
Articular Chondrocyte Phenotype Regulation through the Cytoskeleton and the Signaling Processes That Originate from or Converge on the Cytoskeleton: Towards a Novel Understanding of the Intersection between Actin Dynamics and Chondrogenic Function




Abstract
1. Introduction
2. Actin Isoforms, Classes, Nucleation, and Polymerization
3. Actin Stress Fiber Classification Systems
4. Regulation of Actin Dynamics
5. Cytoskeletal Proteins and Associated Signaling Pathways in Passaging-Induced Chondrocyte Dedifferentiation
6. Stress Fibers in Chondrocyte Differentiation and Dedifferentiation
7. Cytoskeletal Differences between Healthy and OA Chondrocytes

8. Pro-Inflammatory Cytokine Signaling Associated with the Chondrocyte Actin Cytoskeleton
9. Growth Factor Signaling Associated with the Chondrocyte Actin Cytoskeleton
10. TGF-β-Induced Stress Fiber Formation and Cell Stiffening
11. Cell Stiffness Is Related to the Effects of Stress Fiber Distribution vs. Amount
12. Regulation of TGF-β-Dependent SZP Production through the Level of Actin Polymerization
13. Fibrogenic vs. Chondrogenic Chondrocyte Expression Profiles and Catabolic Cleavage Fragment Formation Associated with the Chondrocyte Actin Cytoskeleton
14. Regulation of Chondrogenic SOX9 Expression and Phosphorylation
15. The Role of the Cytoskeleton in Studies Pertaining to 2D vs. 3D Culture Dimensionality, the Microenvironment, and Osmoregulation
16. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 2D | Two-dimensional |
| 3D | Three-dimensional |
| α-SMA | Alpha smooth muscle actin |
| AC | Articular cartilage |
| ACVRL1/ALK1 | Activin A receptor type II-like kinase 1 |
| ADAMTS | A disintegrin and metalloproteinase with thrombospondin motifs |
| ADF | Actin-depolymerizing factor |
| AIP1 | Actin-interacting protein 1 |
| ALK5 | Activin receptor-like kinase 5 |
| Arp2/3 | Actin-related protein complex 2/3 |
| bCHs | Bovine chondrocytes |
| BMP | Bone morphogenetic protein |
| CAP | Cyclase-associated protein |
| CAT | Chloramphenicol acetyltransferase |
| CBP | CREB-binding protein |
| cCHs | Chicken chondrocytes |
| caCHs | Canine chondrocytes |
| CCN2/CTGF | Connective tissue growth factor |
| CECs | Corneal endothelial cells |
| CH(s) | Chondrocyte(s) |
| CHX | Cycloheximide |
| CIN | Chronophin |
| COL1/2/3/10 | Type I/ II/ III/ X collagen |
| cSOX9 | Chicken SRY-box transcription factor 9 |
| DEX | Dexamethasone |
| DRF1 | Dehydration responsive factor 1 |
| ECM | Extracellular matrix |
| EGF | Epidermal growth factor |
| EGFR | Epidermal growth factor receptor |
| ENaC | Epithelial sodium channel |
| ERK | Extracellular signal-regulated kinase |
| ERM | Ezrin/radixin/moesin |
| F-actin | Filamentous actin |
| FAK | Focal adhesion kinase |
| FAs | Focal adhesions |
| FGF-1 / 2 | Fibroblast growth factor 1 / 2 |
| FGFR1 / 2 / 3 / 4 | Fibroblast growth factor receptor 1 / 2 / 3 / 4 |
| G-actin | Globular actin |
| GAG | Glycosaminoglycan |
| GAP | GTPase-activating protein |
| gCHs | Goat chondrocytes |
| G-CSF | Granulocyte colony-stimulating factor |
| GEF | Guanine-nucleotide exchange factor |
| GM-CSF | Granulocyte-macrophage colony-stimulating factor |
| GRAF | GTPase regulator associated with FAK |
| GSK-3 | Glycogen synthase kinase 3 |
| hCHs | Human chondrocytes |
| hSOX9 | Human SRY-box transcription factor 9 |
| HSP-27 | Heat shock protein 27 |
| IFN-γ | Interferon gamma |
| IGF-1 | Insulin growth factor 1 |
| IL-1(α/β) | Interleukin 1 (alpha/beta) |
| IL-6 | Interleukin 6 |
| IL-8 | Interleukin 8 |
| JNK | JUN N-terminal kinase |
| LIMK | LIM domain kinase |
| MAL | Megakaryoblastic leukemia 1 |
| MAPK | Mitogen-activated protein kinase |
| MAPKAPK2 | Mitogen-activated protein kinase-activated protein kinase 2 |
| mCHs | Murine chondrocytes |
| MCPJ | Metacarpophalangeal joints |
| mDia1/DIAPH1/DRF1 | Mammalian Diaphanous 1 |
| MEK1/2 | Mitogen-activated protein kinase / ERK kinase |
| MKK | Mitogen-activated protein kinase kinase |
| MLC | Myosin light chain |
| MLCK | Myosin light chain kinase |
| MLCP | Myosin light chain phosphatase |
| MMP | Matrix metalloproteinase |
| MPs | Micropattern |
| MRCKα | Myotonic dystrophy-related Cdc42-binding kinase alpha |
| mRNA | Messenger ribonucleic acid |
| MRTF | Myocardin-related transcription factor |
| MSCs | Mesenchymal stem cells |
| NMMII(A/B) | Non-muscle myosin II A/B |
| NO | Nitric oxide |
| NPFs | Nucleation-promoting factors |
| OA | Osteoarthritis / Osteoarthritic |
| OSM | Osmo-sensing scaffold for MEKK3 |
| P | Passage |
| Pa | Pascal |
| PAA | Polyacrylamide |
| PAK | p21-activated kinase |
| pCHs | Pig / porcine chondrocytes |
| PDMS | Polydimethylsiloxane |
| PGE2 | Prostaglandin E2 |
| PI3K | Phosphatidylinositol 3-kinase |
| PI4P | Phosphatidylinositol-4-phosphate |
| PIP2 | Phosphatidylinositol (4,5)-bisphosphate |
| PIP3 | Phosphatidylinositol (3,4,5)-trisphosphate |
| PIP5K | Phosphatidylinositol 5-kinase |
| PKA | Protein kinase A |
| PKC | Protein kinase C |
| PKD | Protein kinase D |
| PLCγ | Phospholipase C gamma |
| PP1 | Protein phosphatase type 1 |
| PP2 | 4-amino-5-(4-chlorophenyl)-7-(dimethylethyl)pyrazolo[3,4-d]pyrimidine |
| PP2A | Protein phosphatase type 2A |
| PP2B | Protein phosphatase type 2B |
| PRG4 | Proteoglycan 4 |
| PSGAP | PH- and SH3-domain-containing RhoGAP protein |
| PTHrP | Parathyroid hormone-related protein |
| PTOA | Post-traumatic osteoarthritis |
| PVA | Poly(vinyl alcohol) |
| rabCHs | Rabbit chondrocytes |
| rCHs | Rat chondrocytes |
| RCS | Rat chondrosarcoma |
| ROCK | Rho-associated kinase |
| RVD | Regulatory volume decrease |
| SAPK | Stress-activated protein kinase |
| SF | Stress fiber |
| siRNA | Small interfering ribonucleic acid |
| SMC | Smooth muscle cell |
| SOX4 / 5 / 6 / 9 | SRY-box transcription factor 4 / 5 / 6 / 9 |
| SRF | Serum response factor |
| SSH | Slingshot |
| SZP | Superficial zone protein |
| TESK1 / 2 | Testicular protein kinase 1 / 2 |
| TGF-α | Transforming growth factor alpha |
| TGF-β(1) | Transforming growth factor beta (1) |
| TNF-α | Tumor necrosis factor alpha |
| TNFR-1 | Tumor necrosis factor receptor 1 |
| TonEBP | Tonicity-responsive enhancer binding protein |
| TRPV4 | Vanilloid type 4 channel |
| VASP | Vasodilator-stimulated phosphoprotein |
| VEGF | Vascular endothelial growth factor |
| YAP | Yes-associated protein |
References
- Sun, Y.; Chen, C.S.; Fu, J. Forcing stem cells to behave: A biophysical perspective of the cellular microenvironment. Annu. Rev. Biophys. 2012, 41, 519–542. [Google Scholar] [CrossRef]
- Mammoto, T.; Ingber, D.E. Mechanical control of tissue and organ development. Development 2010, 137, 1407–1420. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, M.A.; Chen, C.S. Mechanotransduction in development: A growing role for contractility. Nat. Rev. Mol. Cell Biol. 2009, 10, 34–43. [Google Scholar] [CrossRef]
- Ingber, D.E. Mechanobiology and diseases of mechanotransduction. Ann. Med. 2003, 35, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Rolauffs, B.; Williams, J.M.; Grodzinsky, A.J.; Kuettner, K.E.; Cole, A.A. Distinct horizontal patterns in the spatial organization of superficial zone chondrocytes of human joints. J. Struct. Biol. 2008, 162, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Aicher, W.K.; Rolauffs, B. The spatial organisation of joint surface chondrocytes: Review of its potential roles in tissue functioning, disease and early, preclinical diagnosis of osteoarthritis. Ann. Rheum. Dis. 2014, 73, 645–653. [Google Scholar] [CrossRef]
- Felka, T.; Rothdiener, M.; Bast, S.; Uynuk-Ool, T.; Zouhair, S.; Ochs, B.G.; De Zwart, P.; Stoeckle, U.; Aicher, W.K.; Hart, M.L.; et al. Loss of spatial organization and destruction of the pericellular matrix in early osteoarthritis in vivo and in a novel in vitro methodology. Osteoarthr. Cartil. 2016, 24, 1200–1209. [Google Scholar] [CrossRef] [PubMed]
- Rolauffs, B.; Rothdiener, M.; Bahrs, C.; Badke, A.; Weise, K.; Kuettner, K.E.; Kurz, B.; Aurich, M.; Grodzinsky, A.J.; Aicher, W.K. Onset of preclinical osteoarthritis: The angular spatial organization permits early diagnosis. Arthritis. Rheum. 2011, 63, 1637–1647. [Google Scholar] [CrossRef]
- Rolauffs, B.; Williams, J.M.; Aurich, M.; Grodzinsky, A.J.; Kuettner, K.E.; Cole, A.A. Proliferative remodeling of the spatial organization of human superficial chondrocytes distant from focal early osteoarthritis. Arthritis. Rheum. 2010, 62, 489–498. [Google Scholar]
- Meinhardt, M.; Lück, S.; Martin, P.; Felka, T.; Aicher, W.; Rolauffs, B.; Schmidt, V. Modeling chondrocyte patterns by elliptical cluster processes. J. Struct. Biol. 2012, 177, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Tschaikowsky, M.; Selig, M.; Brander, S.; Balzer, B.N.; Hugel, T.; Rolauffs, B. Proof-of-concept for the detection of early osteoarthritis pathology by clinically applicable endomicroscopy and quantitative AI-supported optical biopsy. Osteoarthr. Cartil. 2021, 29, 269–279. [Google Scholar] [CrossRef]
- Kuettner, K.E.; Aydelotte, M.B.; Thonar, E.J. Articular cartilage matrix and structure: A minireview. J. Rheumatol. Suppl. 1991, 27, 46–48. [Google Scholar]
- Singh, P.; Marcu, K.B.; Goldring, M.B.; Otero, M. Phenotypic instability of chondrocytes in osteoarthritis: On a path to hypertrophy. Ann. N. Y. Acad. Sci. 2019, 1442, 17–34. [Google Scholar] [CrossRef]
- Woolf, A.D.; Pfleger, B. Burden of major musculoskeletal conditions. Bull. World. Health. Organ. 2003, 81, 646–656. [Google Scholar] [PubMed]
- Felson, D.T. The epidemiology of knee osteoarthritis: Results from the Framingham Osteoarthritis Study. Semin. Arthritis. Rheum. 1990, 20, 42–50. [Google Scholar] [CrossRef]
- Brown, T.D.; Johnston, R.C.; Saltzman, C.L.; Marsh, J.L.; Buckwalter, J.A. Posttraumatic osteoarthritis: A first estimate of incidence, prevalence, and burden of disease. J. Orthop. Trauma. 2006, 20, 739–744. [Google Scholar] [CrossRef]
- van der Kraan, P.M.; van den Berg, W.B. Chondrocyte hypertrophy and osteoarthritis: Role in initiation and progression of cartilage degeneration? Osteoarthr. Cartil. 2012, 20, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Lv, M.; Zhou, Y.; Polson, S.W.; Wan, L.Q.; Wang, M.; Han, L.; Wang, L.; Lu, X.L. Identification of Chondrocyte Genes and Signaling Pathways in Response to Acute Joint Inflammation. Sci. Rep. 2019, 9, 93. [Google Scholar] [CrossRef]
- Loeser, R.F. Aging and osteoarthritis: The role of chondrocyte senescence and aging changes in the cartilage matrix. Osteoarthr. Cartil. 2009, 17, 971–979. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Tambe, D.T.; Deng, L.; Yang, L. Biomechanical properties and mechanobiology of the articular chondrocyte. Am J Physiol. Cell. Physiol. 2013, 305, C1202–C1208. [Google Scholar] [CrossRef] [PubMed]
- Grad, S.; Eglin, D.; Alini, M.; Stoddart, M.J. Physical stimulation of chondrogenic cells in vitro: A review. Clin. Orthop. Relat. Res. 2011, 469, 2764–2772. [Google Scholar] [CrossRef]
- Selig, M.; Lauer, J.C.; Hart, M.L.; Rolauffs, B. Mechanotransduction and Stiffness-Sensing: Mechanisms and Opportunities to Control Multiple Molecular Aspects of Cell Phenotype as a Design Cornerstone of Cell-Instructive Biomaterials for Articular Cartilage Repair. Int. J. Mol. Sci. 2020, 21, 5399. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.R.; Jagga, S.; Lee, S.-S.; Nam, J.-S. Interplay between cartilage and subchondral bone contributing to pathogenesis of osteoarthritis. Int. J. Mol. Sci. 2013, 14, 19805–19830. [Google Scholar] [CrossRef] [PubMed]
- Barter, M.J.; Bui, C.; Young, D.A. Epigenetic mechanisms in cartilage and osteoarthritis: DNA methylation, histone modifications and microRNAs. Osteoarthr. Cartil. 2012, 20, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Bobick, B.E.; Chen, F.H.; Le, A.M.; Tuan, R.S. Regulation of the chondrogenic phenotype in culture. Birth. Defects. Res. C. Embryo. Today. 2009, 87, 351–371. [Google Scholar] [CrossRef]
- Graceffa, V.; Vinatier, C.; Guicheux, J.; Stoddart, M.; Alini, M.; Zeugolis, D.I. Chasing Chimeras—The elusive stable chondrogenic phenotype. Biomaterials 2019, 192, 199–225. [Google Scholar] [CrossRef]
- Aurich, M.; Hofmann, G.O.; Best, N.; Rolauffs, B. Induced Redifferentiation of Human Chondrocytes from Articular Cartilage Lesion in Alginate Bead Culture After Monolayer Dedifferentiation: An Alternative Cell Source for Cell-Based Therapies? Tissue. Eng. Part. A 2018, 24, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Aurich, M.; Hofmann, G.O.; Gras, F.; Rolauffs, B. Human osteochondritis dissecans fragment-derived chondrocyte characteristics ex vivo, after monolayer expansion-induced de-differentiation, and after re-differentiation in alginate bead culture. BMC Musculoskelet. Disord. 2018, 19, 168. [Google Scholar] [CrossRef]
- Behrendt, P.; Feldheim, M.; Preusse-Prange, A.; Weitkamp, J.T.; Haake, M.; Eglin, D.; Rolauffs, B.; Fay, J.; Seekamp, A.; Grodzinsky, A.J.; et al. Chondrogenic potential of IL-10 in mechanically injured cartilage and cellularized collagen ACI grafts. Osteoarthr. Cartil. 2018, 26, 264–275. [Google Scholar] [CrossRef]
- Lodish, H.; Berk, A.; Zipursky, S.L.; Matsudaira, P.; Baltimore, D.; Darnell, J. The Actin Cytoskeleton. In Molecular Cell Biology; W.H. Freeman: New York, NY, USA, 2000. [Google Scholar]
- Bearer, E.L. Role of actin polymerization in cell locomotion: Molecules and models. Am. J. Respir. Cell Mol. Biol. 1993, 8, 582–591. [Google Scholar] [CrossRef]
- Tojkander, S.; Gateva, G.; Lappalainen, P. Actin stress fibers—Assembly, dynamics and biological roles. J. Cell Sci. 2012, 125, 1855–1864. [Google Scholar] [CrossRef] [PubMed]
- Na, S.; Collin, O.; Chowdhury, F.; Tay, B.; Ouyang, M.; Wang, Y.; Wang, N. Rapid signal transduction in living cells is a unique feature of mechanotransduction. Proc. Natl. Acad. Sci. USA. 2008, 105, 6626–6631. [Google Scholar] [CrossRef] [PubMed]
- Vandekerckhove, J.; Weber, K. At least six different actins are expressed in a higher mammal: An analysis based on the amino acid sequence of the amino-terminal tryptic peptide. J. Mol. Biol. 1978, 126, 783–802. [Google Scholar] [CrossRef]
- Khaitlina, S.Y. Functional specificity of actin isoforms. Int. Rev. Cytol. 2001, 202, 35–98. [Google Scholar]
- Dugina, V.; Zwaenepoel, I.; Gabbiani, G.; Clement, S.; Chaponnier, C. Beta and gamma-cytoplasmic actins display distinct distribution and functional diversity. J. Cell Sci. 2009, 122, 2980–2988. [Google Scholar] [CrossRef]
- Firat-Karalar, E.N.; Welch, M.D. New mechanisms and functions of actin nucleation. Curr. Opin. Cell. Biol. 2011, 23, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Graceffa, P.; Dominguez, R. Crystal structure of monomeric actin in the ATP state. Structural basis of nucleotide-dependent actin dynamics. J. Biol. Chem. 2003, 278, 34172–34180. [Google Scholar] [CrossRef]
- Pollard, T.D.; Cooper, J.A. Actin, a central player in cell shape and movement. Science 2009, 326, 1208–1212. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, R.; Holmes, K.C. Actin structure and function. Annu. Rev. Biophys. 2011, 40, 169–186. [Google Scholar] [CrossRef] [PubMed]
- Cramer, L.P.; Siebert, M.; Mitchison, T.J. Identification of novel graded polarity actin filament bundles in locomoting heart fibroblasts: Implications for the generation of motile force. J. Cell. Biol. 1997, 136, 1287–1305. [Google Scholar] [CrossRef]
- Lazarides, E.; Burridge, K. Alpha-actinin: Immunofluorescent localization of a muscle structural protein in nonmuscle cells. Cell 1975, 6, 289–298. [Google Scholar] [CrossRef]
- Wang, K.; Ash, J.F.; Singer, S.J. Filamin, a new high-molecular-weight protein found in smooth muscle and non-muscle cells. Proc. Natl. Acad. Sci. USA 1975, 72, 4483–4486. [Google Scholar] [CrossRef] [PubMed]
- Weber, K.; Groeschel-Stewart, U. Antibody to myosin: The specific visualization of myosin-containing filaments in nonmuscle cells. Proc. Natl. Acad. Sci. USA 1974, 71, 4561–4564. [Google Scholar] [CrossRef] [PubMed]
- Kovac, B.; Teo, J.L.; Makela, T.P.; Vallenius, T. Assembly of non-contractile dorsal stress fibers requires alpha-actinin-1 and Rac1 in migrating and spreading cells. J. Cell. Sci. 2013, 126, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Hotulainen, P.; Lappalainen, P. Stress fibers are generated by two distinct actin assembly mechanisms in motile cells. J. Cell. Biol. 2006, 173, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Burnette, D.T.; Shao, L.; Ott, C.; Pasapera, A.M.; Fischer, R.S.; Baird, M.A.; Der Loughian, C.; Delanoe-Ayari, H.; Paszek, M.J.; Davidson, M.W.; et al. A contractile and counterbalancing adhesion system controls the 3D shape of crawling cells. J. Cell Biol. 2014, 205, 83–96. [Google Scholar] [CrossRef]
- Maninová, M.; Vomastek, T.T.; Maninova, M.; Vomastek, T.T. Dorsal stress fibers, transverse actin arcs, and perinuclear actin fibers form an interconnected network that induces nuclear movement in polarizing fibroblasts. FEBS J. 2016, 283, 3676–3693. [Google Scholar] [CrossRef]
- Pellegrin, S.; Mellor, H. Actin stress fibres. J. Cell Sci. 2007, 120, 3491–3499. [Google Scholar] [CrossRef]
- Wood, S.T.; Dean, B.C.; Dean, D. A computational approach to understand phenotypic structure and constitutive mechanics relationships of single cells. Ann. Biomed. Eng. 2013, 41, 630–644. [Google Scholar] [CrossRef]
- Small, J.V.; Rottner, K.; Kaverina, I.; Anderson, K.I. Assembling an actin cytoskeleton for cell attachment and movement. Biochim. Biophys. Acta. 1998, 1404, 271–281. [Google Scholar] [CrossRef]
- Vallenius, T. Actin stress fibre subtypes in mesenchymal-migrating cells. Open. Biol. 2013, 3, 130001. [Google Scholar] [CrossRef] [PubMed]
- Kassianidou, E.; Kumar, S. A biomechanical perspective on stress fiber structure and function. Biochim. Biophys. Acta. 2015, 1853, 3065–3074. [Google Scholar] [CrossRef]
- Lee, S.; Kumar, S. Actomyosin stress fiber mechanosensing in 2D and 3D. F1000Research 2016, 5, 2261. [Google Scholar] [CrossRef]
- Burridge, K. Substrate adhesions in normal and transformed fibroblasts: Organization and regulation of cytoskeletal, membrane and extracellular matrix components at focal contacts. Cancer. Rev. 1986, 4, 18–78. [Google Scholar]
- Livne, A.; Geiger, B. The inner workings of stress fibers—From contractile machinery to focal adhesions and back. J. Cell. Sci. 2016, 129, 1293–1304. [Google Scholar] [CrossRef] [PubMed]
- Heath, J.P.; Dunn, G.A. Cell to substratum contacts of chick fibroblasts and their relation to the microfilament system. A correlated interference-reflexion and high-voltage electron-microscope study. J. Cell Sci. 1978, 29, 197–212. [Google Scholar]
- Campanale, J.P.; Sun, T.Y.; Montell, D.J. Development and dynamics of cell polarity at a glance. J. Cell Sci. 2017, 130, 1201–1207. [Google Scholar] [CrossRef]
- Tee, Y.H.; Shemesh, T.; Thiagarajan, V.; Hariadi, R.F.; Anderson, K.L.; Page, C.; Volkmann, N.; Hanein, D.; Sivaramakrishnan, S.; Kozlov, M.M.; et al. Cellular chirality arising from the self-organization of the actin cytoskeleton. Nat. Cell Biol. 2015, 17, 445–457. [Google Scholar] [CrossRef]
- Maninova, M.; Caslavsky, J.; Vomastek, T. The assembly and function of perinuclear actin cap in migrating cells. Protoplasma 2017, 254, 1207–1218. [Google Scholar] [CrossRef]
- Chang, C.W.; Kumar, S. Differential contributions of nonmuscle myosin II isoforms and functional domains to stress fiber mechanics. Sci. Rep. 2015, 5, 13736. [Google Scholar] [CrossRef]
- Totsukawa, G.; Yamakita, Y.; Yamashiro, S.; Hartshorne, D.J.; Sasaki, Y.; Matsumura, F. Distinct roles of ROCK (Rho-kinase) and MLCK in spatial regulation of MLC phosphorylation for assembly of stress fibers and focal adhesions in 3T3 fibroblasts. J. Cell Biol. 2000, 150, 797–806. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Maxwell, I.Z.; Heisterkamp, A.; Polte, T.R.; Lele, T.P.; Salanga, M.; Mazur, E.; Ingber, D.E. Viscoelastic retraction of single living stress fibers and its impact on cell shape, cytoskeletal organization, and extracellular matrix mechanics. Biophys. J. 2006, 90, 3762–3773. [Google Scholar] [CrossRef]
- Tanner, K.; Boudreau, A.; Bissell, M.J.; Kumar, S. Dissecting regional variations in stress fiber mechanics in living cells with laser nanosurgery. Biophys. J. 2010, 99, 2775–2783. [Google Scholar] [CrossRef]
- Chang, C.W.; Kumar, S. Vinculin tension distributions of individual stress fibers within cell-matrix adhesions. J. Cell Sci. 2013, 126, 3021–3030. [Google Scholar] [CrossRef] [PubMed]
- Albert, P.J.; Schwarz, U.S. Dynamics of cell shape and forces on micropatterned substrates predicted by a cellular Potts model. Biophys. J. 2014, 106, 2340–2352. [Google Scholar] [CrossRef]
- Kumar, A.; Ouyang, M.; Van den Dries, K.; McGhee, E.J.; Tanaka, K.; Anderson, M.D.; Groisman, A.; Goult, B.T.; Anderson, K.I.; Schwartz, M.A. Talin tension sensor reveals novel features of focal adhesion force transmission and mechanosensitivity. J. Cell Biol. 2016, 213, 371–383. [Google Scholar] [CrossRef]
- Kovács, M.; Wang, F.; Hu, A.; Zhang, Y.; Sellers, J.R. Functional divergence of human cytoplasmic myosin II: Kinetic characterization of the non-muscle IIA isoform. J. Biol. Chem. 2003, 278, 38132–38140. [Google Scholar] [CrossRef]
- Wang, F.; Kovacs, M.; Hu, A.; Limouze, J.; Harvey, E.V.; Sellers, J.R. Kinetic mechanism of non-muscle myosin IIB: Functional adaptations for tension generation and maintenance. J. Biol. Chem. 2003, 278, 27439–27448. [Google Scholar] [CrossRef]
- Pritchard, S.; Guilak, F. Effects of interleukin-1 on calcium signaling and the increase of filamentous actin in isolated and in situ articular chondrocytes. Arthritis. Rheum. 2006, 54, 2164–2174. [Google Scholar] [CrossRef] [PubMed]
- Parreno, J.; Nabavi Niaki, M.; Andrejevic, K.; Jiang, A.; Wu, P.H.; Kandel, R.A. Interplay between cytoskeletal polymerization and the chondrogenic phenotype in chondrocytes passaged in monolayer culture. J. Anat. 2017, 230, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Lassar, A.B. The Transcriptional Activity of Sox9 in Chondrocytes Is Regulated by RhoA Signaling and Actin Polymerization. Mol. Cell Biol. 2009, 29, 4262–4273. [Google Scholar] [CrossRef]
- Heasman, S.J.; Ridley, A.J. Mammalian Rho GTPases: New insights into their functions from in vivo studies. Nat. Rev. Mol. Cell Biol. 2008, 9, 690–701. [Google Scholar] [CrossRef] [PubMed]
- Burridge, K.; Wittchen, E.S. The tension mounts: Stress fibers as force-generating mechanotransducers. J. Cell Biol. 2013, 200, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Leung, T.; Chen, X.Q.; Manser, E.; Lim, L. The p160 RhoA-binding kinase ROK alpha is a member of a kinase family and is involved in the reorganization of the cytoskeleton. Mol. Cell Biol. 1996, 16, 5313–5327. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, N.; Madaule, P.; Reid, T.; Ishizaki, T.; Watanabe, G.; Kakizuka, A.; Saito, Y.; Nakao, K.; Jockusch, B.M.; Narumiya, S. p140mDia, a mammalian homolog of Drosophila diaphanous, is a target protein for Rho small GTPase and is a ligand for profilin. EMBO J. 1997, 16, 3044–3056. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.S.; Wynne, J.; Treisman, R. The Rho family GTPases RhoA, Rac1, and CDC42Hs regulate transcriptional activation by SRF. Cell 1995, 81, 1159–1170. [Google Scholar] [CrossRef]
- Miralles, F.; Posern, G.; Zaromytidou, A.-I.; Treisman, R. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell 2003, 113, 329–342. [Google Scholar] [CrossRef]
- Tominaga, T.; Sahai, E.; Chardin, P.; McCormick, F.; Courtneidge, S.A.; Alberts, A.S. Diaphanous-related formins bridge Rho GTPase and Src tyrosine kinase signaling. Mol. Cell 2000, 5, 13–25. [Google Scholar] [CrossRef]
- Higashida, C.; Kiuchi, T.; Akiba, Y.; Mizuno, H.; Maruoka, M.; Narumiya, S.; Mizuno, K.; Watanabe, N. F-and G-actin homeostasis regulates mechanosensitive actin nucleation by formins. Nat. Cell Biol. 2013, 15, 395–405. [Google Scholar] [CrossRef]
- Mizuno, H.; Tanaka, K.; Yamashiro, S.; Narita, A.; Watanabe, N. Helical rotation of the diaphanous-related formin mDia1 generates actin filaments resistant to cofilin. Proc. Natl. Acad. Sci. USA 2018, 115, E5000–E5007. [Google Scholar] [CrossRef]
- Hu, J.; Lu, J.; Lian, G.; Ferland, R.J.; Dettenhofer, M.; Sheen, V.L. Formin 1 and filamin B physically interact to coordinate chondrocyte proliferation and differentiation in the growth plate. Hum. Mol. Genet. 2014, 23, 4663–4673. [Google Scholar] [CrossRef] [PubMed]
- Vartiainen, M.K.; Guettler, S.; Larijani, B.; Treisman, R. Nuclear Actin Regulates Dynamic Subcellular Localization and Activity of the SRF Cofactor MAL. Science (80- ) 2007, 316, 1749–1752. [Google Scholar] [CrossRef] [PubMed]
- Gau, D.; Roy, P. SRF’ing and SAP’ing—The role of MRTF proteins in cell migration. J. Cell Sci. 2018, 131, jcs218222. [Google Scholar] [CrossRef]
- Parreno, J.; Raju, S.; Niaki, M.N.; Andrejevic, K.; Jiang, A.; Delve, E.; Kandel, R. Expression of type I collagen and tenascin C is regulated by actin polymerization through MRTF in dedifferentiated chondrocytes. FEBS Lett. 2014, 588, 3677–3684. [Google Scholar] [CrossRef]
- Maekawa, M.; Ishizaki, T.; Boku, S.; Watanabe, N.; Fujita, A.; Iwamatsu, A.; Obinata, T.; Ohashi, K.; Mizuno, K.; Narumiya, S. Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science (80- ) 1999, 285, 895–898. [Google Scholar] [CrossRef] [PubMed]
- Narumiya, S.; Tanji, M.; Ishizaki, T. Rho signaling, ROCK and mDia1, in transformation, metastasis and invasion. Cancer Metastasis Rev. 2009, 28, 65–76. [Google Scholar] [CrossRef]
- Sumi, T.; Matsumoto, K.; Takai, Y.; Nakamura, T. Cofilin phosphorylation and actin cytoskeletal dynamics regulated by rho- and Cdc42-activated LIM-kinase 2. J. Cell Biol. 1999, 147, 1519–1532. [Google Scholar] [CrossRef]
- Arber, S.; Barbayannis, F.A.; Hanser, H.; Schnelder, C.; Stanyon, C.A.; Bernards, O.; Caroni, P. Regulation of actin dynamics through phosphorylation of cofilin by LIM- kinase. Nature 1998, 393, 805–809. [Google Scholar] [CrossRef]
- Yang, N.; Higuchi, O.; Ohashi, K.; Nagata, K.; Wada, A.; Kangawa, K.; Nishida, E.; Mizuno, K. Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature 1998, 393, 809–812. [Google Scholar] [CrossRef]
- Gohla, A.; Bokoch, G.M. 14-3-3 Regulates Actin Dynamics by Stabilizing Phosphorylated Cofilin. Curr. Biol. 2002, 12, 1704–1710. [Google Scholar] [CrossRef]
- Nagata-Ohashi, K.; Ohta, Y.; Goto, K.; Chiba, S.; Mori, R.; Nishita, M.; Ohashi, K.; Kousaka, K.; Iwamatsu, A.; Niwa, R.; et al. A pathway of neuregulin-induced activation of cofilin-phosphatase Slingshot and cofilin in lamellipodia. J. Cell Biol. 2004, 165, 465–471. [Google Scholar] [CrossRef]
- Sumi, T.; Matsumoto, K.; Shibuya, A.; Nakamura, T. Activation of LIM kinases by myotonic dystrophy kinase-related Cdc42-binding kinase alpha. J. Biol. Chem. 2001, 276, 23092–23096. [Google Scholar] [CrossRef]
- Sumi, T.; Matsumoto, K.; Nakamura, T. Specific activation of LIM kinase 2 via phosphorylation of threonine 505 by ROCK, a Rho-dependent protein kinase. J. Biol. Chem. 2001, 276, 670–676. [Google Scholar] [CrossRef]
- Mackay, D.J.; Esch, F.; Furthmayr, H.; Hall, A. Rho- and rac-dependent assembly of focal adhesion complexes and actin filaments in permeabilized fibroblasts: An essential role for ezrin/radixin/moesin proteins. J. Cell Biol. 1997, 138, 927–938. [Google Scholar] [CrossRef]
- Yonemura, S.; Matsui, T.; Tsukita, S.; Tsukita, S. Rho-dependent and -independent activation mechanisms of ezrin/radixin/moesin proteins: An essential role for polyphosphoinositides in vivo. J. Cell Sci. 2002, 115, 2569–2580. [Google Scholar]
- Song, Y.; Wong, C.; Chang, D.D. Overexpression of wild-type RhoA produces growth arrest by disrupting actin cytoskeleton and microtubules. J. Cell Biochem. 2000, 80, 229–240. [Google Scholar] [CrossRef]
- Yin, L.-M.; Duan, T.-T.; Ulloa, L.; Yang, Y.-Q. Ezrin orchestrates signal transduction in airway cells. Rev. Physiol. Biochem. Pharmacol. 2018, 174, 1–23. [Google Scholar]
- Riento, K.; Ridley, A.J. Rocks: Multifunctional kinases in cell behaviour. Nat. Rev. Mol. Cell. Biol. 2003, 4, 446–456. [Google Scholar] [CrossRef]
- Kawano, Y.; Fukata, Y.; Oshiro, N.; Amano, M.; Nakamura, T.; Ito, M.; Matsumura, F.; Inagaki, M.; Kaibuchi, K. Phosphorylation of myosin-binding subunit (MBS) of myosin phosphatase by Rho-kinase in vivo. J. Cell Biol. 1999, 147, 1023–1038. [Google Scholar] [CrossRef]
- Pollard, T.D. Regulation of actin filament assembly by Arp2/3 complex and formins. Annu. Rev. Biophys. Biomol. Struct. 2007, 36, 451–477. [Google Scholar] [CrossRef]
- Landry, J.; Huot, J. Regulation of actin dynamics by stress-activated protein kinase 2 (SAPK2)-dependent phosphorylation of heat-shock protein of 27 kDa (Hsp27). Biochem. Soc. Symp. 1999, 64, 79–89. [Google Scholar]
- Lavoie, J.N.; Lambert, H.; Hickey, E.; Weber, L.A.; Landry, J. Modulation of cellular thermoresistance and actin filament stability accompanies phosphorylation-induced changes in the oligomeric structure of heat shock protein 27. Mol. Cell Biol. 1995, 15, 505–516. [Google Scholar] [CrossRef]
- Rousseau, S.; Houle, F.; Landry, J.; Huot, J. P38 MAP kinase activation by vascular endothelial growth factor mediates actin reorganization and cell migration in human endothelial cells. Oncogene 1997, 15, 2169–2177. [Google Scholar] [CrossRef]
- Graceffa, P. Hsp27-actin interaction. Biochem. Res. Int. 2011, 2011, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Han, J.; Sells, M.A.; Chernoff, J.; Knaus, U.G.; Ulevitch, R.J.; Bokoch, G.M. Rho Family GTPases Regulate p38 Mitogen-activated Protein Kinase through the Downstream Mediator Pak1. J. Biol. Biochem. 1995, 270, 23934–23936. [Google Scholar] [CrossRef]
- Kobayashi, M.; Nishita, M.; Mishima, T.; Ohashi, K.; Mizuno, K. MAPKAPK-2-mediated LIM-kinase activation is critical for VEGF-induced actin remodeling and cell migration. EMBO J. 2006, 25, 713–726. [Google Scholar] [CrossRef]
- Singh, S.; Powell, D.W.; Rane, M.J.; Millard, T.H.; Trent, J.O.; Pierce, W.M.; Klein, J.B.; Machesky, L.M.; McLeish, K.R. Identification of the p16-Arc subunit of the Arp 2/3 complex as a substrate of MAPK-activated protein kinase 2 by proteomic analysis. J. Biol. Chem. 2003, 278, 36410–36417. [Google Scholar] [CrossRef]
- Huang, C.; Jacobson, K.; Schaller, M.D. MAP kinases and cell migration. J. Cell Sci. 2004, 117, 4619–4628. [Google Scholar] [CrossRef]
- Han, Q.; Leng, J.; Bian, D.; Mahanivong, C.; Carpenter, K.A.; Pan, Z.K.; Han, J.; Huang, S. Rac1-MKK3-p38-MAPKAPK2 pathway promotes urokinase plasminogen activator mRNA stability in invasive breast cancer cells. J. Biol. Chem. 2002, 277, 48379–48385. [Google Scholar] [CrossRef]
- Cuenda, A.; Rousseau, S. p38 MAP-Kinases pathway regulation, function and role in human diseases. Biochim. Biophys. Acta. Mol. Cell Res. 2007, 1773, 1358–1375. [Google Scholar] [CrossRef]
- Corre, I.; Paris, F.; Huot, J. The p38 pathway, a major pleiotropic cascade that transduces stress and metastatic signals in endothelial cells. Oncotarget 2017, 8, 55684–55714. [Google Scholar] [CrossRef]
- Lamalice, L.; Houle, F.; Jourdan, G.; Huot, J. Phosphorylation of tyrosine 1214 on VEGFR2 is required for VEGF-induced activation of Cdc42 upstream of SAPK2/p38. Oncogene 2004, 23, 434–445. [Google Scholar] [CrossRef]
- Soga, N.; Namba, N.; McAllister, S.; Cornelius, L.; Teitelbaum, S.L.; Dowdy, S.F.; Kawamura, J.; Hruska, K.A. Rho family GTPases regulate VEGF-stimulated endothelial cell motility. Exp. Cell Res. 2001, 269, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Niwa, R.; Nagata-Ohashi, K.; Takeichi, M.; Mizuno, K.; Uemura, T. Control of actin reorganization by Slingshot, a family of phosphatases that dephosphorylate ADF/cofilin. Cell 2002, 108, 233–246. [Google Scholar] [CrossRef]
- Soosairajah, J.; Maiti, S.; Wiggan, O.; Sarmiere, P.; Moussi, N.; Sarcevic, B.; Sampath, R.; Bamburg, J.R.; Bernard, O. Interplay between components of a novel LIM kinase-slingshot phosphatase complex regulates cofilin. EMBO J. 2005, 24, 473–486. [Google Scholar] [CrossRef]
- Ambach, A.; Saunus, J.; Konstandin, M.; Wesselborg, S.; Meuer, S.C.; Samstag, Y. The serine phosphatases PP1 and PP2A associate with and activate the actin-binding protein cofilin in human T lymphocytes. Eur. J. Immunol. 2000, 30, 3422–3431. [Google Scholar] [CrossRef]
- Meberg, P.J.; Ono, S.; Minamide, L.S.; Takahashi, M.; Bamburg, J.R. Actin depolymerizing factor and cofilin phosphorylation dynamics: Response to signals that regulate neurite extension. Cell Motil. Cytoskeleton. 1998, 39, 172–190. [Google Scholar] [CrossRef]
- Zhan, Q.; Bamburg, J.R.; Badwey, J.A. Products of phosphoinositide specific phospholipase C can trigger dephosphorylation of cofilin in chemoattractant stimulated neutrophils. Cell Motil. Cytoskeleton. 2003, 54, 1–15. [Google Scholar] [CrossRef]
- Gohla, A.; Birkenfeld, J.; Bokoch, G.M. Chronophin, a novel HAD-type serine protein phosphatase, regulates cofilin-dependent actin dynamics. Nat. Cell Biol. 2005, 7, 21–29. [Google Scholar] [CrossRef]
- Aizawa, H.; Katadae, M.; Maruya, M.; Sameshima, M.; Murakami-Murofushi, K.; Yahara, I. Hyperosmotic stress-induced reorganization of actin bundles in Dictyostelium cells over-expressing cofilin. Genes Cells 1999, 4, 311–324. [Google Scholar] [CrossRef]
- Okada, K.; Obinata, T.; Abe, H. XAIP1: A Xenopus homologue of yeast actin interacting protein 1 (AIP1), which induces disassembly of actin filaments cooperatively with ADF/cofilin family proteins. J. Cell Sci. 1999, 112, 1553–1565. [Google Scholar] [PubMed]
- Rodal, A.A.; Tetreault, J.W.; Lappalainen, P.; Drubin, D.G.; Amberg, D.C. Aip1p interacts with cofilin to disassemble actin filaments. J. Cell Biol. 1999, 145, 1251–1264. [Google Scholar] [CrossRef] [PubMed]
- DesMarais, V.; Ghosh, M.; Eddy, R.; Condeelis, J. Cofilin takes the lead. J. Cell Sci. 2005, 118, 19–26. [Google Scholar] [CrossRef]
- Kueh, H.Y.; Charras, G.T.; Mitchison, T.J.; Brieher, W.M. Actin disassembly by cofilin, coronin, and Aip1 occurs in bursts and is inhibited by barbed-end cappers. J. Cell Biol. 2008, 182, 341–353. [Google Scholar] [CrossRef]
- Kotila, T.; Wioland, H.; Enkavi, G.; Kogan, K.; Vattulainen, I.; Jégou, A.; Romet-Lemonne, G.; Lappalainen, P. Mechanism of synergistic actin filament pointed end depolymerization by cyclase-associated protein and cofilin. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Shekhar, S.; Chung, J.; Kondev, J.; Gelles, J.; Goode, B.L. Synergy between Cyclase-associated protein and Cofilin accelerates actin filament depolymerization by two orders of magnitude. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Johnston, A.B.; Collins, A.; Goode, B.L. High-speed depolymerization at actin filament ends jointly catalysed by Twinfilin and Srv2/CAP. Nat. Cell Biol. 2015, 17, 1504–1511. [Google Scholar] [CrossRef]
- Hakala, M.; Wioland, H.; Tolonen, M.; Kotila, T.; Jegou, A.; Romet-Lemonne, G.; Lappalainen, P. Twinfilin uncaps filament barbed ends to promote turnover of lamellipodial actin networks. Nat. Cell Biol. 2021, 23, 147–159. [Google Scholar] [CrossRef]
- Allen, P.G.; Janmey, P.A. Gelsolin displaces phalloidin from actin filaments. A new fluorescence method shows that both Ca2+ and Mg2+ affect the rate at which gelsolin severs F-actin. J. Biol. Chem. 1994, 269, 32916–32923. [Google Scholar] [CrossRef]
- Sun, H.Q.; Yamamoto, M.; Mejillano, M.; Yin, H.L. Gelsolin, A Multifunctional Actin Regulatory Protein. J. Biol. Chem. 1999, 274, 33179–33183. [Google Scholar] [CrossRef]
- Janmey, P.A.; Stossel, T.P. Modulation of gelsolin function by phosphatidylinositol 4,5-bisphosphate. Nature 1987, 325, 362–364. [Google Scholar] [CrossRef]
- Patel, V.B.; Zhabyeyev, P.; Chen, X.; Wang, F.; Paul, M.; Fan, D.; McLean, B.A.; Basu, R.; Zhang, P.; Shah, S.; et al. PI3Kα-regulated gelsolin activity is a critical determinant of cardiac cytoskeletal remodeling and heart disease. Nat. Commun. 2018, 9, 5390. [Google Scholar] [CrossRef]
- Hartwig, J.H.; Bokoch, G.M.; Carpenter, C.L.; Janmey, P.A.; Taylor, L.A.; Toker, A.; Stossel, T.P. Thrombin receptor ligation and activated rac uncap actin filament barbed ends through phosphoinositide synthesis in permeabilized human platelets. Cell 1995, 82, 643–653. [Google Scholar] [CrossRef]
- Lind, S.E.; Janmey, P.A.; Chaponnier, C.; Herbert, T.J.; Stossel, T.P. Reversible binding of actin to gelsolin and profilin in human platelet extracts. J. Cell Biol. 1987, 105, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Weernink, P.A.O.; Meletiadis, K.; Hommeltenberg, S.; Hinz, M.; Ishihara, H.; Schmidt, M.; Jakobs, K.H. Activation of Type I Phosphatidylinositol 4-Phosphate 5-Kinase Isoforms by the Rho GTPases, RhoA, Rac1, and Cdc42. J. Biol. Chem. 2004, 279, 7840–7849. [Google Scholar] [CrossRef]
- Oude Weernink, P.A.; Schmidt, M.; Jakobs, K.H. Regulation and cellular roles of phosphoinositide 5-kinases. Eur. J. Pharmacol. 2004, 500, 87–99. [Google Scholar] [CrossRef]
- Czech, M.P. PIP2 and PIP3: Complex roles at the cell surface. Cell 2000, 100, 603–606. [Google Scholar] [CrossRef]
- Chou, J.; Stolz, D.B.; Burke, N.A.; Watkins, S.C.; Wells, A. Distribution of gelsolin and phosphoinositol 4,5-bisphosphate in lamellipodia during EGF-induced motility. Int. J. Biochem. Cell. Biol. 2002, 34, 776–790. [Google Scholar] [CrossRef]
- Tang, W.; Zhang, Y.; Xu, W.; Harden, T.K.; Sondek, J.; Sun, L.; Li, L.; Wu, D. A PLCβ/PI3Kγ-GSK3 Signaling Pathway Regulates Cofilin Phosphatase Slingshot2 and Neutrophil Polarization and Chemotaxis. Dev. Cell 2011, 21, 1038–1050. [Google Scholar] [CrossRef]
- Eiseler, T.; Döppler, H.; Yan, I.K.; Kitatani, K.; Mizuno, K.; Storz, P. Protein kinase D1 regulates cofilin-mediated F-actin reorganization and cell motility through slingshot. Nat. Cell Biol. 2009, 11, 545–556. [Google Scholar] [CrossRef]
- Peterburs, P.; Heering, J.; Link, G.; Pfizenmaier, K.; Olayioye, M.A.; Hausser, A. Protein kinase D regulates cell migration by direct phosphorylation of the cofilin phosphatase slingshot 1 like. Cancer. Res. 2009, 69, 5634–5638. [Google Scholar] [CrossRef]
- Hayakawa, K.; Tatsumi, H.; Sokabe, M. Actin filaments function as a tension sensor by tension-dependent binding of cofilin to the filament. J. Cell Biol. 2011, 195, 721–727. [Google Scholar] [CrossRef]
- Wioland, H.; Jegou, A.; Romet-Lemonne, G. Torsional stress generated by ADF/cofilin on cross-linked actin filaments boosts their severing. Proc. Natl. Acad. Sci. USA 2019, 116, 2595–2602. [Google Scholar] [CrossRef]
- Reymann, A.-C.; Boujemaa-Paterski, R.; Martiel, J.-L.; Guérin, C.; Cao, W.; Chin, H.F.; De La Cruz, E.M.; Théry, M.; Blanchoin, L. Actin Network Architecture Can Determine Myosin Motor Activity. Science (80- ) 2012, 336, 1310–1314. [Google Scholar] [CrossRef]
- Yamashiro, S.; Tanaka, S.; McMillen, L.M.; Taniguchi, D.; Vavylonis, D.; Watanabe, N. Myosin-dependent actin stabilization as revealed by single-molecule imaging of actin turnover. Mol. Biol. Cell 2018, 29, 1941–1947. [Google Scholar] [CrossRef] [PubMed]
- Amano, M.; Ito, M.; Fukata, Y.; Chihara, K.; Nakano, T.; Matsuura, Y.; Kaibuchi, K. Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase). J. Biol. Chem. 1996, 271, 20246–20249. [Google Scholar] [CrossRef]
- Chrzanowska-Wodnicka, M.; Burridge, K. Rho-stimulated contractility drives the formation of stress fibers and focal adhesions. J. Cell Biol. 1996, 133, 1403–1415. [Google Scholar] [CrossRef]
- Kimura, K.; Ito, M.; Amano, M.; Chihara, K.; Fukata, Y.; Nakafuku, M.; Yamamori, B.; Feng, J.; Nakano, T.; Okawa, K.; et al. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase). Science 1996, 273, 245–248. [Google Scholar] [CrossRef]
- Watanabe, N.; Kato, T.; Fujita, A.; Ishizaki, T.; Narumiya, S. Cooperation between mDia1 and ROCK in Rho-induced actin reorganization. Nat. Cell Biol. 1999, 1, 136–143. [Google Scholar] [CrossRef]
- Amano, M.; Nakayama, M.; Kaibuchi, K. Rho-kinase/ROCK: A key regulator of the cytoskeleton and cell polarity. Cytoskeleton 2010, 67, 545–554. [Google Scholar] [CrossRef]
- Oakes, P.W.; Beckham, Y.; Stricker, J.; Gardel, M.L. Tension is required but not sufficient for focal adhesion maturation without a stress fiber template. J. Cell Biol. 2012, 196, 363–374. [Google Scholar] [CrossRef]
- Schaller, M.D.; Hildebrand, J.D.; Shannon, J.D.; Fox, J.W.; Vines, R.R.; Parsons, J.T. Autophosphorylation of the focal adhesion kinase, pp125FAK, directs SH2-dependent binding of pp60src. Mol. Cell Biol. 1994, 14, 1680–1688. [Google Scholar] [CrossRef]
- Calalb, M.B.; Polte, T.R.; Hanks, S.K. Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity: A role for Src family kinases. Mol. Cell Biol. 1995, 15, 954–963. [Google Scholar] [CrossRef] [PubMed]
- Schaller, M.D. Cellular functions of FAK kinases: Insight into molecular mechanisms and novel functions. J. Cell Sci. 2010, 123, 1007–1013. [Google Scholar] [CrossRef]
- Tomar, A.; Schlaepfer, D.D. Focal adhesion kinase: Switching between GAPs and GEFs in the regulation of cell motility. Curr. Opin. Cell. Biol. 2009, 21, 676–683. [Google Scholar] [CrossRef]
- Ward, Y.; Yap, S.-F.; Ravichandran, V.; Matsumura, F.; Ito, M.; Spinelli, B.; Kelly, K. The GTP binding proteins Gem and Rad are negative regulators of the Rho-Rho kinase pathway. J. Cell Biol. 2002, 157, 291–302. [Google Scholar] [CrossRef]
- Riento, K.; Guasch, R.M.; Garg, R.; Jin, B.; Ridley, A.J. RhoE binds to ROCK I and inhibits downstream signaling. Mol. Cell. Biol. 2003, 23, 4219–4229. [Google Scholar] [CrossRef]
- Huang, X.; Yang, N.; Fiore, V.F.; Barker, T.H.; Sun, Y.; Morris, S.W.; Ding, Q.; Thannickal, V.J.; Zhou, Y. Matrix stiffness-induced myofibroblast differentiation is mediated by intrinsic mechanotransduction. Am. J. Respir. Cell Mol. Biol. 2012, 47, 340–348. [Google Scholar] [CrossRef]
- Guilluy, C.; Swaminathan, V.; Garcia-Mata, R.; O’Brien, E.T.; Superfine, R.; Burridge, K. The Rho GEFs LARG and GEF-H1 regulate the mechanical response to force on integrins. Nat. Cell Biol. 2011, 13, 722–727. [Google Scholar] [CrossRef]
- Krendel, M.; Zenke, F.T.; Bokoch, G.M. Nucleotide exchange factor GEF-H1 mediates cross-talk between microtubules and the actin cytoskeleton. Nat. Cell Biol. 2002, 4, 294–301. [Google Scholar] [CrossRef]
- Estrada, L.; Caron, E.; Gorski, J.L. Fgd1, the Cdc42 guanine nucleotide exchange factor responsible for faciogenital dysplasia, is localized to the subcortical actin cytoskeleton and Golgi membrane. Hum. Mol. Genet. 2001, 10, 485–495. [Google Scholar] [CrossRef]
- Pasteris, N.G.; Cadle, A.; Logie, L.J.; Porteous, M.E.; Schwartz, C.E.; Stevenson, R.E.; Glover, T.W.; Wilroy, R.S.; Gorski, J.L. Isolation and characterization of the faciogenital dysplasia (Aarskog-Scott syndrome) gene: A putative Rho/Rac guanine nucleotide exchange factor. Cell 1994, 79, 669–678. [Google Scholar] [CrossRef]
- Gorski, J.L.; Estrada, L.; Hu, C.; Liu, Z. Skeletal-specific expression of Fgd1 during bone formation and skeletal defects in faciogenital dysplasia (FGDY; Aarskog syndrome). Dev. Dyn. 2000, 218, 573–586. [Google Scholar] [CrossRef]
- Benya, P.D.; Padilla, S.R.; Nimni, M.E. Independent regulation of collagen types by chondrocytes during the loss of differentiated function in culture. Cell 1978, 15, 1313–1321. [Google Scholar] [CrossRef]
- Darling, E.M.; Pritchett, P.E.; Evans, B.A.; Superfine, R.; Zauscher, S.; Guilak, F. Mechanical properties and gene expression of chondrocytes on micropatterned substrates following dedifferentiation in monolayer. Cell Mol. Bioeng. 2009, 2, 395–404. [Google Scholar] [CrossRef]
- Kang, S.-W.; Yoo, S.P.; Kim, B.-S. Effect of chondrocyte passage number on histological aspects of tissue-engineered cartilage. Biomed. Mater. Eng. 2007, 17, 269–276. [Google Scholar]
- Schulze-Tanzil, G.; de Souza, P.; Villegas Castrejon, H.; John, T.; Merker, H.J.; Scheid, A.; Shakibaei, M. Redifferentiation of dedifferentiated human chondrocytes in high-density cultures. Cell Tissue. Res. 2002, 308, 371–379. [Google Scholar]
- Sasazaki, Y.; Seedhom, B.B.; Shore, R. Morphology of the bovine chondrocyte and of its cytoskeleton in isolation and in situ: Are chondrocytes ubiquitously paired through the entire layer of articular cartilage? Rheumatology 2008, 47, 1641–1646. [Google Scholar] [CrossRef]
- Shin, H.; Lee, M.N.; Choung, J.S.; Kim, S.; Choi, B.H.; Noh, M.; Shin, J.H. Focal adhesion assembly induces phenotypic changes and dedifferentiation in chondrocytes. J. Cell Physiol. 2016, 231, 1822–1831. [Google Scholar] [CrossRef]
- Liang, J.; Feng, J.; Wu, W.K.K.; Xiao, J.; Wu, Z.; Han, D.; Zhu, Y.; Qiu, G. Leptin-mediated cytoskeletal remodeling in chondrocytes occurs via the RhoA/ROCK pathway. J. Orthop. Res. 2011, 29, 369–374. [Google Scholar] [CrossRef]
- Thompson, C.L.; Plant, J.C.T.; Wann, A.K.T.; Bishop, C.L.; Novak, P.; Mitchison, H.M.; Beales, P.L.; Chapple, J.P.; Knight, M.M. Chondrocyte expansion is associated with loss of primary cilia and disrupted hedgehog signalling. Eur. Cells Mater. 2017, 34, 128–141. [Google Scholar] [CrossRef] [PubMed]
- Schulze-Tanzil, G.; Mobasheri, A.; de Souza, P.; Johns, T.; Shakibaei, M. Loss of chondrogenic potential in dedifferentiated chondrocytes correlates with deficient Shc-Erk interaction and apoptosis. Osteoarthr. Cartil. 2004, 12, 448–458. [Google Scholar] [CrossRef] [PubMed]
- Benya, P.D. Modulation and reexpression of the chondrocyte phenotype; Mediation by cell shape and microfilament modification. Pathol. Immunopathol. Res. 1988, 7, 51–54. [Google Scholar] [CrossRef]
- Mallein-Gerin, F.; Garrone, R.; van der Rest, M. Proteoglycan and collagen synthesis are correlated with actin organization in dedifferentiating chondrocytes. Eur. J. Cell Biol. 1991, 56, 364–373. [Google Scholar]
- McNary, S.M.; Athanasiou, K.A.; Reddi, A.H. Transforming growth factor β-induced superficial zone protein accumulation in the surface zone of articular cartilage is dependent on the cytoskeleton. Tissue. Eng. Part A 2014, 20, 921–929. [Google Scholar] [CrossRef]
- Chan, B.; Parreno, J.; Glogauer, M.; Wang, Y.; Kandel, R. Adseverin, an actin binding protein, regulates articular chondrocyte phenotype. J. Tissue. Eng. Regen. Med. 2019, 13, 1438–1452. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, V.; Dumitriu, B.; Penzo-Méndez, A.; Han, Y.; Pallavi, B. Control of cell fate and differentiation by Sry-related high-mobility-group box (Sox) transcription factors. Int. J. Biochem. Cell. Biol. 2007, 39, 2195–2214. [Google Scholar] [CrossRef]
- Nurminsky, D.; Magee, C.; Faverman, L.; Nurminskaya, M. Regulation of chondrocyte differentiation by actin-severing protein adseverin. Dev. Biol. 2007, 302, 427–437. [Google Scholar] [CrossRef]
- Sakurai, T.; Kurokawa, H.; Nonomura, Y. Comparison between the gelsolin and adseverin domain structure. J. Biol. Chem. 1991, 266, 15979–15983. [Google Scholar] [CrossRef]
- Li, G.; Song, X.; Li, R.; Sun, L.; Gong, X.; Chen, C.; Yang, L. Zyxin-involved actin regulation is essential in the maintenance of vinculin focal adhesion and chondrocyte differentiation status. Cell Prolif. 2019, 52, 1–10. [Google Scholar] [CrossRef]
- Yoshigi, M.; Hoffman, L.M.; Jensen, C.C.; Yost, H.J.; Beckerle, M.C. Mechanical force mobilizes zyxin from focal adhesions to actin filaments and regulates cytoskeletal reinforcement. J. Cell Biol. 2005, 171, 209–215. [Google Scholar] [CrossRef]
- Smith, M.A.; Blankman, E.; Gardel, M.L.; Luettjohann, L.; Waterman, C.M.; Beckerle, M.C. A zyxin-mediated mechanism for actin stress fiber maintenance and repair. Dev. Cell. 2010, 19, 365–376. [Google Scholar] [CrossRef]
- Nguyen, T.N.; Uemura, A.; Shih, W.; Yamada, S. Zyxin-mediated actin assembly is required for efficient wound closure. J. Biol. Chem. 2010, 285, 35439–35445. [Google Scholar] [CrossRef] [PubMed]
- Oakes, P.W.; Wagner, E.; Brand, C.A.; Probst, D.; Linke, M.; Schwarz, U.S.; Glotzer, M.; Gardel, M.L. Optogenetic control of RhoA reveals zyxin-mediated elasticity of stress fibres. Nat. Commun. 2017, 8, 15817. [Google Scholar] [CrossRef]
- Ren, X.D.; Kiosses, W.B.; Sieg, D.J.; Otey, C.A.; Schlaepfer, D.D.; Schwartz, M.A. Focal adhesion kinase suppresses Rho activity to promote focal adhesion turnover. J. Cell Sci. 2000, 113, 3673–3678. [Google Scholar]
- Chen, B.-H.; Tzen, J.T.C.; Bresnick, A.R.; Chen, H.-C. Roles of Rho-associated kinase and myosin light chain kinase in morphological and migratory defects of focal adhesion kinase-null cells. J. Biol. Chem. 2002, 277, 33857–33863. [Google Scholar] [CrossRef] [PubMed]
- Hsia, D.A.; Mitra, S.K.; Hauck, C.R.; Streblow, D.N.; Nelson, J.A.; Ilic, D.; Huang, S.; Li, E.; Nemerow, G.R.; Leng, J.; et al. Differential regulation of cell motility and invasion by FAK. J. Cell Biol. 2003, 160, 753–767. [Google Scholar] [CrossRef]
- Tew, S.R.; Hardingham, T.E. Regulation of SOX9 mRNA in human articular chondrocytes involving p38 MAPK activation and mRNA stabilization. J. Biol. Chem. 2006, 281, 39471–39479. [Google Scholar] [CrossRef] [PubMed]
- Woods, A.; Wang, G.; Beier, F. RhoA/ROCK signaling regulates Sox9 expression and actin organization during chondrogenesis. J. Biol. Chem. 2005, 280, 11626–11634. [Google Scholar] [CrossRef]
- Hershko, D.D.; Robb, B.W.; Wray, C.J.; Luo, G.J.; Hasselgren, P.O. Superinduction of IL-6 by cycloheximide is associated with mRNA stabilization and sustained activation of p38 map kinase and NF-κB in cultured Caco-2 cells. J. Cell Biochem. 2004, 91, 951–961. [Google Scholar] [CrossRef]
- Itani, O.A.; Cornish, K.L.; Liu, K.Z.; Thomas, C.P. Cycloheximide increases glucocorticoid-stimulated alpha -ENaC mRNA in collecting duct cells by p38 MAPK-dependent pathway. Am. J. Physiol. Renal. Physiol. 2003, 284, F778–F787. [Google Scholar] [CrossRef]
- Moriguchi, T.; Toyoshima, F.; Gotoh, Y.; Iwamatsu, A.; Irie, K.; Mori, E.; Kuroyanagi, N.; Hagiwara, M.; Matsumoto, K.; Nishida, E. Purification and identification of a major activator for p38 from osmotically shocked cells. Activation of mitogen-activated protein kinase kinase 6 by osmotic shock, tumor necrosis factor-alpha, and H2O2. J. Biol. Chem. 1996, 271, 26981–26988. [Google Scholar] [CrossRef]
- Newton, R.; Stevens, D.A.; Hart, L.A.; Lindsay, M.; Adcock, I.M.; Barnes, P.J. Superinduction of COX-2 mRNA by cycloheximide and interleukin-1beta involves increased transcription and correlates with increased NF-kappaB and JNK activation. FEBS Lett. 1997, 418, 135–138. [Google Scholar] [CrossRef]
- Geng, Y.; Valbracht, J.; Lotz, M. Selective activation of the mitogen-activated protein kinase subgroups c-Jun NH2 terminal kinase and p38 by IL-1 and TNF in human articular chondrocytes. J. Clin. Invest. 1996, 98, 2425–2430. [Google Scholar] [CrossRef][Green Version]
- Park, E.H.; Kang, S.-S.S.; Lee, Y.-S.S.; Kim, S.-J.J.; Jin, E.-J.J.; Tak, E.N.; Sonn, J.K. Integrity of the cortical actin ring is required for activation of the PI3K/Akt and p38 MAPK signaling pathways in redifferentiation of chondrocytes on chitosan. Cell Biol. Int. 2008, 32, 1272–1278. [Google Scholar] [CrossRef]
- Saraswat, R.; Ratnayake, I.; Perez, E.C.; Schutz, W.M.; Zhu, Z.; Ahrenkiel, S.P.; Wood, S.T. Micropatterned Biphasic Nanocomposite Platform for Maintaining Chondrocyte Morphology. ACS Appl. Mater. Interfaces. 2020, 12, 14814–14824. [Google Scholar] [CrossRef]
- Koskinen-Kolasa, A.; Vuolteenaho, K.; Korhonen, R.; Moilanen, T.; Moilanen, E. Catabolic and proinflammatory effects of leptin in chondrocytes are regulated by suppressor of cytokine signaling-3. Arthritis. Res. Ther. 2016, 18, 215. [Google Scholar] [CrossRef]
- Pascarelli, N.A.; Collodel, G.; Moretti, E.; Cheleschi, S.; Fioravanti, A. Changes in ultrastructure and cytoskeletal aspects of human normal and osteoarthritic chondrocytes exposed to interleukin-1β and cyclical hydrostatic pressure. Int. J. Mol. Sci. 2015, 16, 26019–26034. [Google Scholar] [CrossRef]
- Trickey, W.R.; Vail, T.P.; Guilak, F. The role of the cytoskeleton in the viscoelastic properties of human articular chondrocytes. J. Orthop. Res. 2004, 22, 131–139. [Google Scholar] [CrossRef]
- Kouri, J.B.; Argüello, C.; Luna, J.; Mena, R. Use of microscopical techniques in the study of human chondrocytes from osteoarthritic cartilage: An overview. Microsc. Res. Tech. 1998, 40, 22–36. [Google Scholar] [CrossRef]
- Eckert, B.S. Alteration of intermediate filament distribution in PtK1 cells by acrylamide. Eur J Cell Biol 1985, 37, 169–174. [Google Scholar]
- Haudenschild, D.R.; Chen, J.; Pang, N.; Steklov, N.; Grogan, S.P.; Lotz, M.K.; D’Lima, D.D. Vimentin contributes to changes in chondrocyte stiffness in osteoarthritis. Orthop. Res. 2011, 29, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Lambrecht, S.; Verbruggen, G.; Verdonk, P.C.M.; Elewaut, D.; Deforce, D. Differential proteome analysis of normal and osteoarthritic chondrocytes reveals distortion of vimentin network in osteoarthritis. Osteoarthr. Cartil. 2008, 16, 163–173. [Google Scholar] [CrossRef]
- Rollín, R.; Tornero, P.; Marco, F.; Camafeita, E.; Calvo, E.; López-Durán, L.; Jover, J.Á.; López, J.A.; Lamas, J.R.; Fernández-Gutiérrez, B. Differential Proteome of Articular Chondrocytes From Patients with Osteoarthritis. J. Proteomics. Bioinform. 2008, 1, 267–280. [Google Scholar] [CrossRef]
- Vartiainen, M.K.; Mustonen, T.; Mattila, P.K.; Ojala, P.J.; Thesleff, I.; Partanen, J.; Lappalainen, P. The three mouse actin-depolymerizing factor/cofilins evolved to fulfill cell-type-specific requirements for actin dynamics. Mol. Biol. Cell. 2002, 13, 183–194. [Google Scholar] [CrossRef]
- Chen, C.; Xie, J.; Rajappa, R.; Deng, L.; Fredberg, J.; Yang, L. Interleukin-1β and tumor necrosis factor-α increase stiffness and impair contractile function of articular chondrocytes. Acta. Biochim. Biophys. Sin. (Shanghai) 2015, 47, 121–129. [Google Scholar] [CrossRef]
- Clancy, R.M.; Rediske, J.; Tang, X.; Nijher, N.; Frenkel, S.; Philips, M.; Abramson, S.B. Outside-in signaling in the chondrocyte. Nitric oxide disrupts fibronectin-induced assembly of a subplasmalemmal actin/rho a/focal adhesion kinase signaling complex. J. Clin. Invest. 1997, 100, 1789–1796. [Google Scholar] [CrossRef]
- Novakofski, K.D.; Torre, C.J.; Fortier, L.A. Interleukin-1α, -6, and -8 decrease Cdc42 activity resulting in loss of articular chondrocyte phenotype. J. Orthop. Res. 2012, 30, 246–251. [Google Scholar] [CrossRef]
- Li, R.; Song, X.; Li, G.; Hu, Z.; Sun, L.; Chen, C.; Yang, L. Ibuprofen attenuates interleukin-1β-induced inflammation and actin reorganization via modulation of RhoA signaling in rabbit chondrocytes. Acta. Biochim. Biophys. Sin. 2019, 51, 1026–1033. [Google Scholar] [CrossRef]
- Chen, C.; Xie, J.; Deng, L.; Yang, L. Substrate stiffness together with soluble factors affects chondrocyte mechanoresponses. ACS Appl. Mater. Interfaces. 2014, 6, 16106–16116. [Google Scholar] [CrossRef]
- Vinall, R.L.; Lo, S.H.; Reddi, A.H. Regulation of articular chondrocyte phenotype by bone morphogenetic protein 7, interleukin 1, and cellular context is dependent on the cytoskeleton. Exp. Cell. Res. 2002, 272, 32–44. [Google Scholar] [CrossRef]
- Plotnikov, S.V.; Pasapera, A.M.; Sabass, B.; Waterman, C.M. Force fluctuations within focal adhesions mediate ECM-rigidity sensing to guide directed cell migration. Cell 2012, 151, 1513–1527. [Google Scholar] [CrossRef]
- Goldblum, S.E.; Ding, X.; Campbell-Washington, J. TNF-α induces endothelial cell F-actin depolymerization, new actin synthesis, and barrier dysfunction. Am. J. Physiol. Cell Physiol. 1993, 264, C894–C905. [Google Scholar] [CrossRef]
- Koukouritaki, S.B.; Vardaki, E.A.; Papakonstanti, E.A.; Lianos, E.; Stournaras, C.; Emmanouel, D.S. TNF-α induces actin cytoskeleton reorganization in glomerular epithelial cells involving tyrosine phosphorylation of paxillin and focal adhesion kinase. Mol. Med. 1999, 5, 382–392. [Google Scholar] [CrossRef]
- Kutsuna, H.; Suzuki, K.; Kamata, N.; Kato, T.; Hato, F.; Mizuno, K.; Kobayashi, H.; Ishii, M.; Kitagawa, S. Actin reorganization and morphological changes in human neutrophils stimulated by TNF, GM-CSF, and G-CSF: The role of MAP kinases. Am. J. Physiol. Cell Physiol. 2004, 286, 55–64. [Google Scholar] [CrossRef]
- Mitra, A.; Venkatachalapathy, S.; Ratna, P.; Wang, Y.; Jokhun, D.S.; Shivashankar, G.V. Cell geometry dictates TNFα-induced genome response. Proc. Natl. Acad. Sci. USA 2017, 114, E3882–E3891. [Google Scholar] [CrossRef] [PubMed]
- Papakonstanti, E.A.; Stournaras, C. Tumor necrosis factor-alpha promotes survival of opossum kidney cells via Cdc42-induced phospholipase C-gamma1 activation and actin filament redistribution. Mol. Biol. Cell 2004, 15, 1273–1286. [Google Scholar] [CrossRef]
- Peppelenbosch, M.; Boone, E.; Jones, G.E.; van Deventer, S.J.H.; Haegeman, G.; Fiers, W.; Grooten, J.; Ridley, A.J. Multiple signal transduction pathways regulate TNF-induced actin reorganization in macrophages: Inhibition of Cdc42-mediated filopodium formation by TNF. J. Immunol. 1999, 162, 837–845. [Google Scholar]
- Campbell, I.K.; Novak, U.; Cebon, J.; Layton, J.E.; Hamilton, J.A. Human articular cartilage and chondrocytes produce hemopoietic colony-stimulating factors in culture in response to IL-1. J. Immunol. 1991, 147, 1238–1246. [Google Scholar]
- Lie, P.P.Y.; Yan Cheng, C.; Mruk, D.D. The biology of interleukin-1: Emerging concepts in the regulation of the actin cytoskeleton and cell junction dynamics. Cell Mol. Life. Sci. 2012, 69, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Clahsen, T.; Schaper, F. Interleukin-6 acts in the fashion of a classical chemokine on monocytic cells by inducing integrin activation, cell adhesion, actin polymerization, chemotaxis, and transmigration. J. Leukoc. Biol. 2008, 84, 1521–1529. [Google Scholar] [CrossRef]
- Campos, S.B.; Ashworth, S.L.; Wean, S.; Hosford, M.; Sandoval, R.M.; Hallett, M.A.; Atkinson, S.J.; Molitoris, B.A. Cytokine-induced F-actin reorganization in endothelial cells involves RhoA activation. Am. J. Physiol. Ren. Physiol. 2009, 296, 487–495. [Google Scholar] [CrossRef]
- Khella, C.M.; Asgarian, R.; Horvath, J.M.; Rolauffs, B.; Hart, M.L. An evidence-based systematic review of human knee post-traumatic osteoarthritis (PTOA): Timeline of clinical presentation and disease markers, comparison of knee joint PTOA models and early disease implications. Int. J. Mol. Sci. 2021, 22, 1–48. [Google Scholar] [CrossRef]
- Appleton, C.T.G.; Usmani, S.E.; Mort, J.S.; Beier, F. Rho/ROCK and MEK/ERK activation by transforming growth factor-α induces articular cartilage degradation. Lab. Investig. 2010, 90, 20–30. [Google Scholar] [CrossRef]
- Appleton, C.T.G.; McErlain, D.D.; Pitelka, V.; Schwartz, N.; Bernier, S.M.; Henry, J.L.; Holdsworth, D.W.; Beier, F. Forced mobilization accelerates pathogenesis: Characterization of a preclinical surgical model of osteoarthritis. Arthritis. Res. Ther. 2007, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Appleton, C.T.G.; Usmani, S.E.; Bernier, S.M.; Aigner, T.; Beier, F. Transforming growth factor alpha suppression of articular chondrocyte phenotype and Sox9 expression in a rat model of osteoarthritis. Arthritis. Rheum. 2007, 56, 3693–3705. [Google Scholar] [CrossRef]
- Appleton, C.T.G.; Pitelka, V.; Henry, J.; Beier, F. Global analyses of gene expression in early experimental osteoarthritis. Arthritis. Rheum. 2007, 56, 1854–1868. [Google Scholar] [CrossRef] [PubMed]
- Leipzig, N.D.; Eleswarapu, S.V.; Athanasiou, K.A. The effects of TGF-β1 and IGF-I on the biomechanics and cytoskeleton of single chondrocytes. Osteoarthr. Cartil. 2006, 14, 1227–1236. [Google Scholar] [CrossRef]
- Martin, I.; Vunjak-Novakovic, G.; Yang, J.; Langer, R.; Freed, L.E. Mammalian chondrocytes expanded in the presence of fibroblast growth factor 2 maintain the ability to differentiate and regenerate three-dimensional cartilaginous tissue. Exp. Cell Res. 1999, 253, 681–688. [Google Scholar] [CrossRef]
- Holbourn, K.P.; Acharya, K.R.; Perbal, B. The CCN family of proteins: Structure-function relationships. Trends. Biochem. Sci. 2008, 33, 461–473. [Google Scholar] [CrossRef]
- Woods, A.; Pala, D.; Kennedy, L.; McLean, S.; Rockel, J.S.; Wang, G.; Leask, A.; Beier, F. Rac1 signaling regulates CTGF/CCN2 gene expression via TGFβ/Smad signaling in chondrocytes. Osteoarthr. Cartil. 2009, 17, 406–413. [Google Scholar] [CrossRef]
- Nishida, T.; Kubota, S.; Aoyama, E.; Janune, D.; Maeda, A.; Takigawa, M. Effect of CCN2 on FGF2-induced proliferation and MMP9 and MMP13 productions by chondrocytes. Endocrinology 2011, 152, 4232–4241. [Google Scholar] [CrossRef]
- Takigawa, M. CCN2: A master regulator of the genesis of bone and cartilage. J. Cell Commun. Signal. 2013, 7, 191–201. [Google Scholar] [CrossRef]
- Nishida, T.; Kubota, S. Roles of CCN2 as a mechano-sensing regulator of chondrocyte differentiation. Jpn. Dent. Sci. Rev. 2020, 56, 119–126. [Google Scholar] [CrossRef]
- Eberlein, M.; Heusinger-Ribeiro, J.; Goppelt-Struebe, M. Rho-dependent inhibition of the induction of connective tissue growth factor (CTGF) by HMG CoA reductase inhibitors (statins). Br. J. Pharmacol. 2001, 133, 1172–1180. [Google Scholar] [CrossRef]
- Hahn, A.; Heusinger-Ribeiro, J.; Lanz, T.; Zenkel, S.; Goppelt-Struebe, M. Induction of connective tissue growth factor by activation of heptahelical receptors. Modulation by Rho proteins and the actin cytoskeleton. J. Biol. Chem. 2000, 275, 37429–37435. [Google Scholar] [CrossRef]
- Nishida, T.; Kubota, S.; Aoyama, E.; Yamanaka, N.; Lyons, K.M.; Takigawa, M. Low-intensity pulsed ultrasound (LIPUS) treatment of cultured chondrocytes stimulates production of CCN family protein 2 (CCN2), a protein involved in the regeneration of articular cartilage: Mechanism underlying this stimulation. Osteoarthr. Cartil. 2017, 25, 759–769. [Google Scholar] [CrossRef]
- Allen, J.L.; Cooke, M.E.; Alliston, T. ECM stiffness primes the TGFβ pathway to promote chondrocyte differentiation. Mol. Biol. Cell 2012, 23, 3731–3742. [Google Scholar] [CrossRef]
- Finnson, K.W.; Chi, Y.; Bou-Gharios, G.; Leask, A.; Philip, A. TGF-b signaling in cartilage homeostasis and osteoarthritis. Front. Biosci. 2012, 4, 251–268. [Google Scholar] [CrossRef]
- Thielen, N.G.M.; van der Kraan, P.M.; van Caam, A.P.M. TGFβ/BMP signaling pathway in cartilage homeostasis. Cells 2019, 8, 969. [Google Scholar] [CrossRef] [PubMed]
- Plaas, A.; Velasco, J.; Gorski, D.J.; Li, J.; Cole, A.; Christopherson, K.; Sandy, J.D. The relationship between fibrogenic TGFβ1 signaling in the joint and cartilage degradation in post-injury osteoarthritis. Osteoarthr. Cartil. 2011, 19, 1081–1090. [Google Scholar] [CrossRef]
- van der Kraan, P.M.; Goumans, M.-J.; Blaney Davidson, E.; ten Dijke, P. Age-dependent alteration of TGF-beta signalling in osteoarthritis. Cell Tissue. Res. 2012, 347, 257–265. [Google Scholar] [CrossRef]
- Boehme, K.A.; Rolauffs, B. Onset and progression of human osteoarthritis—Can growth factors, inflammatory cytokines, or differential miRNA expression concomitantly induce proliferation, ECM degradation, and inflammation in articular cartilage? Int. J. Mol. Sci. 2018, 19, 2282. [Google Scholar] [CrossRef] [PubMed]
- Blaney Davidson, E.N.; Remst, D.F.G.; Vitters, E.L.; van Beuningen, H.M.; Blom, A.B.; Goumans, M.-J.; van den Berg, W.B.; van der Kraan, P.M. Increase in ALK1/ALK5 ratio as a cause for elevated MMP-13 expression in osteoarthritis in humans and mice. J. Immunol. 2009, 182, 7937–7945. [Google Scholar] [CrossRef] [PubMed]
- van der Kraan, P.M.; Blaney Davidson, E.N.; Blom, A.; van den Berg, W.B. TGF-beta signaling in chondrocyte terminal differentiation and osteoarthritis: Modulation and integration of signaling pathways through receptor-Smads. Osteoarthr. Cartil. 2009, 17, 1539–1545. [Google Scholar] [CrossRef] [PubMed]
- Vardouli, L.; Vasilaki, E.; Papadimitriou, E.; Kardassis, D.; Stournaras, C. A novel mechanism of TGFbeta-induced actin reorganization mediated by Smad proteins and Rho GTPases. FEBS J. 2008, 275, 4074–4087. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.C.; Spector, M. Distribution of chondrocytes containing alpha-smooth muscle actin in human articular cartilage. J. Orthop. Res. 2000, 18, 749–755. [Google Scholar] [CrossRef]
- Xu, T.; Wu, M.; Feng, J.; Lin, X.; Gu, Z. RhoA/Rho kinase signaling regulates transforming growth factor-β1-induced chondrogenesis and actin organization of synovium-derived mesenchymal stem cells through interaction with the Smad pathway. Int. J. Mol. Med. 2012, 30, 1119–1125. [Google Scholar] [CrossRef]
- Furumatsu, T.; Ozaki, T.; Asahara, H. Smad3 activates the Sox9-dependent transcription on chromatin. Int. J. Biochem. Cell Biol. 2009, 41, 1198–1204. [Google Scholar] [CrossRef]
- Rys, J.P.; DuFort, C.C.; Monteiro, D.A.; Baird, M.A.; Oses-Prieto, J.A.; Chand, S.; Burlingame, A.L.; Davidson, M.W.; Alliston, T.N. Discrete spatial organization of TGFβ receptors couples receptor multimerization and signaling to cellular tension. eLife 2015, 4, e09300. [Google Scholar] [CrossRef] [PubMed]
- Mandriota, N.; Friedsam, C.; Jones-Molina, J.A.; Tatem, K.V.; Ingber, D.E.; Sahin, O. Cellular nanoscale stiffness patterns governed by intracellular forces. Nat. Mater. 2019, 18, 1071–1077. [Google Scholar] [CrossRef]
- Fletcher, D.A.; Mullins, R.D. Cell mechanics and the cytoskeleton. Nature 2010, 463, 485–492. [Google Scholar] [CrossRef]
- Gavara, N.; Chadwick, R.S. Relationship between cell stiffness and stress fiber amount, assessed by simultaneous atomic force microscopy and live-cell fluorescence imaging. Biomech. Model. Mechanobiol. 2016, 15, 511–523. [Google Scholar] [CrossRef]
- Tee, S.Y.; Fu, J.; Chen, C.S.; Janmey, P.A. Cell shape and substrate rigidity both regulate cell stiffness. Biophys. J. 2011, 100, L25–L27. [Google Scholar] [CrossRef]
- Flannery, C.R.; Hughes, C.E.; Schumacher, B.L.; Tudor, D.; Aydelotte, M.B.; Kuettner, K.E.; Caterson, B. Articular cartilage superficial zone protein (SZP) is homologous to megakaryocyte stimulating factor precursor and Is a multifunctional proteoglycan with potential growth-promoting, cytoprotective, and lubricating properties in cartilage metabolism. Biochem. Biophys. Res. Commun. 1999, 254, 535–541. [Google Scholar] [CrossRef]
- Jay, G.D.; Tantravahi, U.; Britt, D.E.; Barrach, H.J.; Cha, C.J. Homology of lubricin and superficial zone protein (SZP): Products of megakaryocyte stimulating factor (MSF) gene expression by human synovial fibroblasts and articular chondrocytes localized to chromosome 1q25. J. Orthop. Res. 2001, 19, 677–687. [Google Scholar] [CrossRef]
- Schmid, T.M.; Su, J.-L.; Lindley, K.M.; Soloveychik, V.; Madsen, L.; Block, J.A.; Kuettner, K.E.; Schumacher, B.L. Superficial zone protein (SZP) is an abundant glycoprotein in human synovial fluid with lubricating properties. In The Many Faces of Osteoarthritis; Hascall, V.C., Kuettner, K.E., Eds.; Birkhäuser: Basel, Switzerland, 2002; pp. 159–161. [Google Scholar]
- Schumacher, B.L.; Block, J.A.; Schmid, T.M.; Aydelotte, M.B.; Kuettner, K.E. A novel proteoglycan synthesized and secreted by chondrocytes of the superficial zone of articular cartilage. Arch. Biochem. Biophys. 1994, 311, 144–152. [Google Scholar] [CrossRef]
- Swann, D.A.; Hendren, R.B.; Radin, E.L.; Sotman, S.L.; Duda, E.A. The lubricating activity of synovial fluid glycoproteins. Arthritis. Rheum. 1981, 24, 22–30. [Google Scholar] [CrossRef]
- Schmidt, T.A.; Sah, R.L. Effect of synovial fluid on boundary lubrication of articular cartilage. Osteoarthr. Cartil. 2007, 15, 35–47. [Google Scholar] [CrossRef]
- Jay, G.D.; Harris, D.A.; Cha, C.J. Boundary lubrication by lubricin is mediated by O-linked beta(1-3)Gal-GalNAc oligosaccharides. Glycoconj. J. 2001, 18, 807–815. [Google Scholar] [CrossRef]
- Chan, S.M.T.; Neu, C.P.; DuRaine, G.; Komvopoulos, K.; Reddi, A.H. Atomic force microscope investigation of the boundary-lubricant layer in articular cartilage. Osteoarthr. Cartil. 2010, 18, 956–963. [Google Scholar] [CrossRef]
- Jay, G.D.; Cha, C.J. The effect of phospholipase digestion upon the boundary lubricating ability of synovial fluid. J. Rheumatol. 1999, 26, 2454–2457. [Google Scholar]
- Peng, G.; McNary, S.M.; Athanasiou, K.A.; Reddi, A.H. The distribution of superficial zone protein (SZP)/lubricin/PRG4 and boundary mode frictional properties of the bovine diarthrodial joint. J. Biomech. 2015, 48, 3406–3412. [Google Scholar] [CrossRef]
- Borge, L.; Lemare, F.; Demignot, S.; Adolphe, M. Restoration of the differentiated functions of serially passaged chondrocytes using staurosporine. Vitr. Cell Dev. Biol. Anim. 1997, 33, 703–709. [Google Scholar] [CrossRef]
- Tamaoki, T.; Nomoto, H.; Takahashi, I.; Kato, Y.; Morimoto, M.; Tomita, F. Staurosporine, a potent inhibitor of phospholipid/Ca++dependent protein kinase. Biochem. Biophys. Res. Commun. 1986, 135, 397–402. [Google Scholar] [CrossRef]
- Matta, C.; Mobasheri, A. Regulation of chondrogenesis by protein kinase C: Emerging new roles in calcium signalling. Cell Signal. 2014, 26, 979–1000. [Google Scholar] [CrossRef]
- Wermuth, P.J.; Addya, S.; Jimenez, S.A. Effect of protein kinase C delta (PKC-δ) inhibition on the transcriptome of normal and systemic sclerosis human dermal fibroblasts in vitro. PLoS ONE 2011, 6, e27110. [Google Scholar] [CrossRef]
- Lengfeld, J.; Wang, Q.; Zohlman, A.; Salvarezza, S.; Morgan, S.; Ren, J.; Kato, K.; Rodriguez-Boulan, E.; Liu, B. Protein kinase C δ regulates the release of collagen type I from vascular smooth muscle cells via regulation of Cdc42. Mol. Biol. Cell 2012, 23, 1955–1963. [Google Scholar] [CrossRef]
- Aigner, T.; Fundel, K.; Saas, J.; Gebhard, P.M.; Haag, J.; Weiss, T.; Zien, A.; Obermayr, F.; Zimmer, R.; Bartnik, E. Large-scale gene expression profiling reveals major pathogenetic pathways of cartilage degeneration in osteoarthritis. Arthritis. Rheum. 2006, 54, 3533–3544. [Google Scholar] [CrossRef]
- Wei, T.; Kulkarni, N.H.; Zeng, Q.Q.; Helvering, L.M.; Lin, X.; Lawrence, F.; Hale, L.; Chambers, M.G.; Lin, C.; Harvey, A.; et al. Analysis of early changes in the articular cartilage transcriptisome in the rat meniscal tear model of osteoarthritis: Pathway comparisons with the rat anterior cruciate transection model and with human osteoarthritic cartilage. Osteoarthr. Cartil. 2010, 18, 992–1000. [Google Scholar] [CrossRef]
- Brew, C.J.; Clegg, P.D.; Boot-Handford, R.P.; Andrew, J.G.; Hardingham, T. Gene expression in human chondrocytes in late osteoarthritis is changed in both fibrillated and intact cartilage without evidence of generalised chondrocyte hypertrophy. Ann. Rheum. Dis. 2010, 69, 234–240. [Google Scholar] [CrossRef]
- Barley, R.D.C.; Adesida, A.B.; Bagnall, K.M.; Jomha, N.M. Immunohistochemical characterization of reparative tissue present in human osteoarthritic tissue. Virchows. Arch. 2010, 456, 561–569. [Google Scholar] [CrossRef]
- Lahm, A.; Mrosek, E.; Spank, H.; Erggelet, C.; Kasch, R.; Esser, J.; Merk, H. Changes in content and synthesis of collagen types and proteoglycans in osteoarthritis of the knee joint and comparison of quantitative analysis with Photoshop-based image analysis. Arch. Orthop. Trauma. Surg. 2010, 130, 557–564. [Google Scholar] [CrossRef]
- Lorenz, H.; Wenz, W.; Ivancic, M.; Steck, E.; Richter, W. Early and stable upregulation of collagen type II, collagen type I and YKL40 expression levels in cartilage during early experimental osteoarthritis occurs independent of joint location and histological grading. Arthritis. Res. Ther. 2005, 7, R156–R165. [Google Scholar] [CrossRef]
- Li, J.; Anemaet, W.; Diaz, M.A.; Buchanan, S.; Tortorella, M.; Malfait, A.M.; Mikecz, K.; Sandy, J.D.; Plaas, A. Knockout of ADAMTS5 does not eliminate cartilage aggrecanase activity but abrogates joint fibrosis and promotes cartilage aggrecan deposition in murine osteoarthritis models. J. Orthop. Res. 2011, 29, 516–522. [Google Scholar] [CrossRef]
- Verrecchia, F.; Chu, M.L.; Mauviel, A. Identification of Novel TGF-β/Smad Gene Targets in Dermal Fibroblasts using a Combined cDNA Microarray/Promoter Transactivation Approach. J. Biol. Chem. 2001, 276, 17058–17062. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Yuan, W.; Mori, Y.; Varga, J. Smad-dependent stimulation of type I collagen gene expression in human skin fibroblasts by TGF-β involves functional cooperation with p300/CBP transcriptional coactivators. Oncogene 2000, 19, 3546–3555. [Google Scholar] [CrossRef]
- Chen, S.J.; Yuan, W.; Mori, Y.; Levenson, A.; Trojanowska, M.; Varga, J. Stimulation of type I collagen transcription in human skin fibroblasts by TGF-β: Involvement of Smad 3. J. Invest. Dermatol. 1999, 112, 49–57. [Google Scholar] [CrossRef]
- Chen, S.J.; Yuan, W.; Lo, S.; Trojanowska, M.; Varga, J. Interaction of Smad3 with a proximal smad-binding element of the human α2(I) procollagen gene promoter required for transcriptional activation by TGF-β. J. Cell Physiol. 2000, 183, 381–392. [Google Scholar] [CrossRef]
- Takahata, Y.; Nakamura, E.; Hata, K.; Wakabayashi, M.; Murakami, T.; Wakamori, K.; Yoshikawa, H.; Matsuda, A.; Fukui, N.; Nishimura, R. Sox4 is involved in osteoarthritic cartilage deterioration through induction of ADAMTS4 and ADAMTS5. FASEB J. 2019, 33, 619–630. [Google Scholar] [CrossRef]
- Tetsunaga, T.; Nishida, K.; Furumatsu, T.; Naruse, K.; Hirohata, S.; Yoshida, A.; Saito, T.; Ozaki, T. Regulation of mechanical stress-induced MMP-13 and ADAMTS-5 expression by RUNX-2 transcriptional factor in SW1353 chondrocyte-like cells. Osteoarthr. Cartil. 2011, 19, 222–232. [Google Scholar] [CrossRef]
- Wang, P.; Mao, Z.; Pan, Q.; Lu, R.; Huang, X.; Shang, X.; Zhang, R.; You, H. Histone deacetylase-4 and histone deacetylase-8 regulate interleukin-1β-induced cartilage catabolic degradation through MAPK/JNK and ERK pathways. Int. J. Mol. Med. 2018, 41, 2117–2127. [Google Scholar] [CrossRef]
- Yatabe, T.; Mochizuki, S.; Takizawa, M.; Chijiiwa, M.; Okada, A.; Kimura, T.; Fujita, Y.; Matsumoto, H.; Toyama, Y.; Okada, Y. Hyaluronan inhibits expression of ADAMTS4 (aggrecanase-1) in human osteoarthritic chondrocytes. Ann. Rheum. Dis. 2009, 68, 1051–1058. [Google Scholar] [CrossRef]
- Bau, B.; Gebhard, P.M.; Haag, J.; Knorr, T.; Bartnik, E.; Aigner, T. Relative messenger RNA expression profiling of collagenases and aggrecanases in human articular chondrocytes in vivo and in vitro. Arthritis Rheum 2002, 46, 2648–2657. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, T.T.; Schulz, R.M.; Rai, S.S.; Thuemmler, C.B.; Wuestneck, N.; Bader, A.; Homandberg, G.A. Biomechanical modulation of collagen fragment-induced anabolic and catabolic activities in chondrocyte/agarose constructs. Arthritis. Res. Ther. 2010, 12, R82. [Google Scholar] [CrossRef]
- Jennings, L.; Wu, L.; King, K.B.; Hämmerle, H.; Cs-Szabo, G.; Mollenhauer, J. The effects of collagen fragments on the extracellular matrix metabolism of bovine and human chondrocytes. Connect. Tissue. Res. 2001, 42, 71–86. [Google Scholar] [CrossRef]
- Billinghurst, R.C.; Dahlberg, L.; Ionescu, M.; Reiner, A.; Bourne, R.; Rorabeck, C.; Mitchell, P.; Hambor, J.; Diekmann, O.; Tschesche, H.; et al. Enhanced cleavage of type II collagen by collagenases in osteoarthritic articular cartilage. J. Clin. Invest. 1997, 99, 1534–1545. [Google Scholar] [CrossRef]
- Luchsinger, L.L.; Patenaude, C.A.; Smith, B.D.; Layne, M.D. Myocardin-related transcription factor-A complexes activate type I collagen expression in lung fibroblasts. J. Biol. Chem. 2011, 286, 44116–44125. [Google Scholar] [CrossRef]
- Akiyama, H. Control of chondrogenesis by the transcription factor Sox9. Mod. Rheumatol. 2008, 18, 213–219. [Google Scholar] [CrossRef]
- Murakami, S.; Kan, M.; McKeehan, W.L.; De Crombrugghe, B. Up-regulation of the chondrogenic Sox9 gene by fibroblast growth factors is mediated by the mitogen-activated protein kinase pathway. Proc. Natl. Acad. Sci. USA 2000, 97, 1113–1118. [Google Scholar] [CrossRef]
- Yan, D.; Chen, D.; Cool, S.M.; van Wijnen, A.J.; Mikecz, K.; Murphy, G.; Im, H.J. Fibroblast growth factor receptor 1 is principally responsible for fibroblast growth factor 2-induced catabolic activities in human articular chondrocytes. Arthritis. Res. Ther. 2011, 13, R130. [Google Scholar] [CrossRef]
- Li, X.; Ellman, M.B.; Kroin, J.S.; Chen, D.; Yan, D.; Mikecz, K.; Ranjan, K.; Xiao, G.; Stein, G.S.; Kim, S.G.; et al. Species-specific biological effects of FGF-2 in articular cartilage: Implication for distinct roles within the FGF receptor family. J. Cell Biochem. 2012, 113, 2532–2542. [Google Scholar] [CrossRef]
- Ellman, M.B.; Yan, D.; Ahmadinia, K.; Chen, D.; An, H.S.; Im, H.J. Fibroblast growth factor control of cartilage homeostasis. J. Cell Biochem. 2013, 114, 735–742. [Google Scholar] [CrossRef]
- Cucchiarini, M.; Terwilliger, E.F.; Kohn, D.; Madry, H. Remodelling of human osteoarthritic cartilage by FGF-2, alone or combined with Sox9 via rAAV gene transfer. J. Cell Mol. Med. 2009, 13, 2476–2488. [Google Scholar] [CrossRef]
- Sekiya, I.; Koopman, P.; Tsuji, K.; Mertin, S.; Harley, V.; Yamada, Y.; Shinomiya, K.; Nifuji, A.; Noda, M. Dexamethasone enhances SOX9 expression in chondrocytes. J. Endocrinol. 2001, 169, 573–579. [Google Scholar] [CrossRef]
- Du, J.; Zu, Y.; Li, J.; Du, S.; Xu, Y.; Zhang, L.; Jiang, L.; Wang, Z.; Chien, S.; Yang, C. Extracellular matrix stiffness dictates Wnt expression through integrin pathway. Sci. Rep. 2016, 6, 20395. [Google Scholar] [CrossRef]
- Zhong, W.; Li, Y.; Li, L.; Zhang, W.; Wang, S.; Zheng, X. YAP-mediated regulation of the chondrogenic phenotype in response to matrix elasticity. J. Mol. Histol. 2013, 44, 587–595. [Google Scholar] [CrossRef]
- Rhee, J.; Ryu, J.H.; Kim, J.H.; Chun, C.H.; Chun, J.S. α-catenin inhibits β-catenin-T-cell factor/lymphoid enhancing factor transcriptional activity and collagen type II expression in articular chondrocytes through formation of Gli3R·α-catenin·β- catenin ternary complex. J. Biol. Chem. 2012, 287, 11751–11760. [Google Scholar] [CrossRef]
- Williams, C.A.C.; Soufi, A.; Pollard, S.M. Post-translational modification of SOX family proteins: Key biochemical targets in cancer? Semin. Cancer. Biol. 2020, 67, 30–38. [Google Scholar] [CrossRef]
- Huang, W.; Zhou, X.; Lefebvre, V.; de Crombrugghe, B. Phosphorylation of SOX9 by cyclic AMP-dependent protein kinase A enhances SOX9’s ability to transactivate a Col2a1 chondrocyte-specific enhancer. Mol. Cell Biol. 2000, 20, 4149–4158. [Google Scholar] [CrossRef]
- Haudenschild, D.R.; Chen, J.; Pang, N.; Lotz, M.K.; D’Lima, D.D. Rho kinase-dependent activation of SOX9 in chondrocytes. Arthritis. Rheum. 2010, 62, 191–200. [Google Scholar] [CrossRef]
- Huang, W.; Chung, U.I.; Kronenberg, H.M.; De Crombrugghe, B. The chondrogenic transcription factor Sox9 is a target of signaling by the parathyroid hormone-related peptide in the growth plate of endochondral bones. Proc. Natl. Acad. Sci. USA 2001, 98, 160–165. [Google Scholar] [CrossRef]
- Pelosi, M.; Lazzarano, S.; Thoms, B.L.; Murphy, C.L. Parathyroid hormone-related protein is induced by hypoxia and promotes expression of the differentiated phenotype of human articular chondrocytes. Clin. Sci. 2013, 125, 461–470. [Google Scholar] [CrossRef]
- Coricor, G.; Serra, R. TGF-β regulates phosphorylation and stabilization of Sox9 protein in chondrocytes through p38 and Smad dependent mechanisms. Sci. Rep. 2016, 6, 38616. [Google Scholar] [CrossRef]
- Haudenschild, D.R.; Chen, J.; Steklov, N.; Lotz, M.K.; D’Lima, D.D. Characterization of the Chondrocyte Actin Cytoskeleton in Living Three-Dimensional Culture: Response to Anabolic and Catabolic Stimuli. Mol. Cell Biochem. 2009, 6, 135–144. [Google Scholar]
- Kanamoto, T.; Hikida, M.; Sato, S.; Oyama, S.; Tachi, Y.; Kuroda, S.; Mazuka, T.; Ebina, K.; Nakai, T.; Nakata, K. Integrin α2β1 plays an important role in the interaction between human articular cartilage-derived chondrocytes and atelocollagen gel. Sci. Rep. 2021, 11, 1–12. [Google Scholar] [CrossRef]
- Ochiya, T.; Takahama, Y.; Nagahara, S.; Sumita, Y.; Hisada, A.; Itoh, H.; Nagai, Y.; Terada, M. New delivery system for plasmid DNA in vivo using atelocollagen as a carrier material: The Minipellet. Nat. Med. 1999, 5, 707–710. [Google Scholar] [CrossRef]
- Jokinen, J.; Dadu, E.; Nykvist, P.; Käpylä, J.; White, D.J.; Ivaska, J.; Vehviläinen, P.; Reunanen, H.; Larjava, H.; Häkkinen, L.; et al. Integrin-mediated cell adhesion to type I collagen fibrils. J. Biol. Chem. 2004, 279, 31956–31963. [Google Scholar] [CrossRef]
- Yuan, X.; Zhou, M.; Gough, J.; Glidle, A.; Yin, H. A novel culture system for modulating single cell geometry in 3D. Acta. Biomater. 2015, 24, 228–240. [Google Scholar] [CrossRef]
- Xu, X.; Urban, J.P.G.; Tirlapur, U.K.; Cui, Z. Osmolarity effects on bovine articular chondrocytes during three-dimensional culture in alginate beads. Osteoarthr. Cartil. 2010, 18, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Tessier, S.; Tran, V.A.; Ottone, O.K.; Novais, E.J.; Doolittle, A.; DiMuzio, M.J.; Shapiro, I.M.; Risbud, M. V TonEBP-deficiency accelerates intervertebral disc degeneration underscored by matrix remodeling, cytoskeletal rearrangements, and changes in proinflammatory gene expression. Matrix. Biol. 2020, 87, 94–111. [Google Scholar] [CrossRef]
- Tessier, S.; Doolittle, A.C.; Sao, K.; Rotty, J.D.; Bear, J.E.; Ulici, V.; Loeser, R.F.; Shapiro, I.M.; Diekman, B.O.; Risbud, M. V Arp2/3 inactivation causes intervertebral disc and cartilage degeneration with dysregulated TonEBP-mediated osmoadaptation. JCI Insight. 2020, 5, e131382. [Google Scholar] [CrossRef]
- Wang, Z.; Irianto, J.; Kazun, S.; Wang, W.; Knight, M.M. The rate of hypo-osmotic challenge influences regulatory volume decrease (RVD) and mechanical properties of articular chondrocytes. Osteoarthr. Cartil. 2015, 23, 289–299. [Google Scholar] [CrossRef]
- Jones, W.R.; Ting-Beall, H.P.; Lee, G.M.; Kelley, S.S.; Hochmuth, R.M.; Guilak, F. Alterations in the Young’s modulus and volumetric properties of chondrocytes isolated from normal and osteoarthritic human cartilage. J. Biomech. 1999, 32, 119–127. [Google Scholar] [CrossRef]
- Bush, P.G.; Hodkinson, P.D.; Hamilton, G.L.; Hall, A.C. Viability and volume of in situ bovine articular chondrocytes-changes following a single impact and effects of medium osmolarity. Osteoarthr. Cartil. 2005, 13, 54–65. [Google Scholar] [CrossRef]
- Bush, P.G.; Hall, A.C. The volume and morphology of chondrocytes within non-degenerate and degenerate human articular cartilage. Osteoarthr. Cartil. 2003, 11, 242–251. [Google Scholar] [CrossRef]
- Goswami, C.; Kuhn, J.; Heppenstall, P.A.; Hucho, T. Importance of non-selective cation channel TRPV4 interaction with cytoskeleton and their reciprocal regulations in cultured cells. PLoS ONE 2010, 5, e11654. [Google Scholar] [CrossRef]
- Du, G.; Li, L.; Zhang, X.; Liu, J.; Hao, J.; Zhu, J.; Wu, H.; Chen, W.; Zhang, Q. Roles of TRPV4 and piezo channels in stretch-evoked Ca(2+) response in chondrocytes. Exp. Biol. Med. 2020, 245, 180–189. [Google Scholar] [CrossRef]
- Morachevskaya, E.A.; Sudarikova, A. V Actin dynamics as critical ion channel regulator: ENaC and Piezo in focus. Am. J. Physiol. Cell Physiol. 2021. [Google Scholar] [CrossRef]
- Lewis, R.; Feetham, C.H.; Gentles, L.; Penny, J.; Tregilgas, L.; Tohami, W.; Mobasheri, A.; Barrett-Jolley, R. Benzamil sensitive ion channels contribute to volume regulation in canine chondrocytes. Br. J. Pharmacol. 2013, 168, 1584–1596. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Izumi, Y.; Burg, M.B.; Ferraris, J.D. Rac1/osmosensing scaffold for MEKK3 contributes via phospholipase C-gamma1 to activation of the osmoprotective transcription factor NFAT5. Proc. Natl. Acad. Sci. USA 2011, 108, 12155–12160. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Haynes, E.M.; Asokan, S.B.; Simon, J.M.; Sharpless, N.E.; Baldwin, A.S.; Davis, I.J.; Johnson, G.L.; Bear, J.E. Loss of Arp2/3 induces an NF-κB-dependent, nonautonomous effect on chemotactic signaling. J. Cell Biol. 2013, 203, 907–916. [Google Scholar] [CrossRef]
- Di Ciano-Oliveira, C.; Thirone, A.C.P.; Szászi, K.; Kapus, A. Osmotic stress and the cytoskeleton: The R(h)ole of Rho GTPases. Acta. Physiol. 2006, 187, 257–272. [Google Scholar] [CrossRef]
- Kang, J.-H.; Jiang, Y.; Toita, R.; Oishi, J.; Kawamura, K.; Han, A.; Mori, T.; Niidome, T.; Ishida, M.; Tatematsu, K.; et al. Phosphorylation of Rho-associated kinase (Rho-kinase/ROCK/ROK) substrates by protein kinases A and C. Biochimie. 2007, 89, 39–47. [Google Scholar] [CrossRef]
- Kang, J.-H.; Asai, D.; Tsuchiya, A.; Mori, T.; Niidome, T.; Katayama, Y. Peptide substrates for Rho-associated kinase 2 (Rho-kinase 2/ROCK2). PLoS ONE 2011, 6, e22699. [Google Scholar] [CrossRef]
- Takahashi, K.; Sasaki, T.; Mammoto, A.; Takaishi, K.; Kameyama, T.; Tsukita, S.; Takai, Y. Direct interaction of the Rho GDP dissociation inhibitor with ezrin/radixin/moesin initiates the activation of the Rho small G protein. J. Biol. Chem. 1997, 272, 23371–23375. [Google Scholar] [CrossRef]
- Kawaguchi, K.; Yoshida, S.; Hatano, R.; Asano, S. Pathophysiological roles of ezrin/radixin/moesin proteins. Biol. Pharm. Bull. 2017, 40, 381–390. [Google Scholar] [CrossRef]
- Cai, S.; Pestic-Dragovich, L.; O’Donnell, M.E.; Wang, N.; Ingber, D.; Elson, E.; De Lanerolle, P. Regulation of cytoskeletal mechanics and cell growth by myosin light chain phosphorylation. Am. J. Physiol. 1998, 275, C1349–C1356. [Google Scholar] [CrossRef]
- Zhang, Q.; Ji, Q.; Wang, X.; Kang, L.; Fu, Y.; Yin, Y.; Li, Z.; Liu, Y.; Xu, X.; Wang, Y. SOX9 is a regulator of ADAMTSs-induced cartilage degeneration at the early stage of human osteoarthritis. Osteoarthr. Cartil. 2015, 23, 2259–2268. [Google Scholar] [CrossRef]
- Ouyang, Y.; Wang, W.; Tu, B.; Zhu, Y.; Fan, C.; Li, Y. Overexpression of SOX9 alleviates the progression of human osteoarthritis in vitro and in vivo. Drug. Des. Devel. Ther. 2019, 13, 2833–2842. [Google Scholar] [CrossRef]
- Haag, J.; Gebhard, P.M.; Aigner, T. SOX gene expression in human osteoarthritic cartilage. Pathobiology 2008, 75, 195–199. [Google Scholar] [CrossRef]
- Rottmar, M.; Mhanna, R.; Guimond-lischer, S.; Vogel, V.; Zenobi-wong, M.; Maniura-weber, K. Interference with the contractile machinery of the fibroblastic chondrocyte cytoskeleton induces re-expression of the cartilage phenotype through involvement of PI3K, PKC and MAPKs. Exp. Cell Res. 2014, 320, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Kanellos, G.; Frame, M.C. Cellular functions of the ADF/cofilin family at a glance. J. Cell Sci. 2016, 129, 3211–3218. [Google Scholar] [CrossRef]
- Edwards, D.C.; Sanders, L.C.; Bokoch, G.M.; Gill, G.N. Activation of LIM-kinase by Pak1 couples Rac/Cdc42 GTPase signalling to actin cytoskeletal dynamics. Nat. Cell Biol. 1999, 1, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Manser, E.; Leung, T.; Salihuddin, H.; Zhao, Z.S.; Lim, L. A brain serine/threonine protein kinase activated by Cdc42 and Rac1. Nature 1994, 367, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Romarowski, A.; Battistone, M.A.; La Spina, F.A.; Puga Molina, L.d.C.; Luque, G.M.; Vitale, A.M.; Cuasnicu, P.S.; Visconti, P.E.; Krapf, D.; Buffone, M.G. PKA-dependent phosphorylation of LIMK1 and Cofilin is essential for mouse sperm acrosomal exocytosis. Dev. Biol. 2015, 405, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Tatsumi, E.; Yamanaka, H.; Kobayashi, K.; Yagi, H.; Sakagami, M.; Noguchi, K. RhoA/ROCK pathway mediates p38 MAPK activation and morphological changes downstream of P2Y12/13 receptors in spinal microglia in neuropathic pain. Glia 2015, 63, 216–228. [Google Scholar] [CrossRef]
- Fukata, Y.; Oshiro, N.; Kinoshita, N.; Kawano, Y.; Matsuoka, Y.; Bennett, V.; Matsuura, Y.; Kaibuchi, K. Phosphorylation of adducin by Rho-kinase plays a crucial role in cell motility. J. Cell Biol. 1999, 145, 347–361. [Google Scholar] [CrossRef]
- Huh, G.Y.; Glantz, S.B.; Je, S.; Morrow, J.S.; Kim, J.H. Calpain proteolysis of alpha II-spectrin in the normal adult human brain. Neurosci. Lett. 2001, 316, 41–44. [Google Scholar] [CrossRef]
- Karsdal, M.A.; Sumer, E.U.; Wulf, H.; Madsen, S.H.; Christiansen, C.; Fosang, A.J.; Sondergaard, B.-C. Induction of increased cAMP levels in articular chondrocytes blocks matrix metalloproteinase-mediated cartilage degradation, but not aggrecanase-mediated cartilage degradation. Arthritis. Rheum. 2007, 56, 1549–1558. [Google Scholar] [CrossRef]
- Ito, K.; Shimomura, E.; Iwanaga, T.; Shiraishi, M.; Shindo, K.; Nakamura, J.; Nagumo, H.; Seto, M.; Sasaki, Y.; Takuwa, Y. Essential role of rho kinase in the Ca2+ sensitization of prostaglandin F2α-induced contraction of rabbit aortae. J. Physiol. 2003, 546, 823–836. [Google Scholar] [CrossRef] [PubMed]
- Bonn, S.; Herrero, S.; Breitenlechner, C.B.; Erlbruch, A.; Lehmann, W.; Engh, R.A.; Gassel, M.; Bossemeyer, D. Structural analysis of protein kinase a mutants with Rho-kinase inhibitor specificity. J. Biol. Chem. 2006, 281, 24818–24830. [Google Scholar] [CrossRef]
- Nadella, K.S.; Saji, M.; Jacob, N.K.; Pavel, E.; Ringel, M.D.; Kirschner, L.S. Regulation of actin function by protein kinase A-mediated phosphorylation of Limk1. EMBO Rep. 2009, 10, 599–605. [Google Scholar] [CrossRef]
- Ohashi, K.; Nagata, K.; Maekawa, M.; Ishizaki, T.; Narumiya, S.; Mizuno, K. Rho-associated kinase ROCK activates LIM-kinase 1 by phosphorylation at threonine 508 within the activation loop. J. Biol. Chem. 2000, 275, 3577–3582. [Google Scholar] [CrossRef]
- Amano, T.; Tanabe, K.; Eto, T.; Narumiya, S.; Mizuno, K. LIM-kinase 2 induces formation of stress fibres, focal adhesions and membrane blebs, dependent on its activation by Rho-associated kinase-catalysed phosphorylation at threonine-505. Biochem. J. 2001, 354, 149–159. [Google Scholar] [CrossRef]
- Itoh, S.; Saito, T.; Hirata, M.; Ushita, M.; Ikeda, T.; Woodgett, J.R.; Algül, H.; Schmid, R.M.; Chung, U.-I.; Kawaguchi, H. GSK-3α and GSK-3β proteins are involved in early stages of chondrocyte differentiation with functional redundancy through RelA protein phosphorylation. J. Biol. Chem. 2012, 287, 29227–29236. [Google Scholar] [CrossRef]
- Zhang, Q.; Yu, Y.; Zhao, H. The effect of matrix stiffness on biomechanical properties of chondrocytes. Acta. Biochim. Biophys. Sin. 2016, 48, 958–965. [Google Scholar] [CrossRef]
- Jannatbabaei, A.; Tafazzoli-Shadpour, M.; Seyedjafari, E.; Fatouraee, N. Cytoskeletal remodeling induced by substrate rigidity regulates rheological behaviors in endothelial cells. J. Biomed. Mater. Res A. 2019, 107, 71–80. [Google Scholar] [CrossRef]
- Zaidel-Bar, R.; Itzkovitz, S.; Ma’ayan, A.; Iyengar, R.; Geiger, B. Functional atlas of the integrin adhesome. Nat. Cell Biol. 2007, 9, 858–867. [Google Scholar] [CrossRef]
- Kuo, J.-C.; Han, X.; Hsiao, C.-T.; Yates, J.R., 3rd; Waterman, C.M. Analysis of the myosin-II-responsive focal adhesion proteome reveals a role for β-Pix in negative regulation of focal adhesion maturation. Nat. Cell Biol. 2011, 13, 383–393. [Google Scholar] [CrossRef]
- Burridge, K.; Chrzanowska-Wodnicka, M. Focal adhesions, contractility, and signaling. Annu. Rev. Cell Dev. Biol. 1996, 12, 463–518. [Google Scholar] [CrossRef]
- Zaidel-Bar, R.; Cohen, M.; Addadi, L.; Geiger, B. Hierarchical assembly of cell-matrix adhesion complexes. Biochem. Soc. Trans. 2004, 32, 416–420. [Google Scholar] [CrossRef]
- Pelham, R.J.J.; Wang, Y.I. Cell locomotion and focal adhesions are regulated by substrate flexibility. Proc. Natl. Acad. Sci. USA 1997, 94, 13661–13665. [Google Scholar] [CrossRef]
- Pelham, R.J.J.; Wang, Y.L. Cell locomotion and focal adhesions are regulated by the mechanical properties of the substrate. Biol. Bull. 1998, 194, 348–350. [Google Scholar] [CrossRef]
- Engler, A.J.; Griffin, M.A.; Sen, S.; Bönnemann, C.G.; Sweeney, H.L.; Discher, D.E. Myotubes differentiate optimally on substrates with tissue-like stiffness: Pathological implications for soft or stiff microenvironments. J. Cell Biol. 2004, 166, 877–887. [Google Scholar] [CrossRef] [PubMed]
- Von der Mark, K.; Gauss, V.; von der Mark, H.; Muller, P. Relationship between cell shape and type of collagen synthesised as chondrocytes lose their cartilage phenotype in culture. Nature 1977, 267, 531–532. [Google Scholar] [CrossRef]
- Gupta, M.; Sarangi, B.R.; Deschamps, J.; Nematbakhsh, Y.; Callan-Jones, A.; Margadant, F.; Mège, R.-M.; Lim, C.T.; Voituriez, R.; Ladoux, B. Adaptive rheology and ordering of cell cytoskeleton govern matrix rigidity sensing. Nat. Commun. 2015, 6, 7525. [Google Scholar] [CrossRef]
- Doss, B.L.; Pan, M.; Gupta, M.; Grenci, G.; Mège, R.-M.; Lim, C.T.; Sheetz, M.P.; Voituriez, R.; Ladoux, B. Cell response to substrate rigidity is regulated by active and passive cytoskeletal stress. Proc. Natl. Acad. Sci. USA 2020, 117, 12817–12825. [Google Scholar] [CrossRef]
- Bauer, M.S.; Baumann, F.; Daday, C.; Redondo, P.; Durner, E.; Jobst, M.A.; Milles, L.F.; Mercadante, D.; Pippig, D.A.; Gaub, H.E.; et al. Structural and mechanistic insights into mechanoactivation of focal adhesion kinase. Proc. Natl. Acad. Sci. USA 2019, 116, 6766–6774. [Google Scholar] [CrossRef]
- Knudsen, P.B.; Hanna, B.; Ohl, S.; Sellner, L.; Zenz, T.; Döhner, H.; Stilgenbauer, S.; Larsen, T.O.; Lichter, P.; Seiffert, M. Chaetoglobosin A preferentially induces apoptosis in chronic lymphocytic leukemia cells by targeting the cytoskeleton. Leukemia 2014, 28, 1289–1298. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, K.; Oosawa, M.; Tashiro, A.; Suzuki, T.; Tanikawa, M.; Kikuchi, M.; Sekita, S.; Natori, S. Effects of chaetoglobosin J on the G-F transformation of actin. Biochim. Biophys. Acta. 1986, 874, 137–143. [Google Scholar] [CrossRef]
- Urbanik, E.; Ware, B.R. Actin filament capping and cleaving activity of cytochalasins B, D, E, and H. Arch. Biochem. Biophys. 1989, 269, 181–187. [Google Scholar] [CrossRef]
- Shoji, K.; Ohashi, K.; Sampei, K.; Oikawa, M.; Mizuno, K. Cytochalasin D acts as an inhibitor of the actin-cofilin interaction. Biochem. Biophys. Res. Commun. 2012, 424, 52–57. [Google Scholar] [CrossRef]
- Spector, I.; Braet, F.; Shochet, N.R.; Bubb, M.R. New anti-actin drugs in the study of the organization and function of the actin cytoskeleton. Microsc. Res. Tech. 1999, 47, 18–37. [Google Scholar] [CrossRef]
- Tian, B.; Kaufman, P.L. Comparisons of actin filament disruptors and Rho kinase inhibitors as potential antiglaucoma medications. Expert. Rev. Ophthalmol. 2012, 7, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Alauddin, M.; Okumura, T.; Rajaxavier, J.; Khozooei, S.; Pöschel, S.; Takeda, S.; Singh, Y.; Brucker, S.Y.; Wallwiener, D.; Koch, A.; et al. Gut bacterial metabolite urolithin A decreases actin polymerization and migration in cancer cells. Mol. Nutr. Food. Res. 2020, 64, e1900390. [Google Scholar] [CrossRef] [PubMed]
- Berrino, E.; Supuran, C.T. Rho-kinase inhibitors in the management of glaucoma. Expert. Opin. Ther. Pat. 2019, 29, 817–827. [Google Scholar] [CrossRef]
- Abedi, F.; Hayes, A.W.; Reiter, R.; Karimi, G. Acute lung injury: The therapeutic role of Rho kinase inhibitors. Pharmacol. Res. 2020, 155, 104736. [Google Scholar] [CrossRef]
- Peng, G.E.; Wilson, S.R.; Weiner, O.D. A pharmacological cocktail for arresting actin dynamics in living cells. Mol. Biol. Cell 2011, 22, 3986–3994. [Google Scholar] [CrossRef]
- Feng, Y.; LoGrasso, P.V.; Defert, O.; Li, R. Rho kinase (ROCK) inhibitors and their therapeutic potential. J. Med. Chem. 2016, 59, 2269–2300. [Google Scholar] [CrossRef]
- Kusuhara, S.; Nakamura, M. Ripasudil hydrochloride hydrate in the treatment of glaucoma: Safety, efficacy, and patient selection. Clin. Ophthalmol. 2020, 14, 1229–1236. [Google Scholar] [CrossRef]
- Tanihara, H.; Kakuda, T.; Sano, T.; Kanno, T.; Gunji, R. Safety and efficacy of ripasudil in Japanese patients with glaucoma or ocular hypertension: 12-month interim analysis of ROCK-J, a post-marketing surveillance study. BMC Ophthalmol. 2020, 20, 275. [Google Scholar]
- Hoy, S.M. Netarsudil Ophthalmic Solution 0.02%: First Global Approval. Drugs 2018, 78, 389–396. [Google Scholar] [CrossRef]
- Hayashi, K.; Michiue, H.; Yamada, H.; Takata, K.; Nakayama, H.; Wei, F.Y.; Fujimura, A.; Tazawa, H.; Asai, A.; Ogo, N.; et al. Fluvoxamine, an anti-depressant, inhibits human glioblastoma invasion by disrupting actin polymerization. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef]
- Choi, J.; Lee, Y.J.; Yoon, Y.J.; Kim, C.H.; Park, S.J.; Kim, S.Y.; Doo Kim, N.; Cho Han, D.; Kwon, B.M. Pimozide suppresses cancer cell migration and tumor metastasis through binding to ARPC2, a subunit of the Arp2/3 complex. Cancer Sci. 2019, 110, 3788–3801. [Google Scholar] [CrossRef]
- Dasgupta, S.K.; Le, A.; Vijayan, K.V.; Thiagarajan, P. Dasatinib inhibits actin fiber reorganization and promotes endothelial cell permeability through RhoA-ROCK pathway. Cancer Med. 2017, 6, 809–818. [Google Scholar] [CrossRef]
- Hocking, K.M.; Putumbaka, G.; Wise, E.S.; Cheung-Flynn, J.; Brophy, C.M.; Komalavilas, P. Papaverine prevents vasospasm by regulation of myosin light chain phosphorylation and actin polymerization in human saphenous vein. PLoS ONE 2016, 11, e0154460. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Watabe, S.; Ozaki, H.; Kigoshi, H.; Yamada, K.; Fusetani, N.; Karaki, H. Novel actin depolymerizing macrolide aplyronine A. J. Biochem. 1996, 120, 552–555. [Google Scholar] [CrossRef]
- Statsuk, A.V.; Bai, R.; Baryza, J.L.; Verma, V.A.; Hamel, E.; Wender, P.A.; Kozmin, S.A. Actin is the primary cellular receptor of bistramide A. Nat. Chem. Biol. 2005, 1, 383–388. [Google Scholar] [CrossRef]
- Zhou, C.; Wang, Y.; Pan, D.; Sun, Y.; Cao, J. The effect of Cytochalasin B and Jasplakinolide on depolymerization of actin filaments in goose muscles during postmortem conditioning. Food. Res. Int. 2016, 90, 1–7. [Google Scholar] [CrossRef]
- Wada, S.; Matsunaga, S.; Saito, S.; Fusetani, N.; Watabe, S. Actin-binding specificity of marine macrolide toxins, mycalolide B and kabiramide D. J. Biochem. 1998, 123, 946–952. [Google Scholar] [CrossRef]
- Blain, J.C.; Mok, Y.F.; Kubanek, J.; Allingham, J.S. Two molecules of lobophorolide cooperate to stabilize an actin dimer using both their “ring” and “tail” region. Chem. Biol. 2010, 17, 802–807. [Google Scholar] [CrossRef]
- Allingham, J.S.; Zampella, A.; D’Auria, M.V.; Rayment, I. Structures of microfilament destabilizing toxins bound to actin provide insight into toxin design and activity. Proc. Natl. Acad. Sci. USA 2005, 102, 14527–14532. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.Y.; Watabe, S.; Ozaki, H.; Kobayashi, M.; Suzuki, T.; Kobayashi, H.; Fusetani, N.; Karaki, H. Actin-depolymerizing effect of dimeric macrolides, bistheonellide A and swinholide A. J. Biochem. 1998, 123, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Terry, D.R.; Spector, I.; Higa, T.; Bubb, M.R. Misakinolide A is a marine macrolide that caps but does not sever filamentous actin. J. Biol. Chem. 1997, 272, 7841–7845. [Google Scholar] [CrossRef]
- Saito, S.; Watabe, S.; Ozaki, H.; Fusetani, N.; Karaki, H. Mycalolide B, a novel actin depolymerizing agent. J. Biol. Chem. 1994, 269, 29710–29714. [Google Scholar] [CrossRef]
- Tiago, T.; Ramos, S.; Aureliano, M.; Gutiérrez-Merino, C. Peroxynitrite induces F-actin depolymerization and blockade of myosin ATPase stimulation. Biochem. Biophys. Res. Commun. 2006, 342, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Yeung, K.S.; Paterson, I. Actin-binding marine macrolides: Total synthesis and biological importance. Angew. Chemie. Int. Ed. 2002, 41, 4632–4653. [Google Scholar] [CrossRef]
- Smith, C.D.; Carmeli, S.; Moore, R.E.; Patterson, G.M. Scytophycins, novel microfilament-depolymerizing agents which circumvent P-glycoprotein-mediated multidrug resistance. Cancer Res. 1993, 53, 1343–1347. [Google Scholar]
- Zhang, X.; Minale, L.; Zampella, A.; Smith, C.D. Microfilament depletion and circumvention of multiple drug resistance by sphinxolides. Cancer Res. 1997, 57, 3751–3758. [Google Scholar]
- Patterson, G.M.; Smith, C.D.; Kimura, L.H.; Britton, B.A.; Carmeli, S. Action of tolytoxin on cell morphology, cytoskeletal organization, and actin polymerization. Cell Motil. Cytoskeleton. 1993, 24, 39–48. [Google Scholar] [CrossRef]
- Sumiya, E.; Shimogawa, H.; Sasaki, H.; Tsutsumi, M.; Yoshita, K.; Ojika, M.; Suenaga, K.; Uesugi, M. Cell-morphology profiling of a natural product library identifies bisebromoamide and miuraenamide A as actin filament stabilizers. ACS Chem. Biol. 2011, 6, 425–431. [Google Scholar] [CrossRef]
- Vincent, E.; Saxton, J.; Baker-Glenn, C.; Moal, I.; Hirst, J.D.; Pattenden, G.; Shaw, P.E. Effects of ulapualide A and synthetic macrolide analogues on actin dynamics and gene regulation. Cell Mol. Life. Sci. 2007, 64, 487–497. [Google Scholar] [CrossRef]
- Saito, S.; Feng, J.; Kira, A.; Kobayashi, J.; Ohizumi, Y. Amphidinolide H, a novel type of actin-stabilizing agent isolated from dinoflagellate. Biochem. Biophys. Res. Commun. 2004, 320, 961–965. [Google Scholar] [CrossRef]
- Herrmann, J.; Hüttel, S.; Müller, R. Discovery and biological activity of new chondramides from Chondromyces sp. Chembiochem 2013, 14, 1573–1580. [Google Scholar] [CrossRef]
- Foerster, F.; Braig, S.; Chen, T.; Altmann, K.H.; Vollmar, A.M. Pharmacological characterization of actin-binding (-)-Doliculide. Bioorganic. Med. Chem. 2015, 22, 5117–5122. [Google Scholar] [CrossRef]
- Bubb, M.R.; Senderowicz, A.M.; Sausville, E.A.; Duncan, K.L.; Korn, E.D. Jasplakinolide, a cytotoxic natural product, induces actin polymerization and competitively inhibits the binding of phalloidin to F-actin. J. Biol. Chem. 1994, 269, 14869–14871. [Google Scholar] [CrossRef]
- Colombo, R.; Milzani, A.; Donne, I.D. Lithium increases actin polymerization rates by enhancing the nucleation step. J. Mol. Biol. 1991, 217, 401–404. [Google Scholar] [CrossRef]
- Arndt, H.-D.; Rizzo, S.; Nöcker, C.; Wakchaure, V.N.; Milroy, L.-G.; Bieker, V.; Calderon, A.; Tran, T.T.N.; Brand, S.; Dehmelt, L.; et al. Divergent solid-phase synthesis of natural product-inspired bipartite cyclodepsipeptides: Total synthesis of seragamide A. Chemistry 2015, 21, 5311–5316. [Google Scholar] [CrossRef]
- Yoon, Y.J.; Han, Y.-M.; Choi, J.; Lee, Y.-J.; Yun, J.; Lee, S.-K.; Lee, C.W.; Kang, J.S.; Chi, S.-W.; Moon, J.H.; et al. Benproperine, an ARPC2 inhibitor, suppresses cancer cell migration and tumor metastasis. Biochem. Pharmacol. 2019, 163, 46–59. [Google Scholar] [CrossRef]
- Beauverger, P.; Ozoux, M.L.; Bégis, G.; Glénat, V.; Briand, V.; Philippo, M.C.; Daveu, C.; Tavares, G.; Roy, S.; Corbier, A.; et al. Reversion of cardiac dysfunction by a novel orally available calcium/calmodulin-dependent protein kinase II inhibitor, RA306, in a genetic model of dilated cardiomyopathy. Cardiovasc. Res. 2020, 116, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.D.; Novack, G.D.; van Haarlem, T.; Kopczynski, C. Ocular hypotensive effect of the Rho kinase inhibitor AR-12286 in patients with glaucoma and ocular hypertension. Am. J. Ophthalmol. 2011, 152, 834–841.e1. [Google Scholar] [CrossRef]
- Skaat, A.; Jasien, J.V.; Ritch, R. Efficacy of topically administered Rho-kinase inhibitor AR-12286 in patients with exfoliation syndrome and ocular hypertension or glaucoma. J. Glaucoma. 2016, 25, e807-14. [Google Scholar] [CrossRef]
- Kopczynski, C.; Novack, G.D.; Swearingen, D.; van Haarlem, T. Ocular hypotensive efficacy, safety and systemic absorption of AR-12286 ophthalmic solution in normal volunteers. Br. J. Ophthalmol. 2013, 97, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wu, S. Effects of fasudil on pulmonary hypertension in clinical practice. Pulm. Pharmacol. Ther. 2017, 46, 54–63. [Google Scholar] [CrossRef]
- Raja, S.G. Evaluation of clinical efficacy of fasudil for the treatment of pulmonary arterial hypertension. Recent. Pat. Cardiovasc. Drug Discov. 2012, 7, 100–104. [Google Scholar] [CrossRef]
- Lin, G.; Craig, G.P.; Zhang, L.; Yuen, V.G.; Allard, M.; McNeill, J.H.; MacLeod, K.M. Acute inhibition of Rho-kinase improves cardiac contractile function in streptozotocin-diabetic rats. Cardiovasc. Res. 2007, 75, 51–58. [Google Scholar] [CrossRef][Green Version]
- Petersen, W.M.; Lampe, J.; Navratil, T. Topical administration of a novel and potent Rho kinase (ROK) inhibitor INS117548 alters the actin cytoskeleton, effectively lowers IOP, and is well tolerated on the ocular surface. Invest. Ophthalmol. Vis. Sci. 2008, 49, 3816. [Google Scholar]
- Lin, C.-W.; Sherman, B.; Moore, L.A.; Laethem, C.L.; Lu, D.-W.; Pattabiraman, P.P.; Rao, P.V.; DeLong, M.A.; Kopczynski, C.C. Discovery and preclinical development of Netarsudil, a novel ocular hypotensive agent for the treatment of glaucoma. J. Ocul. Pharmacol. Ther. 2018, 34, 40–51. [Google Scholar] [CrossRef]
- Sturdivant, J.M.; Royalty, S.M.; Lin, C.-W.; Moore, L.A.; Yingling, J.D.; Laethem, C.L.; Sherman, B.; Heintzelman, G.R.; Kopczynski, C.C.; deLong, M.A. Discovery of the ROCK inhibitor netarsudil for the treatment of open-angle glaucoma. Bioorg. Med. Chem. Lett. 2016, 26, 2475–2480. [Google Scholar] [CrossRef]
- Kopczynski, C.C.; Heah, T. Netarsudil ophthalmic solution 0.02% for the treatment of patients with open-angle glaucoma or ocular hypertension. Drugs. Today 2018, 54, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Radell, J.E.; Serle, J.B. Netarsudil/latanoprost fixed-dose combination for the treatment of open-angle glaucoma or ocular hypertension. Drugs Today 2019, 55, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Lee, D.; Kolomeyer, N.N.; Myers, J.S.; Razeghinejad, R. Fixed combination netarsudil-latanoprost for the treatment of glaucoma and ocular hypertension. Expert. Opin. Pharmacother. 2020, 21, 39–45. [Google Scholar] [CrossRef]
- Yamamoto, K.; Maruyama, K.; Himori, N.; Omodaka, K.; Yokoyama, Y.; Shiga, Y.; Morin, R.; Nakazawa, T. The novel Rho kinase (ROCK) inhibitor K-115: A new candidate drug for neuroprotective treatment in glaucoma. Invest. Ophthalmol. Vis. Sci. 2014, 55, 7126–7136. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Tanihara, H. Ripasudil hydrochloride hydrate: Targeting Rho kinase in the treatment of glaucoma. Expert. Opin. Pharmacother. 2017, 18, 1669–1673. [Google Scholar] [CrossRef]
- Tanihara, H.; Inatani, M.; Honjo, M.; Tokushige, H.; Azuma, J.; Araie, M. Intraocular pressure-lowering effects and safety of topical administration of a selective ROCK inhibitor, SNJ-1656, in healthy volunteers. Arch. Ophthalmol. 2008, 126, 309–315. [Google Scholar] [CrossRef] [PubMed]














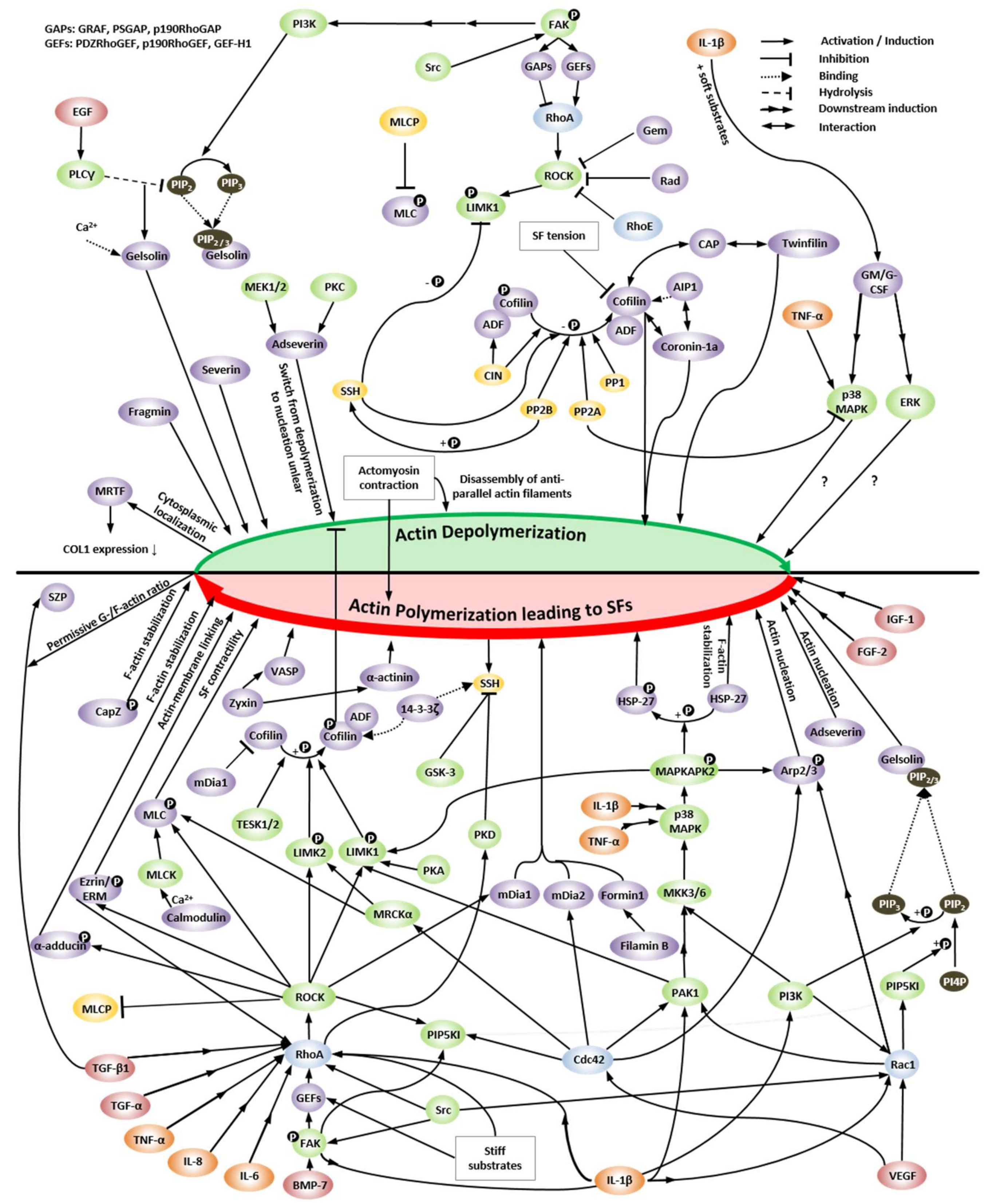{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Term | Definition | Ref. |
|---|---|---|
| Actin nucleation | “Initial assembly of G-actin monomers to form multimers” | [37] |
| Actin polymerization | “Addition of G-actin monomers for the formation of polymerized filaments” | [37] |
| F-actin | “Polymerized actin filaments consisting of two helically coiled chains” | [38] |
| SFs | “Multiple actin filaments crosslinked by α-actinin and/or NMMII and/or other proteins” | [41,42,44] |
| Drug Treatment (Inhibitors) | Effect | Ref. | Clinical Trial Phase | Disease | Studies |
|---|---|---|---|---|---|
| Aplyronine A | Sequesters G-actin, inhibits polymerization, and depolymerizes F-actin | [380] | - | - | 0 |
| Bistramide A | Represses polymerization and depolymerizes F-actin | [381] | - | - | 0 |
| Chaetoglobosin A | Targets F-actin and inhibits actin polymerization | [362] | N/A | Infertility | 1 |
| Chaetoglobosin J | Depolymerizes F-actin | [363] | - | - | 0 |
| Cytochalasin B | Binds to protomers at the actin filament and disrupts them | [364,382] | N/A | Infertility | 2 |
| Cytochalasin D | Caps barbed ends, disrupts actin filaments, binds to G-actin and inhibits interaction with cofilin | [364,365] | N/A | Infertility | 1 |
| Halichondra- mide | Caps barbed ends and severs F-actin | [366] | - | - | 0 |
| Kabiramide A | Binds highly specific to F-actin | [383] | - | - | 0 |
| Latrunculin A | Forms complex with G-actin and thus prevents polymerization and induces F-actin polymerization | [366] | N/A | Infertility | 3 |
| Latrunculin B | Forms complex with G-actin and thus prevents polymerization and induces F-actin polymerization (diminished effect compared to latrunculin A) | [366,367] | N/A | Infertility | 1 |
| Lobophorolide | Inhibits filament growth probably through barbed end capping | [384,385] | - | - | 0 |
| Misakinolide A (Bistheonellide A) | Sequesters G-actin, inhibits polymerization, depolymerizes F-actin, caps but does not sever F-actin | [386,387] | - | - | 0 |
| Mycalolide B | Quickly depolymerizes F-actin | [383,388] | - | - | 0 |
| Pectenotoxin 2 | Sequesters G-actin | [366] | - | - | 0 |
| Peroxynitrite | Causes F-actin depolymerization | [389] | - | - | 0 |
| Scytophycin C | Inhibits polymerization and induces F-actin depolymerization | [390,391] | - | - | 0 |
| Sphinxolide / Reidispongiolide | Inhibits actin polymerization | [390,392] | - | - | 0 |
| Swinholide A | Severs F-actin | [386] | - | - | 0 |
| Tolytoxin | Inhibits actin polymerization and induces depolymerization | [393,394] | - | - | 0 |
| Ulapualide A | Depolymerizes actin | [390,395] | - | - | 0 |
| Urolithin A | Decreases actin polymerization | [368] | N/A | “Healthy aging” | 1 |
| Drug Treatment (Stabilizer / Enhancer) | Effect | Ref. | Clinical Trial Phase | Disease | Studies |
|---|---|---|---|---|---|
| Bisebromoamide | Enhances G-actin polymerization and inhibits F-actin depolymerization | [394] | - | - | 0 |
| Amphidinolide H | Diminishes actin depolymerization | [396] | - | - | 0 |
| Chondramide C | Induce G-actin polymerization | [397] | - | - | 0 |
| Doliculide | Stabilizes and overpolymerizes actin filaments | [398] | - | - | 0 |
| Jasplakinolide (jaspamide) | Causes rapid nucleation of actin polymerization, stabilizes actin filaments | [399] | - | - | 0 |
| Lithium | Increases actin nucleation, enhances actin polymerization | [400] | Early-4 | Several | 439 |
| Lyngbyabellin C | Stabilizes actin filaments | [394] | - | - | 0 |
| Miuraenamide A | Enhances G-actin polymerization and inhibits F-actin depolymerization | [394] | - | - | 0 |
| Phalloidin | Stabilizes actin oligomers | [366] | - | - | 0 |
| Seragamide A | Stabilizes actin filaments | [401] | - | - | 0 |
| Indirect Actin Affecters | Inhibitor of | Effect | Ref. | Clinical Trial Phase | Disease | Studies |
|---|---|---|---|---|---|---|
| Benpro- perine phosphate | ARPC2 | Inhibits actin polymerization | [402] | - | - | 0 |
| Pimozide | Delays the ARP2/3-mediated actin polymerization | [377] | 2–4 | Several | 13 | |
| RA306 | CaMKII | Bundles actin filaments | [403] | - | - | 0 |
| Blebbistatin | NMMIIb | Blocks the ATPase activity of NMMII and thus, depolymerizes actin | [371] | - | - | 0 |
| Papaverine | Phospho- diesterase | Causes F-actin depolymerization | [379] | Early-4 | Several | 22 |
| AR-12286 | ROCK | Block the signaling pathway that induces NMMII activity and thus, depolymerize actin stress fibers [371] | [404,405,406] | 1, 2 | Glaucoma, Ocular hyper- tension | 12 |
| Fasudil 1 (HA-1077) | [407,408] | 2–4 | Several | 10 | ||
| H-1152 | [409] | - | - | 0 | ||
| INS117548 | [410] | 1 | Glaucoma | 1 | ||
| Netarsudil 2 (AR-13503) | [375,411,412,413,414,415] | Early-4 | Several | 24 | ||
| Ripasudil 3 (K-115) | [373,374,416,417] | 2, 4 | Fuchs’ Endothelial Dystrophy | 3 | ||
| SNJ-1656 | [418] | yes (1, stated in [418]) | Glaucoma | 1 | ||
| Y27632 | [371] | - | - | 0 | ||
| Fluvo- xamine 4 | Selective serotonin uptake | Inhibits actin polymerization | [376] | 1–4 | Several | 66 |
| Dasatinib | Tyrosine kinase | Inhibits F-actin reorganization | [378] | Early-4 | Cancer, mainly leukemia | 324 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lauer, J.C.; Selig, M.; Hart, M.L.; Kurz, B.; Rolauffs, B. Articular Chondrocyte Phenotype Regulation through the Cytoskeleton and the Signaling Processes That Originate from or Converge on the Cytoskeleton: Towards a Novel Understanding of the Intersection between Actin Dynamics and Chondrogenic Function. Int. J. Mol. Sci. 2021, 22, 3279. https://doi.org/10.3390/ijms22063279
Lauer JC, Selig M, Hart ML, Kurz B, Rolauffs B. Articular Chondrocyte Phenotype Regulation through the Cytoskeleton and the Signaling Processes That Originate from or Converge on the Cytoskeleton: Towards a Novel Understanding of the Intersection between Actin Dynamics and Chondrogenic Function. International Journal of Molecular Sciences. 2021; 22(6):3279. https://doi.org/10.3390/ijms22063279
Chicago/Turabian StyleLauer, Jasmin C., Mischa Selig, Melanie L. Hart, Bodo Kurz, and Bernd Rolauffs. 2021. "Articular Chondrocyte Phenotype Regulation through the Cytoskeleton and the Signaling Processes That Originate from or Converge on the Cytoskeleton: Towards a Novel Understanding of the Intersection between Actin Dynamics and Chondrogenic Function" International Journal of Molecular Sciences 22, no. 6: 3279. https://doi.org/10.3390/ijms22063279
APA StyleLauer, J. C., Selig, M., Hart, M. L., Kurz, B., & Rolauffs, B. (2021). Articular Chondrocyte Phenotype Regulation through the Cytoskeleton and the Signaling Processes That Originate from or Converge on the Cytoskeleton: Towards a Novel Understanding of the Intersection between Actin Dynamics and Chondrogenic Function. International Journal of Molecular Sciences, 22(6), 3279. https://doi.org/10.3390/ijms22063279