
The Contemporary Approach to CALR-Positive Myeloproliferative Neoplasms


Abstract
1. Introduction
2. CALR Mutations
3. The Calreticulin Protein
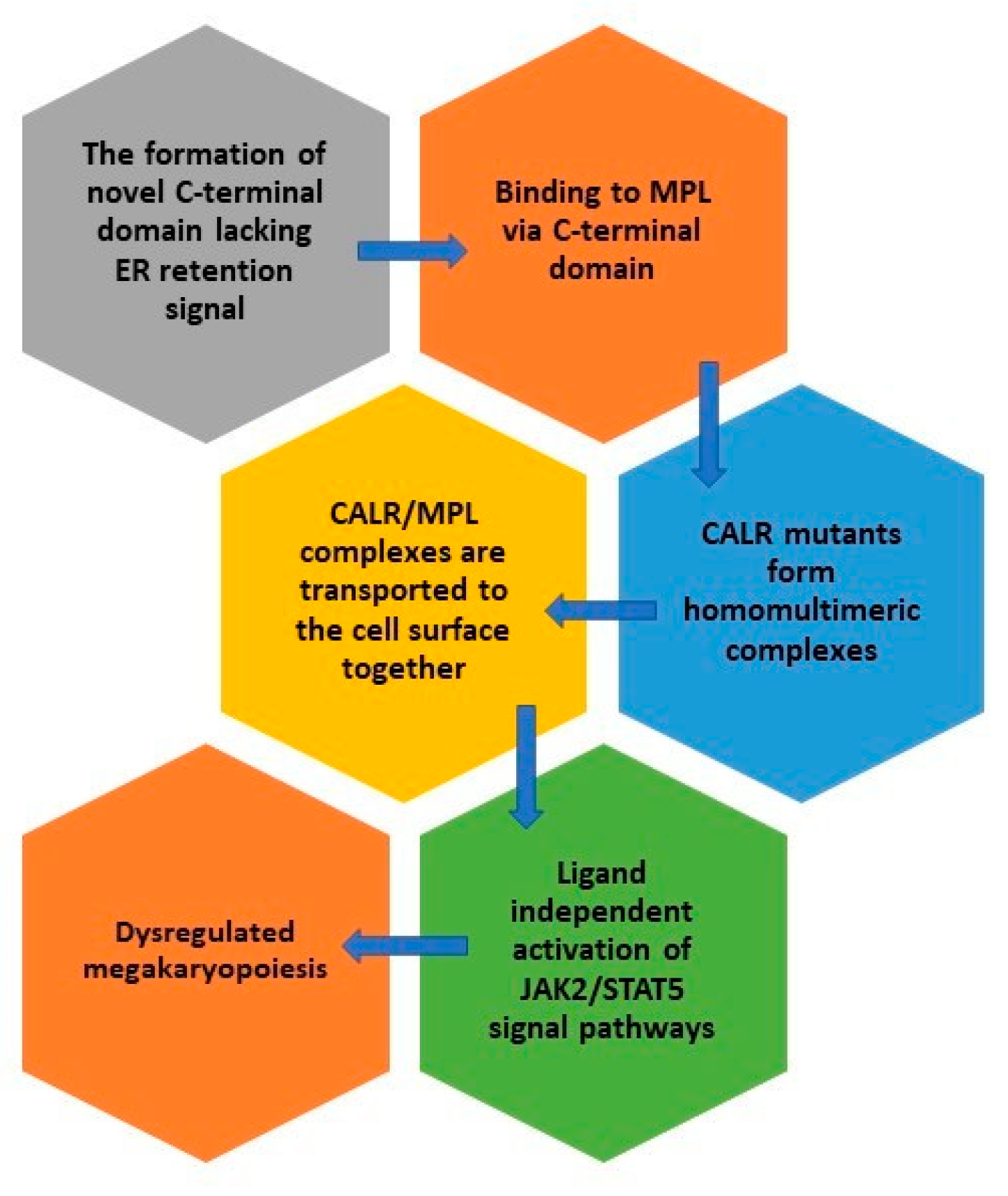
4. Mutant CALR
5. Detection of CALR Mutations and In-Depth Mutational Analysis
6. The Clinical Value of CALR Mutations
7. Machine Based Learning and its Role in MPN
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dameshek, W. Some speculations on the myeloproliferative syndromes [editorial]. Blood 1951, 6, 372–375. [Google Scholar] [CrossRef] [PubMed]
- Nowell, P.C.; Hungerford, D.A. Chromosome Studies on Normal and Leukemic Human Leukocytes. J. Natl. Cancer Inst. 1960, 25, 85–109. [Google Scholar] [CrossRef] [PubMed]
- James, E.C.; Ugo, V.; Le Couédic, J.-P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garçon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nat. Cell Biol. 2005, 434, 1144–1148. [Google Scholar] [CrossRef]
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005, 365, 1054–1061. [Google Scholar] [CrossRef]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.-S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A Gain-of-Function Mutation ofJAK2in Myeloproliferative Disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef]
- Levine, R.L.; Wadleigh, M.; Cools, J.; Ebert, B.L.; Wernig, G.; Huntly, B.J.; Boggon, T.J.; Wlodarska, I.; Clark, J.J.; Moore, S.; et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005, 7, 387–397. [Google Scholar] [CrossRef]
- Zhao, R.; Xing, S.; Li, Z.; Fu, X.; Li, Q.; Krantz, S.B.; Zhao, Z.J. Identification of an Acquired JAK2 Mutation in Polycythemia Vera. J. Biol. Chem. 2005, 280, 22788–22792. [Google Scholar] [CrossRef]
- Pikman, Y.; Lee, B.H.; Mercher, T.; McDowell, E.; Ebert, B.L.; Gozo, M.; Cuker, A.; Wernig, G.; Moore, S.; Galinsky, I.; et al. MPLW515L Is a Novel Somatic Activating Mutation in Myelofibrosis with Myeloid Metaplasia. PLoS Med. 2006, 3, e270. [Google Scholar] [CrossRef] [PubMed]
- Klampfl, T.; Gisslinger, H.; Harutyunyan, A.S.; Nivarthi, H.; Rumi, E.; Milosevic, J.D.; Them, N.C.; Berg, T.; Gisslinger, B.; Pietra, D.; et al. Somatic Mutations of Calreticulin in Myeloproliferative Neoplasms. N. Engl. J. Med. 2013, 369, 2379–2390. [Google Scholar] [CrossRef] [PubMed]
- Nangalia, J.; Massie, C.; Baxter, E.; Nice, F.; Gundem, G.; Wedge, D.; Avezov, E.; Li, J.; Kollmann, K.; Kent, D.; et al. SomaticCALRMutations in Myeloproliferative Neoplasms with NonmutatedJAK2. N. Engl. J. Med. 2013, 369, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Chao, M.P.; Jaiswal, S.; Weissman-Tsukamoto, R.; Alizadeh, A.A.; Gentles, A.J.; Volkmer, J.; Weiskopf, K.; Willingham, S.B.; Raveh, T.; Park, C.Y.; et al. Calreticulin Is the Dominant Pro-Phagocytic Signal on Multiple Human Cancers and Is Counterbalanced by CD47. Sci. Transl. Med. 2010, 2, 63–94. [Google Scholar] [CrossRef] [PubMed]
- Yoon, G.S.; Lee, H.; Jung, Y.; Yu, E.; Moon, H.B.; Song, K.; Lee, I. Nuclear matrix of calreticulin in hepatocellular carcinoma. Cancer Res. 2000, 60, 1117–1120. [Google Scholar] [PubMed]
- Kageyama, S.; Isono, T.; Iwaki, H.; Wakabayashi, Y.; Okada, Y.; Kontani, K.; Yoshimura, K.; Terai, A.; Arai, Y.; Yoshiki, T. Identification by Proteomic Analysis of Calreticulin as a Marker for Bladder Cancer and Evaluation of the Diagnostic Accuracy of Its Detection in Urine. Clin. Chem. 2004, 50, 857–866. [Google Scholar] [CrossRef]
- Hong, S.-H.; Misek, D.E.; Wang, H.; Puravs, E.; Giordano, T.J.; Greenson, J.K.; Brenner, D.E.; Simeone, D.M.; Logsdon, C.D.; Hanash, S.M. An Autoantibody-Mediated Immune Response to Calreticulin Isoforms in Pancreatic Cancer. Cancer Res. 2004, 64, 5504–5510. [Google Scholar] [CrossRef][Green Version]
- Vougas, K.; Gaitanarou, E.; Marinos, E.; Kittas, C.; Voloudakis-Baltatzis, E.I. Two-dimensional electrophoresis and immunohistochemical study of calreticulin in colorectal adenocarcinoma and mirror biopsies. J. BUON 2008, 13, 101–107. [Google Scholar] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Heuser, M.; Panagiota, V.; Koenecke, C.; Fehse, B.; Alchalby, H.; Badbaran, A.; Shahswar, R.; Stadler, M.; Eder, M.; Göhring, G.; et al. Low frequency of calreticulin mutations in MDS patients. Leukemia 2014, 28, 1933–1934. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Im, K.; Park, S.N.; Kwon, J.; Kim, J.-A.; Lee, D.S. CALR, JAK2, and MPL Mutation Profiles in Patients with Four Different Subtypes of Myeloproliferative Neoplasms. Am. J. Clin. Pathol. 2015, 143, 635–644. [Google Scholar] [CrossRef]
- Lin, Y.; Liu, E.; Sun, Q.; Ma, J.; Li, Q.; Cao, Z.; Wang, J.; Jia, Y.; Zhang, H.; Song, Z.; et al. The Prevalence ofJAK2, MPL, and CALR Mutations in Chinese Patients WithBCR-ABL1–Negative Myeloproliferative Neoplasms. Am. J. Clin. Pathol. 2015, 144, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Broséus, J.; Park, J.-H.; Carillo, S.; Hermouet, S.; Girodon, F. Presence of calreticulin mutations in JAK2-negative polycythemia vera. Blood 2014, 124, 3964–3966. [Google Scholar] [CrossRef]
- Broséus, J.; Lippert, E.; Harutyunyan, A.S.; Jeromin, S.; Zipperer, E.; Florensa, L.; Milosevic, J.D.; Haferlach, T.; Germing, U.; Luño, E.; et al. Low rate of calreticulin mutations in refractory anaemia with ring sideroblasts and marked thrombocytosis. Leukemia 2014, 28, 1374–1376. [Google Scholar] [CrossRef]
- Patnaik, M.M.; Belachew, A.; Finke, C.; Lasho, T.L.; Hanson, A.C.; Tefferi, A. CALR mutations are infrequent in WHO-defined refractory anemia with ring sideroblasts. Leukemia 2014, 28, 1370–1371. [Google Scholar] [CrossRef] [PubMed]
- Vainchenker, W.; Kralovics, R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood 2017, 129, 667–679. [Google Scholar] [CrossRef] [PubMed]
- Pietra, D.; Rumi, E.; Ferretti, V.; Di Buduo, A.C.; Milanesi, C.; Cavalloni, C.; Sant’Antonio, E.; Abbonante, V.; Moccia, F.; Casetti, I.; et al. Differential clinical effects of different mutation subtypes in CALR-mutant myeloproliferative neoplasms. Leukemia 2016, 30, 431–438. [Google Scholar] [CrossRef]
- Cabagnols, X.; Defour, J.; Ugo, V.; Ianotto, J.C.; Mossuz, P.; Mondet, J.; Girodon, F.; Alexandre, J.H.; Mansier, O.; Viallard, J.; et al. Differential association of calreticulin type 1 and type 2 mutations with myelofibrosis and essential thrombocytemia: Relevance for disease evolution. Leukemia 2015, 29, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Guglielmelli, P.; Rotunno, G.; Fanelli, T.; Pacilli, A.; Brogi, G.; Calabresi, L.; Pancrazzi, A.; Vannucchi, A.M. Validation of the differential prognostic impact of type 1/type 1-like versus type 2/type 2-like CALR mutations in myelofibrosis. Blood Cancer J. 2015, 5, 360. [Google Scholar] [CrossRef] [PubMed]
- Mikic, T.B.; Pajic, T.; Sever, M. CALR mutations in a cohort of JAK2 V617F negative patients with suspected myeloproliferative neoplasms. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef]
- Cordua, S.; Kjaer, L.; Skov, V.; Pallisgaard, N.; Hasselbalch, H.C.; Ellervik, C. Prevalence and phenotypes of JAK2 V617F and calreticulin mutations in a Danish general population. Blood 2019, 134, 469–479. [Google Scholar] [CrossRef]
- Ostwald, T.J.; MacLennan, D.H. Isolation of a High Affinity Calcium-binding Protein from Sarcoplasmic Reticulum. J. Biol. Chem. 1974, 249, 974–979. [Google Scholar] [CrossRef]
- Michalak, M.; Corbett, E.F.; Mesaeli, N.; Nakamura, K.; Opas, M. Calreticulin: One protein, one gene, many functions. Biochem. J. 1999, 344, 281–292. [Google Scholar] [CrossRef]
- Michalak, M.; Groenendyk, J.; Szabo, E.; Gold, L.I.; Opas, M. Calreticulin, a multi-process calcium-buffering chaperone of the endoplasmic reticulum. Biochem. J. 2009, 417, 651–666. [Google Scholar] [CrossRef]
- Nakamura, K.; Zuppini, A.; Arnaudeau, S.; Lynch, J.; Ahsan, I.; Krause, R.; Papp, S.; De Smedt, H.; Parys, J.B.; Muüller-Esterl, W.; et al. Functional specialization of calreticulin domains. J. Cell Biol. 2001, 154, 961–972. [Google Scholar] [CrossRef] [PubMed]
- Migliaccio, A.R.; Uversky, V.N. Dissecting physical structure of calreticulin, an intrinsically disordered Ca2+-buffering chaperone from endoplasmic reticulum. J. Biomol. Struct. Dyn. 2017, 36, 1617–1636. [Google Scholar] [CrossRef]
- Boelt, S.G.; Norn, C.; Rasmussen, M.I.; André, I.; Čiplys, E.; Slibinskas, R.; Houen, G.; Højrup, P. Mapping the Ca2+ induced structural change in calreticulin. J. Proteom. 2016, 142, 138–148. [Google Scholar] [CrossRef]
- Varricchio, L.; Falchi, M.; Dall’Ora, M.; De Benedittis, C.; Ruggeri, A.; Uversky, V.N.; Migliaccio, A.R. Calreticulin: Challenges Posed by the Intrinsically Disordered Nature of Calreticulin to the Study of Its Function. Front. Cell Dev. Biol. 2017, 5, 96. [Google Scholar] [CrossRef]
- Balligand, T.; Achouri, Y.; Pecquet, C.; Gaudray, G.; Colau, D.; Hug, E.; Rahmani, Y.; Stroobant, V.; Plo, I.; Vainchenker, W.; et al. Knock-in of murine Calr del52 induces essential thrombocythemia with slow-rising dominance in mice and reveals key role of Calr exon 9 in cardiac development. Leukemia 2020, 34, 510–521. [Google Scholar] [CrossRef]
- Raghavan, M.; Wijeyesakere, S.J.; Peters, L.R.; Del Cid, N. Calreticulin in the immune system: Ins and outs. Trends Immunol. 2013, 34, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Houen, G. Commentary: Calreticulin—Oncogene, Anti-oncogene, or Both? Curr. Protein Pept. Sci. 2018, 20, 111–112. [Google Scholar] [CrossRef] [PubMed]
- Stanley, R.F.; Steidl, U. Molecular Mechanism of Mutant CALR–Mediated Transformation. Cancer Discov. 2016, 6, 344–346. [Google Scholar] [CrossRef] [PubMed]
- Elf, S.; Abdelfattah, N.S.; Chen, E.; Perales-Patón, J.; Rosen, E.A.; Ko, A.; Peisker, F.; Florescu, N.; Giannini, S.; Wolach, O.; et al. Mutant Calreticulin Requires Both Its Mutant C-terminus and the Thrombopoietin Receptor for Oncogenic Transformation. Cancer Discov. 2016, 6, 368–381. [Google Scholar] [CrossRef] [PubMed]
- Marty, C.; Pecquet, C.; Nivarthi, H.; El-Khoury, M.; Chachoua, I.; Tulliez, M.; Villeval, J.-L.; Raslova, H.; Kralovics, R.; Constantinescu, S.N.; et al. Calreticulin mutants in mice induce an MPL-dependent thrombocytosis with frequent progression to myelofibrosis. Blood 2016, 127, 1317–1324. [Google Scholar] [CrossRef]
- Elf, S.; Abdelfattah, N.S.; Baral, A.J.; Beeson, D.; Rivera, J.F.; Ko, A.; Florescu, N.; Birrane, G.; Chen, E.; Mullally, A. Defining the requirements for the pathogenic interaction between mutant calreticulin and MPL in MPN. Blood 2018, 131, 782–786. [Google Scholar] [CrossRef]
- Chachoua, I.; Pecquet, C.; El-Khoury, M.; Nivarthi, H.; Albu, R.-I.; Marty, C.; Gryshkova, V.; Defour, J.-P.; Vertenoeil, G.; Ngo, A.; et al. Thrombopoietin receptor activation by myeloproliferative neoplasm associated calreticulin mutants. Blood 2016, 127, 1325–1335. [Google Scholar] [CrossRef]
- Araki, M.; Yang, Y.; Masubuchi, N.; Hironaka, Y.; Takei, H.; Morishita, S.; Mizukami, Y.; Kan, S.; Shirane, S.; Edahiro, Y.; et al. Activation of the thrombopoietin receptor by mutant calreticulin in CALR-mutant myeloproliferative neoplasms. Blood 2016, 127, 1307–1316. [Google Scholar] [CrossRef]
- Lau, W.W.Y.; Hannah, R.; Green, A.R.; Gottgens, B. The JAK-STAT signaling pathway is differentially activated in CALR-positive compared with JAK2V617F-positive ET patients. Blood 2015, 125, 1679–1681. [Google Scholar] [CrossRef] [PubMed]
- Pecquet, C.; Chachoua, I.; Roy, A.; Balligand, T.; Vertenoeil, G.; Leroy, E.; Albu, R.-I.; Defour, J.-P.; Nivarthi, H.; Hug, E.; et al. Calreticulin mutants as oncogenic rogue chaperones for TpoR and traffic-defective pathogenic TpoR mutants. Blood 2019, 133, 2669–2681. [Google Scholar] [CrossRef] [PubMed]
- Araki, M.; Yang, Y.; Imai, M.; Mizukami, Y.; Kihara, Y.; Sunami, Y.; Masubuchi, N.; Edahiro, Y.; Hironaka, Y.; Osaga, S.; et al. Homomultimerization of mutant calreticulin is a prerequisite for MPL binding and activation. Leukemia 2019, 33, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Masubuchi, N.; Araki, M.; Yang, Y.; Hayashi, E.; Imai, M.; Edahiro, Y.; Hironaka, Y.; Mizukami, Y.; Kihara, Y.; Takei, H.; et al. Mutant calreticulin interacts with MPL in the secretion pathway for activation on the cell surface. Leukemia 2020, 34, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Zhao, L.; Loos, F.; Marty, C.; Xie, W.; Martins, I.; Lachkar, S.; Qu, B.; Waeckel-Énée, E.; Plo, I.; et al. Immunosuppression by Mutated Calreticulin Released from Malignant Cells. Mol. Cell 2020, 77, 748–760. [Google Scholar] [CrossRef]
- Dedhar, S.; Rennie, P.S.; Shago, M.; Hagesteijn, C.-Y.L.; Yang, H.; Filmus, J.; Hawley, R.G.; Bruchovsky, N.; Cheng, H.; Matusik, R.J.; et al. Inhibition of nuclear hormone receptor activity by calreticulin. Nat. Cell Biol. 1994, 367, 480–483. [Google Scholar] [CrossRef]
- Burns, K.; Duggan, B.; Atkinson, E.A.; Famulski, K.S.; Nemer, M.; Bleackley, R.C.; Michalak, M. Modulation of gene expression by calreticulin binding to the glucocorticoid receptor. Nat. Cell Biol. 1994, 367, 476–480. [Google Scholar] [CrossRef]
- Roderick, H.; Campbell, A.K.; Llewellyn, D.H. Nuclear localisation of calreticulin in vivo is enhanced by its interaction with glucocorticoid receptors. FEBS Lett. 1997, 405, 181–185. [Google Scholar] [CrossRef]
- Iborra, F.J.; Papadopoulos, P. Calreticulin in Essential Thrombocythemia: Stressing out the Megakaryocyte Nucleus. Front. Oncol. 2017, 7, 103. [Google Scholar] [CrossRef] [PubMed]
- Pronier, E.; Cifani, P.; Merlinsky, T.R.; Berman, K.B.; Somasundara, A.V.H.; Rampal, R.K.; Lacava, J.; Wei, K.E.; Pastore, F.; Maag, J.L.; et al. Targeting the CALR interactome in myeloproliferative neoplasms. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Nivarthi, H.; Chen, D.; Cleary, C.; Kubesova, B.; Jäger, R.; Bogner, E.; Marty, C.; Pecquet, C.; Vainchenker, W.; Constantinescu, S.N.; et al. Thrombopoietin receptor is required for the oncogenic function of CALR mutants. Leukemia 2016, 30, 1759–1763. [Google Scholar] [CrossRef] [PubMed]
- Di Buduo, C.A.; Abbonante, V.; Marty, C.; Moccia, F.; Rumi, E.; Pietra, D.; Soprano, P.M.; Lim, D.; Cattaneo, D.; Iurlo, A.; et al. Defective interaction of mutant calreticulin and SOCE in megakaryocytes from patients with myeloproliferative neoplasms. Blood 2020, 135, 133–144. [Google Scholar] [CrossRef]
- Benlabiod, C.; Cacemiro, M.D.C.; Nédélec, A.; Edmond, V.; Muller, D.; Rameau, P.; Touchard, L.; Gonin, P.; Constantinescu, S.N.; Raslova, H.; et al. Calreticulin del52 and ins5 knock-in mice recapitulate different myeloproliferative phenotypes observed in patients with MPN. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Prins, D.; Park, H.J.; Grinfeld, J.; Gonzalez-Arias, C.; Loughran, S.; Dovey, O.M.; Klampfl, T.; Bennett, C.; Hamilton, T.L.; et al. Mutant calreticulin knockin mice develop thrombocytosis and myelofibrosis without a stem cell self-renewal advantage. Blood 2018, 131, 649–661. [Google Scholar] [CrossRef]
- Holmström, O.M.; Martinenaite, E.; Ahmad, S.M.; Met, Ö.; Friese, C.; Kjær, L.; Riley, C.H.; Straten, P.T.; Svane, I.M.; Hasselbalch, H.C.; et al. The calreticulin (CALR) exon 9 mutations are promising targets for cancer immune therapy. Leukemia 2018, 32, 429–437. [Google Scholar] [CrossRef]
- Jones, A.V.; Ward, D.; Lyon, M.; Leung, W.; Callaway, A.; Chase, A.; Dent, C.L.; White, H.E.; Drexler, H.G.; Nangalia, J.; et al. Evaluation of methods to detect CALR mutations in myeloproliferative neoplasms. Leukemia Res. 2015, 39, 82–87. [Google Scholar] [CrossRef]
- Chi, J.; Nicolaou, A.K.; Nicolaidou, V.; Koumas, L.; Mitsidou, A.; Pierides, C.; Manoloukos, M.; Barbouti, K.; Melanthiou, F.; Prokopiou, C.; et al. Calreticulin gene exon 9 frameshift mutations in patients with thrombocytosis. Leukemia 2013, 28, 1152–1154. [Google Scholar] [CrossRef]
- Kjær, L.; Cordua, S.; Holmström, M.O.; Thomassen, M.; Kruse, A.T.; Pallisgaard, N.; Larsen, T.S.; De Stricker, K.; Skov, V.; Hasselbalch, H.C. Differential Dynamics of CALR Mutant Allele Burden in Myeloproliferative Neoplasms during Interferon Alfa Treatment. PLoS ONE 2016, 11, e0165336. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.-M.; Zhou, J.; Gale, R.P.; Li, J.-L.; Li, L.-D.; Li, N.; Chen, S.-S.; Ruan, G.-R. A rapid, highly accurate method for quantifying CALR mutant allele burden in persons with myeloproliferative neoplasms. Hematology 2015, 20, 517–522. [Google Scholar] [CrossRef]
- Trung, N.T.; Quyen, D.T.; Hoan, N.X.; Giang, D.P.; Trang, T.T.H.; Velavan, T.P.; Bang, M.H.; Song, L.H. Rapid, low cost and sensitive detection of Calreticulin mutations by a PCR based amplicon length differentiation assay for diagnosis of myeloproliferative neoplasms. BMC Med. Genet. 2019, 20, 1–9. [Google Scholar] [CrossRef]
- Giannopoulos, A.; Rougkala, N.; Loupis, T.; Mantzourani, M.; Viniou, N.-A.; Variami, E.; Vassilakopoulos, T.P.; Dryllis, G.; Kotsianidis, I.; Gougopoulou, T.; et al. Detection of Calr Mutations Using High Resolution Melting Curve Analysis (Hrm-A); Application on a Large Cohort of Greek Et and Mf Patients. Mediterr. J. Hematol. Infect. Dis. 2019, 11, e2019009. [Google Scholar] [CrossRef] [PubMed]
- Pajič, T.; Mikič, T.B.; Podgornik, H.; Klun, J.; Šućurović, S.; Zver, S.; Sever, M. Genetic Variant Detection in the CALR gene using High Resolution Melting Analysis. J. Vis. Exp. 2020, 10, e61642. [Google Scholar] [CrossRef]
- Lim, K.-H.; Lin, H.-C.; Chen, C.G.-S.; Wang, W.-T.; Chang, Y.-C.; Chiang, Y.-H.; Lin, C.-S.; Su, N.-W.; Su, Y.-W.; Lin, J.; et al. Rapid and sensitive detection of CALR exon 9 mutations using high-resolution melting analysis. Clin. Chim. Acta 2015, 440, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Bilbao-Sieyro, C.; Santana, G.; Moreno, M.; Torres, L.; Santana-Lopez, G.; Rodríguez-Medina, C.; Perera, M.; Bellosillo, B.; De La Iglesia, S.; Molero, T.; et al. High Resolution Melting Analysis: A Rapid and Accurate Method to Detect CALR Mutations. PLoS ONE 2014, 9, e103511. [Google Scholar] [CrossRef]
- Guglielmelli, P.; Pietra, D.; Pane, F.; Pancrazzi, A.; Cazzola, M.; Vannucchi, A.M.; Tura, S.; Barosi, G. Recommendations for molecular testing in classical Ph1-neg myeloproliferative disorders–A consensus project of the Italian Society of Hematology. Leukemia Res. 2017, 58, 63–72. [Google Scholar] [CrossRef]
- Lasho, T.L.; Elliott, M.A.; Pardanani, A.; Tefferi, A. CALRmutation studies in chronic neutrophilic leukemia. Am. J. Hematol. 2014, 89, 450. [Google Scholar] [CrossRef] [PubMed]
- Chi, J.; Manoloukos, M.; Pierides, C.; Nicolaidou, V.; Nicolaou, K.; Kleopa, M.; Vassiliou, G.; Costeas, P. Calreticulin mutations in myeloproliferative neoplasms and new methodology for their detection and monitoring. Ann. Hematol. 2014, 94, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Mansier, O.; Migeon, M.; Saint-Lézer, A.; James, E.C.; Verger, E.; Robin, M.; Socié, G.; Bidet, A.; Mahon, F.-X.; Cassinat, B.; et al. Quantification of the Mutant CALR Allelic Burden by Digital PCR. J. Mol. Diagn. 2016, 18, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Lasho, T.L.; Finke, C.M.; Tischer, A.; Pardanani, A.; Tefferi, A. Mayo CALR mutation type classification guide using alpha helix propensity. Am. J. Hematol. 2018, 93, 128–129. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, B.; Chen, B.; Zhou, R.-F.; Zhang, Q.-G.; Li, J.; Yang, Y.-G.; Zhou, M.; Shao, X.-Y.; Xu, Y.; et al. JAK2, MPL, and CALR mutations in Chinese Han patients with essential thrombocythemia. Hematology 2016, 22, 145–148. [Google Scholar] [CrossRef][Green Version]
- Kluk, M.J.; Lindsley, R.C.; Aster, J.C.; Lindeman, N.I.; Szeto, D.; Hall, D.; Kuo, F.C. Validation and Implementation of a Custom Next-Generation Sequencing Clinical Assay for Hematologic Malignancies. J. Mol. Diagn. 2016, 18, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Vannucchi, A.M. Genetic Risk Assessment in Myeloproliferative Neoplasms. Mayo Clin. Proc. 2017, 92, 1283–1290. [Google Scholar] [CrossRef]
- Grinfeld, J.; Nangalia, J.; Baxter, E.J.; Wedge, D.C.; Angelopoulos, N.; Cantrill, R.; Godfrey, A.L.; Papaemmanuil, E.; Gundem, G.; MacLean, C.; et al. Classification and Personalized Prognosis in Myeloproliferative Neoplasms. N. Engl. J. Med. 2018, 379, 1416–1430. [Google Scholar] [CrossRef]
- Aguilera-Diaz, A.; Vazquez, I.; Ariceta, B.; Mañú, A.; Blasco-Iturri, Z.; Palomino-Echeverría, S.; Larrayoz, M.J.; García-Sanz, R.; Prieto-Conde, M.I.; Chillón, M.D.C.; et al. Assessment of the clinical utility of four NGS panels in myeloid malignancies. Suggestions for NGS panel choice or design. PLoS ONE 2020, 15, e0227986. [Google Scholar] [CrossRef] [PubMed]
- Jennings, L.J.; Arcila, M.E.; Corless, C.; Kamel-Reid, S.; Lubin, I.M.; Pfeifer, J.; Temple-Smolkin, R.L.; Voelkerding, K.V.; Nikiforova, M.N. Guidelines for Validation of Next-Generation Sequencing–Based Oncology Panels. J. Mol. Diagn. 2017, 19, 341–365. [Google Scholar] [CrossRef] [PubMed]
- Gardner, J.-A.; Peterson, J.D.; Turner, S.A.; Soares, B.L.; Lancor, C.R.; Dos Santos, L.L.; Kaur, P.; Ornstein, D.L.; Tsongalis, G.J.; De Abreu, F.B. Detection ofCALRMutation in Clonal and Nonclonal Hematologic Diseases Using Fragment Analysis and Next-Generation Sequencing. Am. J. Clin. Pathol. 2016, 146, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdóttir, H.; Wenger, A.M.; Zehir, A.; Mesirov, J.P. Variant Review with the Integrative Genomics Viewer. Cancer Res. 2017, 77, 31–34. [Google Scholar] [CrossRef]
- Palumbo, G.A.; Stella, S.; Pennisi, M.S.; Pirosa, C.; Fermo, E.; Fabris, S.; Cattaneo, D.; Iurlo, A. The Role of New Technologies in Myeloproliferative Neoplasms. Front. Oncol. 2019, 9, 321. [Google Scholar] [CrossRef]
- Ross, D.M.; Thomson, C.; Hamad, N.; Lane, S.W.; Manos, K.; Grigg, A.P.; Guo, B.; Erber, W.N.; Scott, A.; Viiala, N.; et al. Myeloid somatic mutation panel testing in myeloproliferative neoplasms. Pathology 2021. [Google Scholar] [CrossRef]
- Barbui, T.; Thiele, J.; Vannucchi, A.M.; Tefferi, A. Myeloproliferative neoplasms: Morphology and clinical practice. Am. J. Hematol. 2016, 91, 430–433. [Google Scholar] [CrossRef] [PubMed]
- Verger, E.; Cassinat, B.; Chauveau, A.; Dosquet, C.; Giraudier, S.; Schlageter, M.-H.; Ianotto, J.-C.; Yassin, M.A.; Al-Dewik, N.; Carillo, S.; et al. Clinical and molecular response to interferon-α therapy in essential thrombocythemia patients with CALR mutations. Blood 2015, 126, 2585–2591. [Google Scholar] [CrossRef]
- Accetta, R.; Elli, L.; Libera, L.; Siracusa, C.; Cassavia, F.; Orsini, F.; Orlandi, L.; Passamonti, F.; Casalone, R.; Pallotti, F. Analysis of three screening methods for the detection of calreticulin gene mutations. Int. J. Lab. Hematol. 2019, 42, 76. [Google Scholar] [CrossRef]
- Badbaran, A.; Fehse, B.; Christopeit, M.; Aranyossy, T.; Ayuk, A.F.; Wolschke, C.; Kröger, N. Digital-PCR assay for screening and quantitative monitoring of calreticulin (CALR) type-2 positive patients with myelofibrosis following allogeneic stem cell transplantation. Bone Marrow Transplant. 2016, 51, 872–873. [Google Scholar] [CrossRef] [PubMed]
- Wolschke, C.; Badbaran, A.; Zabelina, T.; Christopeit, M.; Ayuk, F.; Triviai, I.; Zander, A.; Alchalby, H.; Bacher, U.; Fehse, B.; et al. Impact of molecular residual disease post allografting in myelofibrosis patients. Bone Marrow Transplant. 2017, 52, 1526–1529. [Google Scholar] [CrossRef] [PubMed]
- Michiels, J.J.; Van Genderen, P.J.J.; Lindemans, J.; Van Vliet, H.H.D.M. Erythromelalgic, Thrombotic and Hemorrhagic Manifestations in 50 Cases of Thrombocythemia. Leuk. Lymphoma 1996, 22, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Elliott, M. Thrombosis in Myeloproliferative Disorders: Prevalence, Prognostic Factors, and the Role of Leukocytes and JAK2V617F. Semin. Thromb. Hemost. 2007, 33, 313–320. [Google Scholar] [CrossRef]
- Tefferi, A. Myelofibrosis with Myeloid Metaplasia. N. Engl. J. Med. 2000, 342, 1255–1265. [Google Scholar] [CrossRef] [PubMed]
- Kvasnicka, H.M.; Thiele, J. Prodromal myeloproliferative neoplasms: The 2008 WHO classification. Am. J. Hematol. 2009, 85, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A. Pathogenesis of Myelofibrosis with Myeloid Metaplasia. J. Clin. Oncol. 2005, 23, 8520–8530. [Google Scholar] [CrossRef] [PubMed]
- Jutzi, J.S.; Mullally, A. Remodeling the Bone Marrow Microenvironment—A Proposal for Targeting Pro-inflammatory Contributors in MPN. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Kc, D.; Falchi, L.; Verstovsek, S. The underappreciated risk of thrombosis and bleeding in patients with myelofibrosis: A review. Ann. Hematol. 2017, 96, 1595–1604. [Google Scholar] [CrossRef] [PubMed]
- Barbui, T.; Thiele, J.; Passamonti, F.; Rumi, E.; Boveri, E.; Ruggeri, M.; Rodeghiero, F.; D’Amore, E.S.; Randi, M.L.; Bertozzi, I.; et al. Survival and Disease Progression in Essential Thrombocythemia Are Significantly Influenced by Accurate Morphologic Diagnosis: An International Study. J. Clin. Oncol. 2011, 29, 3179–3184. [Google Scholar] [CrossRef]
- Langabeer, S.E. Chasing down the triple-negative myeloproliferative neoplasms: Implications for molecular diagnostics. Jak-stat 2016, 5, e1248011. [Google Scholar] [CrossRef]
- Rozovski, U.; Verstovsek, S.; Manshouri, T.; Dembitz, V.; Bozinovic, K.; Newberry, K.; Zhang, Y.; Bove, J.E.; Pierce, S.; Kantarjian, H.; et al. An accurate, simple prognostic model consisting of age, JAK2, CALR, and MPL mutation status for patients with primary myelofibrosis. Haematology 2016, 102, 79–84. [Google Scholar] [CrossRef]
- Guglielmelli, P.; Barosi, G.; Specchia, G.; Rambaldi, A.; Coco, F.L.; Antonioli, E.; Pieri, L.; Pancrazzi, A.; Ponziani, V.; Delaini, F.; et al. Identification of patients with poorer survival in primary myelofibrosis based on the burden of JAK2V617F mutated allele. Blood 2009, 114, 1477–1483. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Finke, C.M.; Knudson, A.R.; Ketterling, R.; Hanson, C.H.; Maffioli, M.; Caramazza, D.; Passamonti, F.; Pardanani, A. CALR vs JAK2 vs MPL-mutated or triple-negative myelofibrosis: Clinical, cytogenetic and molecular comparisons. Leukemia 2014, 28, 1472–1477. [Google Scholar] [CrossRef] [PubMed]
- Rumi, E.; Pietra, D.; Ferretti, V.; Klampfl, T.; Harutyunyan, A.S.; Milosevic, J.D.; Them, N.C.C.; Berg, T.; Elena, C.; Casetti, I.C.; et al. JAK2 or CALR mutation status defines subtypes of essential thrombocythemia with substantially different clinical course and outcomes. Blood 2014, 123, 1544–1551. [Google Scholar] [CrossRef] [PubMed]
- Finazzi, M.C.; Carobbio, A.; Cervantes, F.; Isola, I.M.; Vannucchi, A.M.; Guglielmelli, P.; Rambaldi, A.; Finazzi, G.; Barosi, G.; Barbui, T. CALR mutation, MPL mutation and triple negativity identify patients with the lowest vascular risk in primary myelofibrosis. Leukemia 2014, 29, 1209–1210. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Lasho, T.L.; Tischer, A.; Wassie, E.A.; Finke, C.M.; Belachew, A.A.; Ketterling, R.P.; Hanson, C.A.; Pardanani, A.D. The prognostic advantage of calreticulin mutations in myelofibrosis might be confined to type 1 or type 1-like CALR variants. Blood 2014, 124, 2465–2466. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Nicolosi, M.; Mudireddy, M.; Szuber, N.; Finke, C.M.; Lasho, T.L.; Hanson, C.A.; Ketterling, R.P.; Pardanani, A.; Gangat, N.; et al. Driver mutations and prognosis in primary myelofibrosis: Mayo-Careggi MPN alliance study of 1,095 patients. Am. J. Hematol. 2018, 93, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Saki, N.; Shirzad, R.; Rahim, F.; Malehi, A.S. Estimation of diagnosis and prognosis in ET by assessment of CALR and JAK2V617F mutations and laboratory findings: A meta-analysis. Clin. Transl. Oncol. 2017, 19, 874–883. [Google Scholar] [CrossRef]
- Tefferi, A.; Barbui, T. Polycythemia vera and essential thrombocythemia: 2021 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2020, 95, 1599–1613. [Google Scholar] [CrossRef]
- Pardanani, A.; Lasho, T.L.; Finke, C.M.; Oh, S.T.; Gotlib, J.; Tefferi, A. LNK mutation studies in blast-phase myeloproliferative neoplasms, and in chronic-phase disease with TET2, IDH, JAK2 or MPL mutations. Leukemia 2010, 24, 1713–1718. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Pardanani, A. Myeloproliferative Neoplasms. JAMA Oncol. 2015, 1, 97–105. [Google Scholar] [CrossRef]
- Chang, Y.-C.; Lin, H.-C.; Chiang, Y.-H.; Chen, C.G.-S.; Huang, L.; Wang, W.-T.; Cheng, C.-C.; Lin, J.; Chang, Y.-F.; Chang, M.-C.; et al. Targeted next-generation sequencing identified novel mutations in triple-negative myeloproliferative neoplasms. Med. Oncol. 2017, 34, 83. [Google Scholar] [CrossRef]
- Lundberg, P.; Karow, A.; Nienhold, R.; Looser, R.; Hao-Shen, H.; Nissen, I.; Girsberger, S.; Lehmann, T.; Passweg, J.; Stern, M.; et al. Clonal evolution and clinical correlates of somatic mutations in myeloproliferative neoplasms. Blood 2014, 123, 2220–2228. [Google Scholar] [CrossRef]
- Agarwal, R.; Blombery, P.; McBean, M.; Jones, K.; Fellowes, A.; Doig, K.; Forsyth, C.; Westerman, D.A. Clinicopathological differences exist between CALR- and JAK2-mutated myeloproliferative neoplasms despite a similar molecular landscape: Data from targeted next-generation sequencing in the diagnostic laboratory. Ann. Hematol. 2017, 96, 725–732. [Google Scholar] [CrossRef]
- Tefferi, A.; Guglielmelli, P.; Lasho, T.L.; Coltro, G.; Finke, C.M.; Loscocco, G.G.; Sordi, B.; Szuber, N.; Rotunno, G.; Pacilli, A.; et al. Mutation-enhanced international prognostic systems for essential thrombocythaemia and polycythaemia vera. Br. J. Haematol. 2020, 189, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Lasho, T.L.; Guglielmelli, P.; Finke, C.M.; Rotunno, G.; Elala, Y.; Pacilli, A.; Hanson, C.A.; Pancrazzi, A.; Ketterling, R.P.; et al. Targeted deep sequencing in polycythemia vera and essential thrombocythemia. Blood Adv. 2016, 1, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Cervantes, F.; Dupriez, B.; Pereira, A.; Passamonti, F.; Reilly, J.T.; Morra, E.; Vannucchi, A.M.; Mesa, R.A.; Demory, J.-L.; Barosi, G.; et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood 2009, 113, 2895–2901. [Google Scholar] [CrossRef]
- Tefferi, A.; Guglielmelli, P.; Nicolosi, M.; Mannelli, F.; Mudireddy, M.; Bartalucci, N.; Finke, C.M.; Lasho, T.L.; Hanson, C.A.; Ketterling, R.P.; et al. GIPSS: Genetically inspired prognostic scoring system for primary myelofibrosis. Leukemia 2018, 32, 1631–1642. [Google Scholar] [CrossRef]
- Tefferi, A.; Guglielmelli, P.; Lasho, T.L.; Gangat, N.; Ketterling, R.P.; Pardanani, A.; Vannucchi, A.M. MIPSS70+ Version 2.0: Mutation and Karyotype-Enhanced International Prognostic Scoring System for Primary Myelofibrosis. J. Clin. Oncol. 2018, 36, 1769–1770. [Google Scholar] [CrossRef] [PubMed]
- Guglielmelli, P.; Lasho, T.L.; Rotunno, G.; Mudireddy, M.; Mannarelli, C.; Nicolosi, M.; Pacilli, A.; Pardanani, A.; Rumi, E.; Rosti, V.; et al. MIPSS70: Mutation-Enhanced International Prognostic Score System for Transplantation-Age Patients with Primary Myelofibrosis. J. Clin. Oncol. 2018, 36, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Shouval, R.; Fein, J.A.; Savani, B.; Mohty, M.; Nagler, A. Machine learning and artificial intelligence in haematology. Br. J. Haematol. 2021, 192, 239–250. [Google Scholar] [CrossRef]
- Handelman, G.S.; Kok, H.K.; Chandra, R.V.; Razavi, A.H.; Lee, M.J.; Asadi, H. eDoctor: Machine learning and the future of medicine. J. Intern. Med. 2018, 284, 603–619. [Google Scholar] [CrossRef]
- Gupta, V.; Braun, T.M.; Chowdhury, M.; Tewari, M.; Choi, S.W. A Systematic Review of Machine Learning Techniques in Hematopoietic Stem Cell Transplantation (HSCT). Sensors 2020, 20, 6100. [Google Scholar] [CrossRef] [PubMed]
- Sirinukunwattana, K.; Aberdeen, A.; Theissen, H.; Sousos, N.; Psaila, B.; Mead, A.J.; Turner, G.D.H.; Rees, G.; Rittscher, J.; Royston, D. Artificial intelligence–based morphological fingerprinting of megakaryocytes: A new tool for assessing disease in MPN patients. Blood Adv. 2020, 4, 3284–3294. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Li, Z. Bioinformatics Analysis of Key Genes and Pathways Associated with Thrombosis in Essential Thrombocythemia. Med. Sci. Monit. 2019, 25, 9262–9271. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.-J.; Cao, X.-J.; Zhou, C.; Sun, Y.; Lv, Q.-L.; Feng, F.-B.; Zhang, Y.-Y.; Sun, C.-G. Construction of polycythemia vera protein interaction network and prediction of related biological functions. Genet. Mol. Res. 2016, 15. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Ning, P. Predicting pathogenic genes for primary myelofibrosis based on a system‑network approach. Mol. Med. Rep. 2017, 17, 186–192. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Method | Advantage | Critical Remarks | Sensitivity | Reference |
|---|---|---|---|---|
| Sanger sequencing | Known and unknown genetic variant detection. | Low sensitivity; Not quantitative; Moderate cost. | 10 to 20% | [9,10,60] |
| PCR and fragment analysis | Known and unknown genetic variant detection; Qualitative and quantitative; Simple to perform; Low cost and rapid. | Moderate to low sensitivity.; Preferential amplification of shorter amplicons may lead to over- or underestimation of the mutant allele burden; Sanger sequencing needed for correctly genotype the CALR variants. | 1 to 10% | [9,60,61,72,80,85] |
| High-resolution Melt | Known and unknown genetic variant detection; Simple to perform; Low cost and rapid. | Moderate to low sensitivity; Not quantitative; Sanger sequencing needed for correctly genotype the CALR variants. | 1 to 5% | [60,66,67,68] |
| Quantitative PCR (real-time PCR) (qPCR) | High sensitivity; Quantitative; Rapid. | Detects only target genetic variants; Moderate cost. | 0.01 to 1% | [60,62,71,86] |
| Digital PCR | High sensitivity; Quantitative; Rapid. | Detects only target genetic variants; Moderate cost. | 0.01 to 1% | [28,72,86,87,88] |
| NGS | Known and unknown genetic variant detection; Simultaneous screening of multiple genes in multiple samples. | Complex genetic variants and large indels need in some instances confirmation by alternate molecular genetic methods; Complex workflow and result interpretation; Moderate to high cost. | 1 to 5% | [60,75,80] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belčič Mikič, T.; Pajič, T.; Zver, S.; Sever, M. The Contemporary Approach to CALR-Positive Myeloproliferative Neoplasms. Int. J. Mol. Sci. 2021, 22, 3371. https://doi.org/10.3390/ijms22073371
Belčič Mikič T, Pajič T, Zver S, Sever M. The Contemporary Approach to CALR-Positive Myeloproliferative Neoplasms. International Journal of Molecular Sciences. 2021; 22(7):3371. https://doi.org/10.3390/ijms22073371
Chicago/Turabian StyleBelčič Mikič, Tanja, Tadej Pajič, Samo Zver, and Matjaž Sever. 2021. "The Contemporary Approach to CALR-Positive Myeloproliferative Neoplasms" International Journal of Molecular Sciences 22, no. 7: 3371. https://doi.org/10.3390/ijms22073371
APA StyleBelčič Mikič, T., Pajič, T., Zver, S., & Sever, M. (2021). The Contemporary Approach to CALR-Positive Myeloproliferative Neoplasms. International Journal of Molecular Sciences, 22(7), 3371. https://doi.org/10.3390/ijms22073371