcia et al. 2018 [98], Apicella et al. 2017 [100] |
| Nimutuzumab | EGFR | Under clinical trial for EGFRhigh GC | Carrasco-Garcia et al. 2018 [98], Apicella et al. 2017 [100] |
| Everolimus | mTOR pathway | Under clinical trial for MSI type GC with activating mutations of mTOR pathway members | Carrasco-Garcia et al. 2018 [98], Apicella et al. 2017 [100] |
| Anti-HDGF antibodies | HDGF | Potential prognostic marker & target of H. pylori-induced GC | Chu et al. 2019 [101] |
| AMS 337 | c-Met | Positive results in Phase I clinical trial | Andres et al. 2019 [102] |
| MET-binding DARPins | c-Met kinase activity | Potential receptor targeting strategy | Andres et al. 2019 [102] |
| Exosomes-delivered c-Met siRNA | c-Met | Potential use as therapy in combination with chemotherapy | Zhang et al. 2020 [103] |
| BST2 siRNA | BST2 | Inhibits GC cell proliferation and motility – potential anti-GC therapy | Liu et al. 2018 [104] |
| Anti-CEACAM6 antibodies | CEACAM6 | Potential endoscopic marker for early GC diagnosis | Roy et al. 2016 [105] |
| Drug/Molecules | Target/Effects | Known Effects in GC | References |
|---|---|---|---|
| Verapamil (in combination with chemotherapies) | Inhibit calcium-dependent channels | Blocks drug efflux mechanisms of CD44+ALDH+ GCSCs and prevents resistance to conventional therapies | Nguyen et al. 2017 [55] |
| Tretinoin | FDA-approved drug for topical treatment of acne vulgaris; pro-differentiation properties | Forces differentiation and decreases tumorigenic properties of CD44+ALDH+ GCSCs | Nguyen et al. 2016 [107] |
| Metformin | FDA-approved drug for first-line treatment of type 2 diabetes; decreases insulin resistance and hepatic neo-glucogenesis | Decreases tumorigenic properties of CD44+ GCSCs by targeting EMT and metabolism modulation | Courtois et al. 2017 [108] |
| Buparlisib | Pan-class I PI3K inhibitor | Decreases CD44+ GCSC tumorigenic and metastatic capacity | Giraud et al. 2019 [109] |
| Verteporfin | FDA-approved drug for age-related macular degeneration – inhibits Hippo effector YAP/TAZ-TEAD interaction | Decreases CD44+ALDH+ GCSC tumorigenic properties through Hippo pathway oncogenic effectors inhibition | Giraud et al. 2019 [59] |
| LIF cytokine | Pro-differentiation properties | Decreases CD44+ALDH+ GCSCs tumorigenic properties by inducing Hippo tumour suppressor kinases activity | Seeneevassen et al. 2020 [60] |
| Vismodegib (in combination with chemotherapies) | FDA-approved drug for recurrent locally advanced and/or metastatic Basal Cell carcinoma; antagonist of the Shh signalling pathway | Improves patient survival in combination with chemotherapies by targeting CD44+ GCSCs having high Shh pathway activity | Bekaii-Saab et al. 2017 [106] |
| Napabucasin (in combination with chemotherapies) | FDA-approved as orphan drug for treatment of gastroesophageal junction cancer; STAT3 inhibitor | Decreases GCSCs tumorigenic properties in combination with paclitaxel in patients with advanced tumours | Bekaii-Saab et al. 2017 [106] |
| Glutamate-cystine exchange transporters inhibitor (xCT) | xCT inhibition | Sensitizes GCSCs to 5-FU conventional therapy by blocking xCT anti-ROS mechanisms | Miyoshi et al. 2018 [110] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seeneevassen, L.; Bessède, E.; Mégraud, F.; Lehours, P.; Dubus, P.; Varon, C. Gastric Cancer: Advances in Carcinogenesis Research and New Therapeutic Strategies. Int. J. Mol. Sci. 2021, 22, 3418. https://doi.org/10.3390/ijms22073418
Seeneevassen L, Bessède E, Mégraud F, Lehours P, Dubus P, Varon C. Gastric Cancer: Advances in Carcinogenesis Research and New Therapeutic Strategies. International Journal of Molecular Sciences. 2021; 22(7):3418. https://doi.org/10.3390/ijms22073418
Chicago/Turabian StyleSeeneevassen, Lornella, Emilie Bessède, Francis Mégraud, Philippe Lehours, Pierre Dubus, and Christine Varon. 2021. "Gastric Cancer: Advances in Carcinogenesis Research and New Therapeutic Strategies" International Journal of Molecular Sciences 22, no. 7: 3418. https://doi.org/10.3390/ijms22073418
APA StyleSeeneevassen, L., Bessède, E., Mégraud, F., Lehours, P., Dubus, P., & Varon, C. (2021). Gastric Cancer: Advances in Carcinogenesis Research and New Therapeutic Strategies. International Journal of Molecular Sciences, 22(7), 3418. https://doi.org/10.3390/ijms22073418