
A Mitocentric View of the Main Bacterial and Parasitic Infectious Diseases in the Pediatric Population

 ,
,
Abstract
1. Introduction
2. Mitochondria
2.1. Mitochondrial Structure
2.1.1. Outer Mitochondrial Membrane (OMM)
2.1.2. Inner Mitochondrial Membrane (IMM)
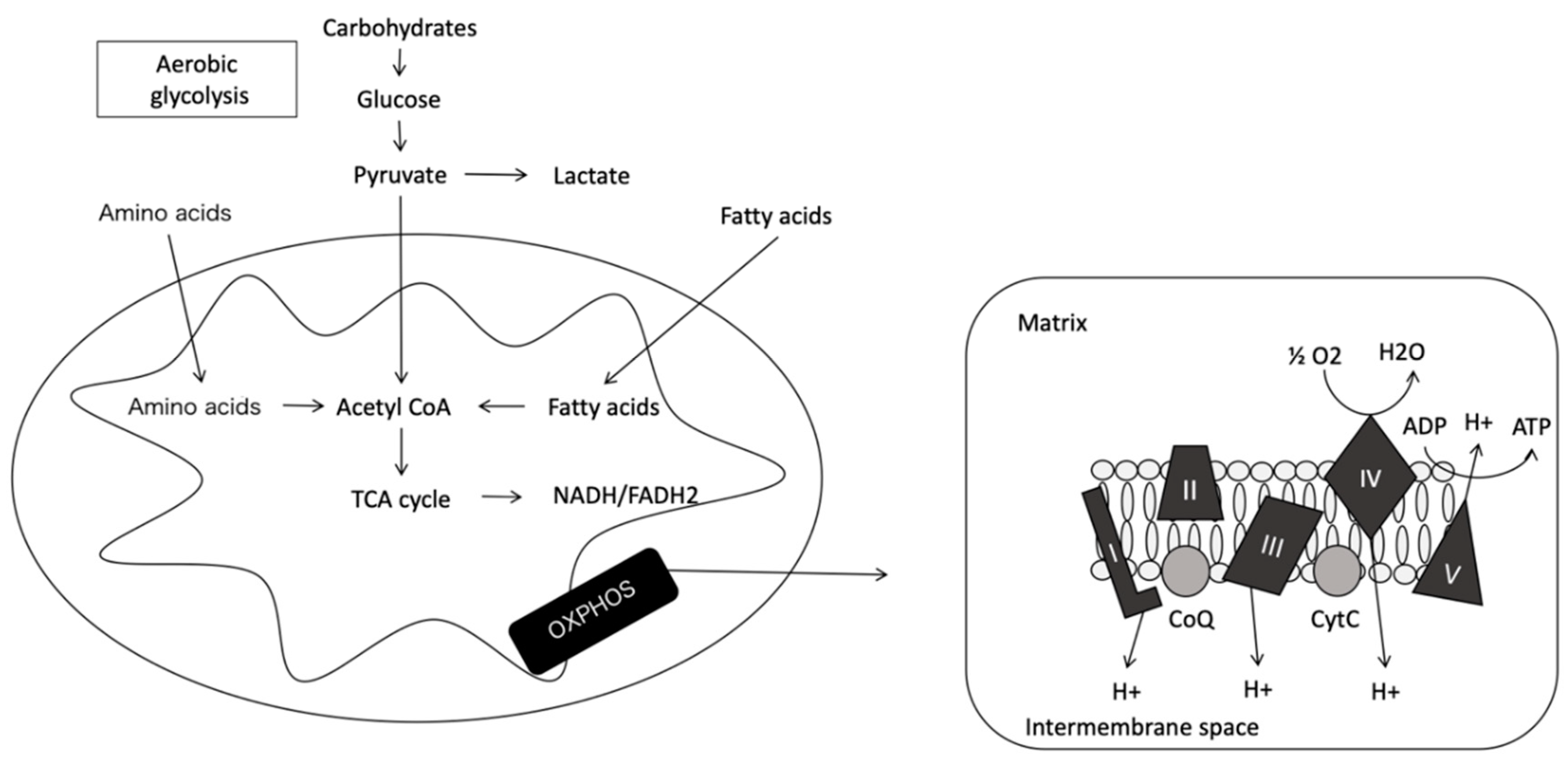
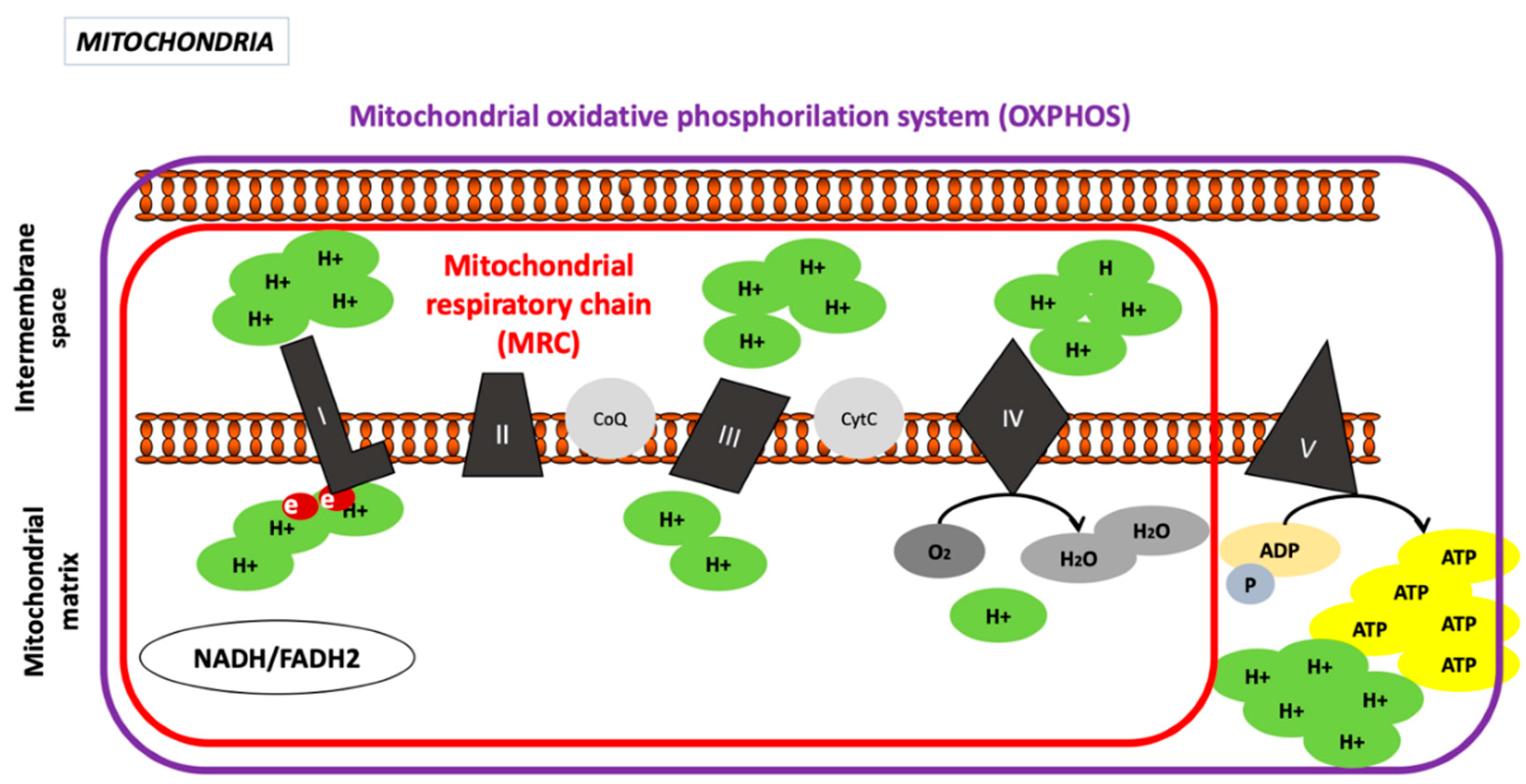
Oxidative Phosphorylation System
Complex I, Nicotinamide Adenine Dinucleotide Hydrogen (NADH) Dehydrogenase or NADH−CoQ Reductase (CI)
Complex II, Succinate Dehydrogenase or Succinate-CoQ Reductase (CII)
Complex III or CoQH2, Known as Cytochrome c Reductase
Complex IV or Cytochrome C Oxidase (COX)
Complex V or ATP Synthase Complex
2.1.3. Intermembrane Space
2.1.4. Mitochondrial Matrix
2.2. Mitochondrial Physiology
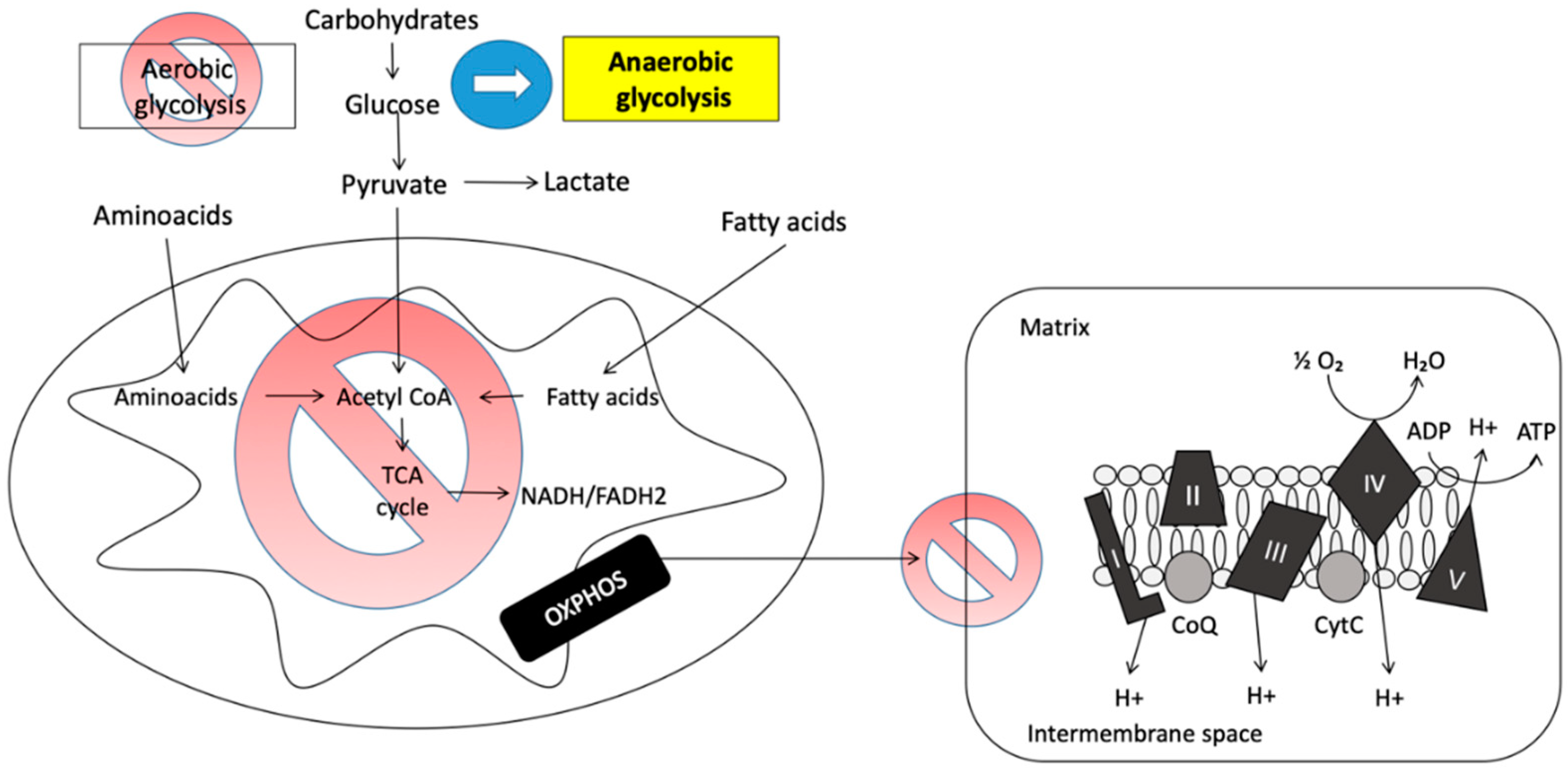
2.3. Mitochondrial Pathology
2.3.1. Anaerobiosis
2.3.2. Reactive Oxygen Species
2.3.3. Apoptosis
3. Bacterial Infectious Processes and Mitochondrial Involvement
3.1. Tuberculosis (TB)
3.1.1. Structure and Replication Cycle
3.1.2. TB in the Pediatric Population
3.1.3. Mitochondrial changes in TB infection
3.1.4. TB Treatment in the Pediatric Population and Mitochondrial Involvement
3.1.5. Pediatric Studies of Mitochondrial Interaction in TB Infection
3.2. Enterobacteria
Pediatric Studies of Mitochondrial Interaction in Enterobacteria
3.3. Staphylococcus aureus
3.3.1. Structure and Replication Cycle
3.3.2. Staphylococcus aureus in the Pediatric Population
3.3.3. Mitochondrial Changes in S. aureus Infection
3.3.4. S. aureus Treatment in the Pediatric Population and Mitochondrial Involvement
3.3.5. Pediatric Studies of Mitochondrial Interactions in S. aureus Infection
3.4. Meningitis
3.4.1. Structure and Replication Cycle
3.4.2. Meningitis in the Pediatric Population
3.4.3. Mitochondrial Changes Derived from Meningitis Pathogens
3.4.4. Meningitis Treatment in the Pediatric Population and Mitochondrial Involvement
3.4.5. Pediatric Studies of Mitochondrial Interaction in Meningitis Infection
4. Parasitic Infectious Processes and Mitochondrial Involvement
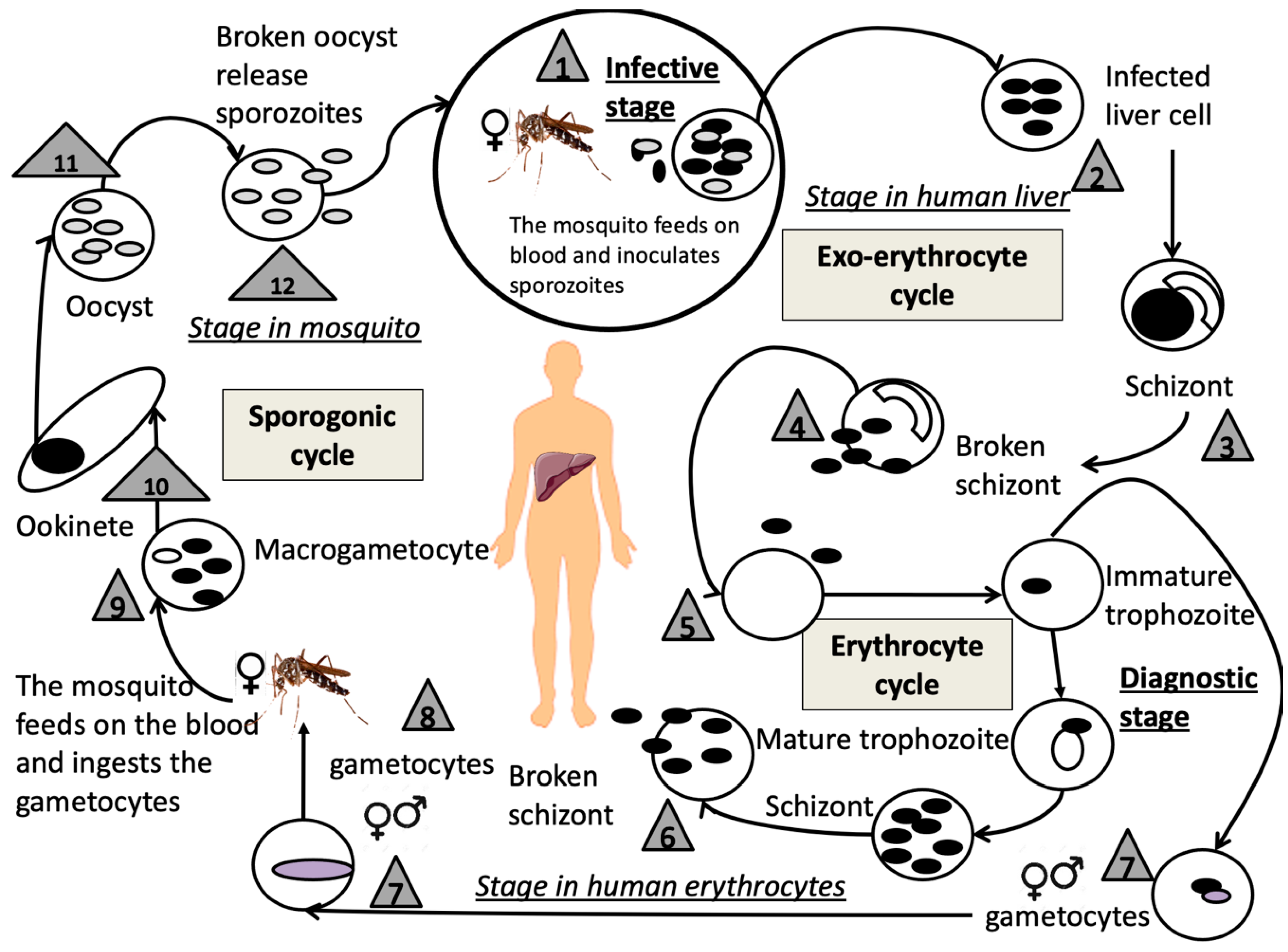
4.1. Malaria
4.1.1. Structure and Replication Cycle
4.1.2. Malaria in the Pediatric Population
4.1.3. Mitochondrial Changes in Malaria Infection
4.1.4. Malaria Treatment in the Pediatric Population and Mitochondrial Involvement
5. Discussion
6. Conclusions
7. Selection Criteria and Outcomes
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| AIF | Apoptosis-Inducing Factor |
| ANT | Adenine Nucleotide Translocase |
| APAF-1 | Apoptosis Protease-Activating Factor-1 |
| ATP | Adenosine Triphosphate |
| ADP | Adenosine Diphosphate |
| EMB | Ethambutol |
| ER | Endoplasmic Reticulum |
| ETC | Electron Transport Chain |
| CoQ | Coenzyme Q |
| CytC | Cytochrome C |
| DNAse | Deoxyribonuclease |
| FADH | Flavin and Adenine Dinucleotide Hydrogen |
| FMN | Flavin Mononucleotide |
| Fp | Flavoprotein |
| CI | Complex I |
| CII | Complex II |
| CIII | Complex III |
| CIV | Complex IV |
| CV | Complex V |
| COX | Cytochrome C Oxidase |
| CypD | Cyclophilin D |
| ESAT-6 | Early Secretory Antigenic Target |
| ETC | Electron Transport Chain |
| HIV | Human Immunodeficiency Virus |
| HSV | Herpes Simplex Virus |
| IGRA | Interferon-Gamma Release Assays |
| IFNγ | Interferon-γ |
| IMM | Inner Mitochondrial Membrane |
| INH | Isoniazid |
| iNOS | Inducible Nitric Oxide Synthase |
| KCN | Potassium Cyanide |
| LOS | Lipooligosaccharide |
| MRC | Mitochondrial Respiratory Chain |
| MRSA | Methicillin Resistant Staphylococcus Aureus |
| Mtb | Mycobacterium tuberculosis |
| mtDNA | Mitochondrial Deoxyribonucleic Acid |
| mtRNA | Mitochondrial Ribonucleic Acid |
| NADH | Nicotinamide Adenine Dinucleotide Hydrogen |
| NHBA | Neisseria Heparin-Binding Antigen |
| NO | Nitric Oxide |
| OMM | Outer Mitochondrial Membrane |
| OXPHOS | Oxidative Phosphorylation System |
| PARP | Poly ADP-Ribose Polymerase |
| Pi | Inorganic Phosphate |
| PTP | Permeability Transition Pore |
| PZA | Pyrazinamide |
| RIF | Rifampin |
| ROS | Reactive Oxygen Species |
| rRNA | Ribosomal Ribonucleic Acid |
| SOD | Superoxide Dismutase |
| TB | Tuberculosis |
| TCA | Tricarboxylic Acid |
| TNFα | Tumor Necrosis Factor-α |
| TNT | TB Necrotizing Toxin |
| tRNA | Transfer Ribonucleic Acid |
| TST | Tuberculin Skin Test |
| Δψm | Mitochondrial Membrane Potential |
References
- World Health Organization (WHO). MCEE-WHO Methods and Data Sources for Child Causes of Death 2000–2017; World Health Organization: Geneva, Switzerland, 2018. [Google Scholar]
- Koopman, W.J.; Beyrath, J.; Fung, C.W.; Koene, S.; Rodenburg, R.J.; Willems, P.H.; Smeitink, J.A. Mitochondrial disorders in children: Toward development of small-molecule treatment strategies. EMBO Mol. Med. 2016, 8, 311–327. [Google Scholar] [CrossRef]
- Morén, C. Mitochondrial Funcionalism in HIV-Infected Children Receiving Antiretroviral Therapy. Ph.D. Thesis, University of Barcelona, Barcelona, Spain, 2012. [Google Scholar]
- Glingston, R.S.; Deb, R.; Kumar, S.; Nagotu, S. Organelle dynamics and viral infections: At cross roads. Microbes Infect. 2019, 21, 20–32. [Google Scholar] [CrossRef]
- Scheffler, I.E. Mitochondria; Wiley: Hoboken, NJ, USA, 2017. [Google Scholar]
- Margulis, L. Origin of Eukaryotic Cells: Evidence and Research Implications for a Theory of the Origin and Evolution of Microbial, Plant and Animal Cells on the Precambrian Earth; Yale University Press: London, UK, 1970. [Google Scholar]
- Van der Bliek, A.M.; Sedensky, M.M.; Morgan, P.G. Cell Biology of the Mitochondrion. Genetics 2017, 207, 843–871. [Google Scholar] [CrossRef] [PubMed]
- Picard, M.; Wallace, D.C.; Burelle, Y. The rise of mitochondria in medicine. Mitochondrion 2016, 30, 105–116. [Google Scholar] [CrossRef]
- Anand, S.K.; Tikoo, S.K. Viruses as modulators of mitochondrial functions. Adv. Virol. 2013, 2013, 738794. [Google Scholar] [CrossRef]
- Molnar, M.J.; Kovacs, G.G. Mitochondrial diseases. Handb. Clin. Neurol. 2017, 145, 147–155. [Google Scholar] [PubMed]
- Menezes, M.J.; Riley, L.G.; Christodoulou, J. Mitochondrial respiratory chain disorders in childhood: Insights into diagnosis and management in the new era of genomic medicine. Biochim. Biophys. Acta 2014, 1840, 1368–1379. [Google Scholar] [CrossRef] [PubMed]
- Nazir, M.; Wani, W.A.; Malik, M.A.; Mir, M.R.; Ashraf, Y.; Kawoosa, K.; Ali, S.W. Cerebrospinal fluid lactate: A differential biomarker for bacterial and viral meningitis in children. J. Pediatrics Versão Port. 2018, 94, 88–92. [Google Scholar] [CrossRef]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants maintain cellular redox homeostasis by elimination of reactive oxygen species. Cell. Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef]
- Dunn, J.D.; Alvarez, L.A.; Zhang, X.; Soldati, T. Reactive oxygen species and mitochondria: A nexus of cellular homeostasis. Redox Biol. 2015, 6, 472–485. [Google Scholar] [CrossRef]
- Akaike, T. Host defense and oxidative stress signaling in bacterial infection. Nihon Saikingaku Zasshi 2015, 70, 339–349. [Google Scholar] [CrossRef][Green Version]
- Lindsay, J.; Esposti, M.D.; Gilmore, A.P. Bcl-2 proteins and mitochondria—Specificity in membrane targeting for death. Biochim. Biophys. Acta 2011, 1813, 532–539. [Google Scholar] [CrossRef]
- Park, H.H. Caspase recruitment domains for protein interactions in cellular signaling. Int. J. Mol. Med. 2019, 43, 1119–1127. [Google Scholar] [CrossRef]
- Valmiki, M.G.; Ramos, J. Death effector domain-containing proteins. Cell. Mol. Life Sci. 2009, 66, 814–830. [Google Scholar] [CrossRef] [PubMed]
- Fielden, L.F.; Kang, Y.; Newton, H.J.; Stojanovski, D. Targeting mitochondria: How intravacuolar bacterial pathogens manipulate mitochondria. Cell Tissue Res. 2017, 367, 141–154. [Google Scholar] [CrossRef]
- Singh, R.; Sripada, L.; Singh, R. Side effects of antibiotics during bacterial infection: Mitochondria, the main target in host cell. Mitochondrion 2014, 16, 50–54. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). Global Tuberculosis Report 2019–2019. Available online: https://www.who.int/tb/publications/global_report/en/ (accessed on 10 February 2021).
- Banuls, A.L.; Sanou, A.; Van Anh, N.T.; Godreuil, S. Mycobacterium tuberculosis: Ecology and evolution of a human bacterium. J. Med. Microbiol. 2015, 64, 1261–1269. [Google Scholar] [CrossRef] [PubMed]
- Dubey, R.K. Assuming the role of mitochondria in mycobacterial infection. Int. J. Mycobacteriol. 2016, 5, 379–383. [Google Scholar] [CrossRef]
- CDC. Tuberculosis: Signs & Symptoms. 2021. Available online: https://www.cdc.gov/tb/topic/basics/signsandsymptoms.htm (accessed on 10 February 2021).
- Cardona, P.J. Pathogenesis of tuberculosis and other mycobacteriosis. Enferm. Infecc. Microbiol. Clin. 2018, 36, 38–46. [Google Scholar] [CrossRef]
- Abarca-Rojano, E.; Rosas-Medina, P.; Zamudio-Cortez, P.; Mondragon-Flores, R.; Sanchez-Garcia, F.J. Mycobacterium tuberculosis virulence correlates with mitochondrial cytochrome c release in infected macrophages. Scand. J. Immunol. 2003, 58, 419–427. [Google Scholar] [CrossRef]
- Escombe, A.R.; Oeser, C.; Gilman, R.H.; Navincopa, M.; Ticona, E.; Martinez, C.; Caviedes, L.; Sheen, P.; Gonzalez, A.; Noakes, C.; et al. The detection of airborne transmission of tuberculosis from HIV-infected patients, using an in vivo air sampling model. Clin. Infect. Dis. 2007, 44, 1349–1357. [Google Scholar] [CrossRef] [PubMed]
- Arcos, J.; Sasindran, S.J.; Fujiwara, N.; Turner, J.; Schlesinger, L.S.; Torrelles, J.B. Human lung hydrolases delineate Mycobacterium tuberculosis-macrophage interactions and the capacity to control infection. J. Immunol. 2011, 187, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Storla, D.G.; Yimer, S.; Bjune, G.A. A systematic review of delay in the diagnosis and treatment of tuberculosis. BMC Public Health 2008, 8, 15. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, G.; Chen, C.; Portnoy, D.A. Strategies Used by Bacteria to Grow in Macrophages. Microbiol. Spectr. 2016, 4, 701–725. [Google Scholar]
- Pajuelo, D.; Gonzalez-Juarbe, N.; Tak, U.; Sun, J.; Orihuela, C.J.; Niederweis, M. NAD(+) Depletion Triggers Macrophage Necroptosis, a Cell Death Pathway Exploited by Mycobacterium tuberculosis. Cell Rep. 2018, 24, 429–440. [Google Scholar] [CrossRef]
- Kremer, L.; Maughan, W.N.; Wilson, R.A.; Dover, L.G.; Besra, G.S. The M. tuberculosis antigen 85 complex and mycolyltransferase activity. Lett. Appl. Microbiol. 2002, 34, 233–237. [Google Scholar] [CrossRef]
- Grange, J.M.; Yates, M.D. The time-table of tuberculosis. Respir Med. 1995, 89, 313–314. [Google Scholar] [CrossRef][Green Version]
- Heemskerk, D.C.M.; Marais, B.; Farrar, J. Tuberculosis in Adults and Children; Springer: Berlin/Heidelberg, Germany, 2015. [Google Scholar]
- Kulchavenya, E. Extrapulmonary tuberculosis: Are statistical reports accurate? Ther. Adv. Infect. Dis. 2014, 2, 61–70. [Google Scholar] [CrossRef]
- Lamb, G.S.; Starke, J.R. Tuberculosis in Infants and Children. Microbiol. Spectr. 2017, 5, 17–27. [Google Scholar] [CrossRef]
- Norouzi, S.; Aghamohammadi, A.; Mamishi, S.; Rosenzweig, S.D.; Rezaei, N. Bacillus Calmette-Guerin (BCG) complications associated with primary immunodeficiency diseases. J. Infect. 2012, 64, 543–554. [Google Scholar] [CrossRef]
- Venturini, E.; Turkova, A.; Chiappini, E.; Galli, L.; de Martino, M.; Thorne, C. Tuberculosis and HIV co-infection in children. BMC Infect Dis. 2014, 14 (Suppl. 1), S5. [Google Scholar] [CrossRef]
- Dangor, Z.; Izu, A.; Hillier, K.; Solomon, F.; Beylis, N.; Moore, D.P.; Nunes, M.C.; Madhi, S.A. Impact of the antiretroviral treatment program on the burden of hospitalization for culture-confirmed tuberculosis in South African children: A time-series analysis. Pediatrics Infect. Dis. J. 2013, 32, 972–977. [Google Scholar] [CrossRef]
- Tzelepis, F.; Blagih, J.; Khan, N.; Gillard, J.; Mendonca, L.; Roy, D.G.; Ma, E.H.; Joubert, P.; Jones, R.G.; Divangahi, M. Mitochondrial cyclophilin D regulates T cell metabolic responses and disease tolerance to tuberculosis. Sci. Immunol. 2018, 3, eaar4135. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Gan, H.; Golan, D.E.; Remold, H.G. Critical role of mitochondrial damage in determining outcome of macrophage infection with Mycobacterium tuberculosis. J. Immunol. 2002, 169, 5181–5187. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Gan, H.; Remold, H.G. A mechanism of virulence: Virulent Mycobacterium tuberculosis strain H37Rv, but not attenuated H37Ra, causes significant mitochondrial inner membrane disruption in macrophages leading to necrosis. J. Immunol. 2006, 176, 3707–3716. [Google Scholar] [CrossRef]
- Jamwal, S.; Midha, M.K.; Verma, H.N.; Basu, A.; Rao, K.V.; Manivel, V. Characterizing virulence-specific perturbations in the mitochondrial function of macrophages infected with Mycobacterium tuberculosis. Sci. Rep. 2013, 3, 1328. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, A.; Khare, N.K.; Bunkar, N.; Lenka, R.K.; Mishra, P.K. Role of mitochondrial oxidative stress on lymphocyte homeostasis in patients diagnosed with extra-pulmonary tuberculosis. Cell Biol. Int. 2016, 40, 166–176. [Google Scholar] [CrossRef]
- Sanchez, A.; Espinosa, P.; Garcia, T.; Mancilla, R. The 19 kDa Mycobacterium tuberculosis lipoprotein (LpqH) induces macrophage apoptosis through extrinsic and intrinsic pathways: A role for the mitochondrial apoptosis-inducing factor. Clin. Dev. Immunol. 2012, 2012, 950503. [Google Scholar] [CrossRef]
- Joseph, S.; Yuen, A.; Singh, V.; Hmama, Z. Mycobacterium tuberculosis Cpn60.2 (GroEL2) blocks macrophage apoptosis via interaction with mitochondrial mortalin. Biol. Open 2017, 6, 481–488. [Google Scholar] [CrossRef]
- Derrick, S.C.; Morris, S.L. The ESAT6 protein of Mycobacterium tuberculosis induces apoptosis of macrophages by activating caspase expression. Cell Microbiol. 2007, 9, 1547–1555. [Google Scholar] [CrossRef]
- Cumming, B.M.; Addicott, K.W.; Adamson, J.H.; Steyn, A.J. Mycobacterium tuberculosis induces decelerated bioenergetic metabolism in human macrophages. Elife 2018, 7, e39169. [Google Scholar] [CrossRef]
- Centers for Disease, Control and Prevention. Treatment for TB Disease 2016. Available online: https://www.cdc.gov/tb/topic/treatment/tbdisease.htm (accessed on 20 May 2020).
- Fernandes, G.; Salgado, H.R.N.; Santos, J.L.D. Isoniazid: A Review of Characteristics, Properties and Analytical Methods. Crit. Rev. Anal. Chem. 2017, 47, 298–308. [Google Scholar] [CrossRef]
- Vosatka, R.; Kratky, M.; Svarcova, M.; Janousek, J.; Stolarikova, J.; Madacki, J.; Vinsova, J. New lipophilic isoniazid derivatives and their 1,3,4-oxadiazole analogues: Synthesis, antimycobacterial activity and investigation of their mechanism of action. Eur. J. Med. Chem. 2018, 151, 824–835. [Google Scholar] [CrossRef]
- Ramachandran, A.; Visschers, R.G.J.; Duan, L.; Akakpo, J.Y.; Jaeschke, H. Mitochondrial dysfunction as a mechanism of drug-induced hepatotoxicity: Current understanding and future perspectives. J. Clin. Transl. Res. 2018, 4, 75–100. [Google Scholar]
- Adeyemo, A.A.; Oluwatosin, O.; Omotade, O.O. Study of streptomycin-induced ototoxicity: Protocol for a longitudinal study. Springerplus 2016, 5, 758. [Google Scholar] [CrossRef]
- Maalej, S.; Drira, I.; Ben Mefteh, R.; Fennira, H.; Bourguiba, M.; Ben Kheder, A. Isoniazid-induced visual hallucinosis. Presse Med. 2006, 35, 425–426. [Google Scholar]
- Grace, S.G. Barriers to the implementation of isoniazid preventive therapy for tuberculosis in children in endemic settings: A review. J. Paediatr. Child Health 2019, 55, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Jhanjhria, S.; Kashyap, B.; Gomber, S.; Gupta, N.; Hyanki, P.; Singh, N.P.; Sharma, A.K. Phenotypic isoniazid resistance and associated mutations in pediatric tuberculosis. Indian J. Tuberc. 2019, 66, 474–479. [Google Scholar] [CrossRef] [PubMed]
- Rifampin. Tuberculosis Edinb. 2008, 88, 151–154. [CrossRef]
- Diallo, T.; Adjobimey, M.; Ruslami, R.; Trajman, A.; Sow, O.; Obeng Baah, J.; Marks, G.B.; Richard Long, F.R.A.C.P.; Elwood, K.; Zielinski, D.; et al. Safety and Side Effects of Rifampin versus Isoniazid in Children. N. Engl. J. Med. 2018, 379, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Gaensbauer, J.; Aiona, K.; Haas, M.; Reves, R.; Young, J.; Belknap, R. Better Completion of Pediatric Latent Tuberculosis Treatment Using 4 Months of Rifampin in a US-based Tuberculosis Clinic. Pediatrics Infect. Dis. J. 2018, 37, 224–228. [Google Scholar] [CrossRef]
- Yendapally, R.; Lee, R.E. Design, synthesis, and evaluation of novel ethambutol analogues. Bioorg. Med. Chem. Lett. 2008, 18, 1607–1611. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wikipedia—La Enciclopedia Libre. Etambutol 2019. Available online: https://es.wikipedia.org/wiki/Etambutol (accessed on 20 May 2020).
- Ethambutol. Tuberculosis Edinb. 2008, 88, 102–105. [CrossRef]
- Chung, H.; Yoon, Y.H.; Hwang, J.J.; Cho, K.S.; Koh, J.Y.; Kim, J.G. Ethambutol-induced toxicity is mediated by zinc and lysosomal membrane permeabilization in cultured retinal cells. Toxicol. Appl. Pharmacol. 2009, 235, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Dotti, M.T.; Plewnia, K.; Cardaioli, E.; Manneschi, L.; Rufa, A.; Alema, G.; Federico, A. A case of ethambutol-induced optic neuropathy harbouring the primary mitochondrial LHON mutation at nt 11778. J. Neurol. 1998, 245, 302–303. [Google Scholar] [CrossRef] [PubMed]
- Heng, J.E.; Vorwerk, C.K.; Lessell, E.; Zurakowski, D.; Levin, L.A.; Dreyer, E.B. Ethambutol is toxic to retinal ganglion cells via an excitotoxic pathway. Investig. Ophthalmol. Vis. Sci. 1999, 40, 190–196. [Google Scholar] [PubMed]
- Yu-Wai-Man, P.; Griffiths, P.G.; Chinnery, P.F. Mitochondrial optic neuropathies—Disease mechanisms and therapeutic strategies. Prog. Retin. Eye Res. 2011, 30, 81–114. [Google Scholar] [CrossRef]
- Guillet, V.; Chevrollier, A.; Cassereau, J.; Letournel, F.; Gueguen, N.; Richard, L.; Desquiret, V.; Verny, C.; Procaccio, V.; Amati-Bonneau, P.; et al. Ethambutol-induced optic neuropathy linked to OPA1 mutation and mitochondrial toxicity. Mitochondrion 2010, 10, 115–124. [Google Scholar] [CrossRef]
- Trebucq, A. Should ethambutol be recommended for routine treatment of tuberculosis in children? A review of the literature. Int. J. Tuberc. Lung Dis. 1997, 1, 12–15. [Google Scholar]
- Donald, P.R.; Maher, D.; Maritz, J.S.; Qazi, S. Ethambutol dosage for the treatment of children: Literature review and recommendations. Int. J. Tuberc. Lung Dis. 2006, 10, 1318–1330. [Google Scholar]
- Wikipedia—La Enciclopedia Libre. Pirazinamida. 2019. Available online: https://es.wikipedia.org/wiki/Pirazinamida (accessed on 20 May 2020).
- Zhang, Y.; Liu, K.; Hassan, H.M.; Guo, H.; Ding, P.; Han, L.; Jiang, Z. Liver Fatty Acid Binding Protein Deficiency Provokes Oxidative Stress, Inflammation, and Apoptosis-Mediated Hepatotoxicity Induced by Pyrazinamide in Zebrafish Larvae. Antimicrob. Agents Chemother. 2016, 60, 7347–7356. [Google Scholar] [PubMed]
- Roy, V.; Tekur, U.; Chopra, K. Pharmacokinetics of pyrazinamide in children suffering from pulmonary tuberculosis. Int. J. Tuberc. Lung Dis. 1999, 3, 133–137. [Google Scholar] [PubMed]
- Thee, S.; Detjen, A.; Wahn, U.; Magdorf, K. Pyrazinamide serum levels in childhood tuberculosis. Int. J. Tuberc. Lung Dis. 2008, 12, 1099–1101. [Google Scholar]
- Arya, D.S.; Ojha, S.K.; Semwal, O.P.; Nandave, M. Pharmacokinetics of pyrazinamide in children with primary progressive disease of lungs. Indian J. Med. Res. 2008, 128, 611–615. [Google Scholar] [PubMed]
- Baciewicz, A.M.; Chrisman, C.R.; Finch, C.K.; Self, T.H. Update on rifampin, rifabutin, and rifapentine drug interactions. Curr. Med. Res. Opin. 2013, 29, 1–12. [Google Scholar] [CrossRef]
- Wikipedia—La Enciclopedia Libre. Rifapentina. 2019. Available online: https://es.wikipedia.org/wiki/Rifapentina (accessed on 20 May 2020).
- Weiner, M.; Savic, R.M.; Kenzie, W.R.; Wing, D.; Peloquin, C.A.; Engle, M.; Bliven, E.; Prihoda, T.J.; Gelfond, J.A.; Scott, N.A.; et al. Rifapentine Pharmacokinetics and Tolerability in Children and Adults Treated Once Weekly with Rifapentine and Isoniazid for Latent Tuberculosis Infection. J. Pediatric Infect. Dis. Soc. 2014, 3, 132–145. [Google Scholar] [CrossRef]
- Villarino, M.E.; Scott, N.A.; Weis, S.E.; Weiner, M.; Conde, M.B.; Jones, B.; Nachman, S.; Oliveira, R.; Moro, R.N.; Shang, N.; et al. Treatment for preventing tuberculosis in children and adolescents: A randomized clinical trial of a 3-month, 12-dose regimen of a combination of rifapentine and isoniazid. JAMA Pediatrics 2015, 169, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Blake, M.J.; Abdel-Rahman, S.M.; Jacobs, R.F.; Lowery, N.K.; Sterling, T.R.; Kearns, G.L. Pharmacokinetics of rifapentine in children. Pediatrics Infect. Dis. J. 2006, 25, 405–409. [Google Scholar] [CrossRef]
- Song, M.; Wu, H.; Wu, S.; Ge, T.; Wang, G.; Zhou, Y.; Sheng, S.; Jiang, J. Antibiotic drug levofloxacin inhibits proliferation and induces apoptosis of lung cancer cells through inducing mitochondrial dysfunction and oxidative damage. Biomed. Pharmacother. 2016, 84, 1137–1143. [Google Scholar] [CrossRef]
- Yu, M.; Li, R.; Zhang, J. Repositioning of antibiotic levofloxacin as a mitochondrial biogenesis inhibitor to target breast cancer. Biochem. Biophys. Res. Commun. 2016, 471, 639–645. [Google Scholar] [CrossRef]
- Li, H.T.; Zhu, S.Y.; Pei, H.S. The effect of moxifloxacin on apoptosis of airway smooth muscle cells and mitochondria membrane potential. Zhonghua Jie He He Hu Xi Za Zhi 2011, 34, 684–687. [Google Scholar]
- Lancella, L.; Vecchio, A.L.; Chiappini, E.; Tadolini, M.; Cirillo, D.; Tortoli, E.; de Martino, M.; Guarino, A.; Principi, N.; Villani, A.; et al. How to manage children who have come into contact with patients affected by tuberculosis. J. Clin. Tuberc. Other. Mycobact. Dis. 2015, 1, 1–12. [Google Scholar] [CrossRef][Green Version]
- Santini, A.; Ronchi, D.; Garbellini, M.; Piga, D.; Protti, A. Linezolid-induced lactic acidosis: The thin line between bacterial and mitochondrial ribosomes. Expert. Opin. Drug Saf. 2017, 16, 833–843. [Google Scholar] [CrossRef] [PubMed]
- De Vriese, A.S.; Coster, R.V.; Smet, J.; Seneca, S.; Lovering, A.; Van Haute, L.L.; Vanopdenbosch, L.J.; Martin, J.J.; Groote, C.C.; Vandecasteele, S.; et al. Linezolid-induced inhibition of mitochondrial protein synthesis. Clin. Infect. Dis. 2006, 42, 1111–1117. [Google Scholar] [CrossRef]
- Aguado García, J.M.; Lumbreras Bermejo, C. Infecciones por enterobacterias. Medicine 1998, 7, 3622–3628. [Google Scholar]
- Isselbacher, K.J.B.E.; Wilson, J.D.; Petersdorf, R.G.; Martin, J.B.; Fauci, A.J.; Root, R.K. Diseases Caused by Gram-Negative Enteric Bacilli. In Harrison’s Principles of Internal Medicine, 13th ed.; McGraw-Hill: New York, NY, USA, 1994; pp. 661–669. [Google Scholar]
- Foley, S.L.; Johnson, T.J.; Ricke, S.C.; Nayak, R.; Danzeisen, J. Salmonella pathogenicity and host adaptation in chicken-associated serovars. Microbiol. Mol. Biol. Rev. 2013, 77, 582–607. [Google Scholar] [CrossRef]
- Ecured. Enterobacter Cloacae. 2007. Available online: https://www.ecured.cu/Enterobacter_cloacae (accessed on 20 May 2020).
- Amin, H.; Zafar, A.; Ejaz, H.; Jameel, N.U. Phenotypic characterization of ESBL producing Enterobacter cloacae among children. Pak. J. Med. Sci. 2013, 29, 144–147. [Google Scholar] [CrossRef]
- Chen, H.; Wang, C.; Wang, X.; Guo, Z.; Xu, Z.; Zhao, Y.; Liu, J. A polysaccharide from Enterobacter cloacae induces apoptosis of human osteosarcoma cells through the activation of p53 and mitochondrial intrinsic pathway. Int. J. Biol. Macromol. 2019, 122, 58–63. [Google Scholar] [CrossRef]
- Cavagnaro, F. Urinary tract infection in childhood. Rev. Chilena Infectol. 2005, 22, 161–168. [Google Scholar]
- Mathoera, R.B.; Kok, D.J.; Verduin, C.M.; Nijman, R.J. Pathological and therapeutic significance of cellular invasion by Proteus mirabilis in an enterocystoplasty infection stone model. Infect. Immun. 2002, 70, 7022–7032. [Google Scholar] [CrossRef]
- Nougayrede, J.P.; Donnenberg, M.S. Enteropathogenic Escherichia coli EspF is targeted to mitochondria and is required to initiate the mitochondrial death pathway. Cell Microbiol. 2004, 6, 1097–1111. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). Salmonella no Tifoidea 2018. Available online: https://www.who.int/es/news-room/fact-sheets/detail/salmonella-(non-typhoidal) (accessed on 20 May 2020).
- Ahmadova, R. Effect of the low intensity laser rays on the functional state of monocytes in early aged children with salmonellosis infection. Azerbaijan Med J. 2003, 4, 9–12. [Google Scholar]
- Lowy, F.D. Staphylococcus aureus infections. N. Engl. J. Med. 1998, 339, 520–532. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, J.R. Evolution of Staphylococcus aureus during human colonization and infection. Infect. Genet. Evol. 2014, 21, 542–547. [Google Scholar] [CrossRef] [PubMed]
- Prava, J.; Pranavathiyani, G.; Pan, A. Functional assignment for essential hypothetical proteins of Staphylococcus aureus N315. Int. J. Biol. Macromol. 2018, 108, 765–774. [Google Scholar] [CrossRef]
- Cervantes-García, E.; García-González, R.; Salazar-Schettino, P.M. Características Generales del Staphylococcus aureus Patología Cínica. Patología de Laboratorio 2014. Available online: https://www.medigraphic.com/pdfs/patol/pt-2014/pt141e.pdf (accessed on 10 February 2021).
- Dayan, G.H.; Mohamed, N.; Scully, I.L.; Cooper, D.; Begier, E.; Eiden, J.; Jansen, K.U.; Gurtman, A.; Anderson, A.S. Staphylococcus aureus: The current state of disease, pathophysiology and strategies for prevention. Expert Rev. Vaccines 2016, 15, 1373–1392. [Google Scholar] [CrossRef]
- Socorro Zendejas-Manzo, G.; Avalos-Flores, H.; Soto-Padilla, M.Y. Microbiología General de Staphylococcus aureus: Generalidades, Patogenicidad y Métodos de Identificación 2014. Available online: https://www.medigraphic.com/pdfs/revbio/bio-2014/bio143d.pdf (accessed on 10 February 2021).
- Grosz, M.; Kolter, J.; Paprotka, K.; Winkler, A.C.; Schafer, D.; Chatterjee, S.S.; Geiger, T.; Wolz, C.; Ohlsen, K.; Otto, M.; et al. Cytoplasmic replication of Staphylococcus aureus upon phagosomal escape triggered by phenol-soluble modulin alpha. Cell Microbiol. 2014, 16, 451–465. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.B.; Weber, D.J.; Goodrich, J.S.; Popowitch, E.B.; Poe, M.D.; Nyugen, V.; Shope, T.R.; Foster, D.T.; Miller, J.R.; Kotch, J. Prevalence and risk factor analysis for methicillin-resistant Staphylococcus aureus nasal colonization in children attending child care centers. J. Clin. Microbiol. 2011, 49, 1041–1047. [Google Scholar] [CrossRef]
- Odutola, A.; Bottomley, C.; Zaman, S.A.; Lindsay, J.; Shah, M.; Hossain, I.; Ndiaye, M.; Osuorah, C.D.I.; Olatunji, Y.; Badji, H.; et al. Staphylococcus aureus Bacteremia in Children of Rural Areas of The Gambia, 2008–2015. Emerg. Infect. Dis. 2019, 25, 701–709. [Google Scholar] [CrossRef] [PubMed]
- Uehara, Y.; Sasaki, T.; Baba, T.; Lu, Y.; Imajo, E.; Sato, Y.; Tanno, S.; Furuichi, M.; Kawada, M.; Hiramatsu, K. Regional outbreak of community-associated methicillin-resistant Staphylococcus aureus ST834 in Japanese children. BMC Infect Dis. 2019, 19, 35. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.K.; Kumar, A. Mitochondria mediates caspase-dependent and independent retinal cell death in Staphylococcus aureus endophthalmitis. Cell Death Discov. 2016, 2, 16034. [Google Scholar] [CrossRef]
- Choe, H.R.; Kim, J.H.; Ma, A.; Jung, H.; Kim, H.Y.; Nam, K.M. Understanding Reaction Kinetics by Tailoring Metal Co-catalysts of the BiVO4 Photocatalyst. ACS Omega 2019, 4, 16597–16602. [Google Scholar] [CrossRef]
- Weglarczyk, K.; Baran, J.; Zembala, M.; Pryjma, J. Caspase-8 activation precedes alterations of mitochondrial membrane potential during monocyte apoptosis induced by phagocytosis and killing of Staphylococcus aureus. Infect. Immun. 2004, 72, 2590–2597. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.B.; Coon, T.A.; Glasser, J.R.; Zou, C.; Ellis, B.; Das, T.; McKelvey, A.C.; Rajbhandari, S.; Lear, T.; Kamga, C.; et al. E3 ligase subunit Fbxo15 and PINK1 kinase regulate cardiolipin synthase 1 stability and mitochondrial function in pneumonia. Cell Rep. 2014, 7, 476–487. [Google Scholar] [CrossRef] [PubMed]
- Genestier, A.L.; Michallet, M.C.; Prevost, G.; Bellot, G.; Chalabreysse, L.; Peyrol, S.; Thivolet, F.; Etienne, J.; Lina, G.; Vallette, F.M.; et al. Staphylococcus aureus Panton-Valentine leukocidin directly targets mitochondria and induces Bax-independent apoptosis of human neutrophils. J. Clin. Investig. 2005, 115, 3117–3127. [Google Scholar] [CrossRef]
- Alonzo, F., 3rd; Torres, V.J. The bicomponent pore-forming leucocidins of Staphylococcus aureus. Microbiol. Mol. Biol. Rev. 2014, 78, 199–230. [Google Scholar] [CrossRef] [PubMed]
- Fraunholz, M.; Sinha, B. Intracellular Staphylococcus aureus: Live-in and let die. Front. Cell Infect. Microbiol. 2012, 2, 43. [Google Scholar] [CrossRef]
- Krause, K.; Daily, K.; Estfanous, S.; Hamilton, K.; Badr, A.; Abu Khweek, A.; Hegazi, R.; Anne, M.N.; Klamer, B.; Zhang, X.; et al. Caspase-11 counteracts mitochondrial ROS-mediated clearance of Staphylococcus aureus in macrophages. EMBO Rep. 2019, 20, e48109. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.S.; Fowler, V.G.; Shukla, S.K.; Rose, W.E.; Proctor, R.A. Development of a vaccine against Staphylococcus aureus invasive infections: Evidence based on human immunity, genetics and bacterial evasion mechanisms. FEMS Microbiol. Rev. 2020, 44, 123–153. [Google Scholar] [CrossRef]
- Kane, T.L.; Carothers, K.E.; Lee, S.W. Virulence Factor Targeting of the Bacterial Pathogen Staphylococcus aureus for Vaccine and Therapeutics. Curr. Drug Targets 2018, 19, 111–127. [Google Scholar] [CrossRef]
- Lakhundi, S.; Zhang, K. Methicillin-Resistant Staphylococcus aureus: Molecular Characterization, Evolution, and Epidemiology. Clin. Microbiol. Rev. 2018, 31. [Google Scholar] [CrossRef]
- Hodille, E.; Rose, W.; Diep, B.A.; Goutelle, S.; Lina, G.; Dumitrescu, O. The Role of Antibiotics in Modulating Virulence in Staphylococcus aureus. Clin. Microbiol. Rev. 2017, 30, 887–917. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, M.; Young, L.; Kirkwood, N.K.; Richardson, G.P.; Kros, C.J.; Moore, A.L. Gentamicin Affects the Bioenergetics of Isolated Mitochondria and Collapses the Mitochondrial Membrane Potential in Cochlear Sensory Hair Cells. Front. Cell Neurosci. 2019, 13, 416. [Google Scholar] [CrossRef] [PubMed]
- Elliott, R.L.; Jiang, X.P. The adverse effect of gentamicin on cell metabolism in three cultured mammary cell lines: “Are cell culture data skewed?”. PLoS ONE 2019, 14, e0214586. [Google Scholar] [CrossRef] [PubMed]
- Jia, P.; Teng, J.; Zou, J.; Fang, Y.; Jiang, S.; Yu, X.; Kriegel, A.J.; Liang, M.; Ding, X. Intermittent exposure to xenon protects against gentamicin-induced nephrotoxicity. PLoS ONE 2013, 8, e64329. [Google Scholar] [CrossRef]
- McWilliam, S.J.; Antoine, D.J.; Smyth, R.L.; Pirmohamed, M. Aminoglycoside-induced nephrotoxicity in children. Pediatrics Nephrol. 2017, 32, 2015–2025. [Google Scholar] [CrossRef]
- Garrabou, G.; Soriano, A.; Lopez, S.; Guallar, J.P.; Giralt, M.; Villarroya, F.; Martinez, J.A.; Casademont, J.; Cardellach, F.; Mensa, J.; et al. Reversible inhibition of mitochondrial protein synthesis during linezolid-related hyperlactatemia. Antimicrob. Agents Chemother. 2007, 51, 962–967. [Google Scholar] [CrossRef]
- Palenzuela, L.; Hahn, N.M.; Nelson, R.P., Jr.; Arno, J.N.; Schobert, C.; Bethel, R.; Ostrowski, L.A.; Sharma, M.R.; Datta, P.P.; Agrawal, R.K.; et al. Does linezolid cause lactic acidosis by inhibiting mitochondrial protein synthesis? Clin. Infect. Dis. 2005, 40, e113–e116. [Google Scholar]
- Su, E.; Crowley, K.; Carcillo, J.A.; Michaels, M.G. Linezolid and lactic acidosis: A role for lactate monitoring with long-term linezolid use in children. Pediatrics Infect. Dis. J. 2011, 30, 804–806. [Google Scholar] [CrossRef]
- Johnson, P.C.; Vaduganathan, M.; Phillips, K.M.; O’Donnell, W.J. A triad of linezolid toxicity: Hypoglycemia, lactic acidosis, and acute pancreatitis. Proc. Bayl. Univ. Med. Cent. 2015, 28, 466–468. [Google Scholar] [CrossRef]
- Vademecum. Doxiciclina Madrid. 2017. Available online: https://www.vademecum.es/principios-activos-doxiciclina-J01AA02 (accessed on 10 February 2021).
- Tan, Q.; Yan, X.; Song, L.; Yi, H.; Li, P.; Sun, G.; Yu, D.; Li, L.; Zeng, Z.; Guo, Z. Induction of Mitochondrial Dysfunction and Oxidative Damage by Antibiotic Drug Doxycycline Enhances the Responsiveness of Glioblastoma to Chemotherapy. Med. Sci. Monit. 2017, 23, 4117–4125. [Google Scholar] [CrossRef]
- Luger, A.L.; Sauer, B.; Lorenz, N.I.; Engel, A.L.; Braun, Y.; Voss, M.; Harter, P.N.; Steinbach, J.P.; Ronellenfitsch, M.W. Doxycycline Impairs Mitochondrial Function and Protects Human Glioma Cells from Hypoxia-Induced Cell Death: Implications of Using Tet-Inducible Systems. Int. J. Mol. Sci. 2018, 19, 1504. [Google Scholar] [CrossRef] [PubMed]
- Thompson, E.J.; Wu, H.; Melloni, C.; Balevic, S.; Sullivan, J.E.; Laughon, M.; Clark, K.M.; Kalra, R.; Mendley, S.; Payne, E.H.; et al. Population Pharmacokinetics of Doxycycline in Children. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef]
- Vademecum. Tigeciclina 2016. Available online: https://www.vademecum.es/medicamento-tigeciclina%20mylan_48058 (accessed on 20 May 2020).
- Dong, Z.; Abbas, M.N.; Kausar, S.; Yang, J.; Li, L.; Tan, L.; Cui, H. Biological Functions and Molecular Mechanisms of Antibiotic Tigecycline in the Treatment of Cancers. Int. J. Mol. Sci. 2019, 20, 3577. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Liu, W.; Huang, Q.; Wang, J.; Wang, Y.; Li, H.; Fu, X. Tigecycline as a dual inhibitor of retinoblastoma and angiogenesis via inducing mitochondrial dysfunctions and oxidative damage. Sci. Rep. 2018, 8, 11747. [Google Scholar] [CrossRef] [PubMed]
- Vademecum. Vancomicina. 2016. Available online: https://www.vademecum.es/principios-activos-vancomicina-J01XA01 (accessed on 20 May 2020).
- Ehrenreich, H.; Spaepen, F. Glycopeptide Antibiotics. In Advances in Research and Application; Academic Press: Cambridge, MA, USA, 2013; pp. 42–43. [Google Scholar]
- McNeil, J.C.; Kok, E.Y.; Forbes, A.R.; Lamberth, L.; Hulten, K.G.; Vallejo, J.G.; Kaplan, S.L. Healthcare-associated Staphylococcus aureus Bacteremia in Children: Evidence for Reverse Vancomycin Creep and Impact of Vancomycin Trough Values on Outcome. Pediatrics Infect. Dis. J. 2016, 35, 263–268. [Google Scholar] [CrossRef]
- Madigan, T.; Sieve, R.M.; Graner, K.K.; Banerjee, R. The effect of age and weight on vancomycin serum trough concentrations in pediatric patients. Pharmacotherapy 2013, 33, 1264–1272. [Google Scholar] [CrossRef]
- Szabo, L.; Toth, T.; Takacs, E.; Wojnarovits, L. One-Electron Reduction of Penicillins in Relation to the Oxidative Stress Phenomenon. Int. J. Mol. Sci. 2015, 16, 29673–29681. [Google Scholar] [CrossRef] [PubMed]
- Kalghatgi, S.; Spina, C.S.; Costello, J.C.; Liesa, M.; Morones-Ramirez, J.R.; Slomovic, S.; Molina, A.; Shirihai, O.S.; Collins, J.J. Bactericidal antibiotics induce mitochondrial dysfunction and oxidative damage in Mammalian cells. Sci. Transl. Med. 2013, 5, 192ra85. [Google Scholar] [CrossRef]
- Syriopoulou, V.; Dailiana, Z.; Dmitriy, N.; Utili, R.; Pathan, R.; Hamed, K. Clinical Experience with Daptomycin for the Treatment of Gram-positive Infections in Children and Adolescents. Pediatrics Infect. Dis. J. 2016, 35, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Haden, D.W.; Suliman, H.B.; Carraway, M.S.; Welty-Wolf, K.E.; Ali, A.S.; Shitara, H.; Yonekawa, H.; Piantadosi, C.A. Mitochondrial biogenesis restores oxidative metabolism during Staphylococcus aureus sepsis. Am. J. Respir. Crit. Care Med. 2007, 176, 768–777. [Google Scholar] [CrossRef] [PubMed]
- Stanford Children’s Health. Meningitis en Niños. 2020. Available online: https://www.stanfordchildrens.org/es/topic/default?id=meningitis-en-nios-90-P05638 (accessed on 20 May 2020).
- Medline. Meningitis. Available online: https://medlineplus.gov/spanish/meningitis.html (accessed on 20 May 2020).
- Roca Goderich, R. Meningitis. Enfermedades del Sistema Nervioso Central; ECIMED: Havana, Cuba, 2002; Volume II, p. 138. [Google Scholar]
- Schoen, C.; Kischkies, L.; Elias, J.; Ampattu, B.J. Metabolism and virulence in Neisseria meningitidis. Front. Cell Infect. Microbiol. 2014, 4, 114. [Google Scholar] [CrossRef]
- Rouphael, N.G.; Stephens, D.S. Neisseria meningitidis: Biology, microbiology, and epidemiology. Methods Mol. Biol. 2012, 799, 1–20. [Google Scholar]
- Stephens, D.S.; Greenwood, B.; Brandtzaeg, P. Epidemic meningitis, meningococcaemia, and Neisseria meningitidis. Lancet 2007, 369, 2196–2210. [Google Scholar] [CrossRef]
- Chang, Q.; Tzeng, Y.L.; Stephens, D.S. Meningococcal disease: Changes in epidemiology and prevention. Clin. Epidemiol. 2012, 4, 237–245. [Google Scholar] [PubMed]
- Plus, M. Meningitis Neumocócica 2020. Available online: https://medlineplus.gov/spanish/ency/article/000607.htm#:~:text=La%20meningitis%20neumoc%C3%B3cica%20es%20causada,ni%C3%B1os%20mayores%20de%202%20a%C3%B1os (accessed on 20 May 2020).
- Woese, C.R. Bacterial evolution. Microbiol Rev. 1987, 51, 221–271. [Google Scholar] [CrossRef]
- Engelmoer, D.J.; Rozen, D.E. Competence increases survival during stress in Streptococcus pneumoniae. Evolution 2011, 65, 3475–3485. [Google Scholar] [CrossRef] [PubMed]
- Tommassen, J.; Arenas, J. Biological Functions of the Secretome of Neisseria meningitidis. Front. Cell Infect. Microbiol. 2017, 7, 256. [Google Scholar] [CrossRef]
- Valmari, P.; Peltola, H.; Ruuskanen, O.; Korvenranta, H. Childhood bacterial meningitis: Initial symptoms and signs related to age, and reasons for consulting a physician. Eur. J. Pediatrics 1987, 146, 515–518. [Google Scholar] [CrossRef]
- Harwood, C.A.; Stevens, J.C.; Orton, D.; Bull, R.C.; Paige, D.; Lessing, M.P.; Mortimer, P.S.; Marsden, R.A.; Cerio, R. Chronic meningococcaemia: A forgotten meningococcal disease. Br. J. Dermatol. 2005, 153, 669–671. [Google Scholar] [CrossRef] [PubMed]
- Bosis, S.; Mayer, A.; Esposito, S. Meningococcal disease in childhood: Epidemiology, clinical features and prevention. J. Prev. Med. Hyg. 2015, 56, E121–E124. [Google Scholar] [PubMed]
- Vacca, I.; Del Tordello, E.; Gasperini, G.; Pezzicoli, A.; Di Fede, M.; Rossi Paccani, S.; Marchi, S.; Mubaiwa, T.D.; Hartley-Tassell, L.E.; Jennings, M.P.; et al. Neisserial Heparin Binding Antigen (NHBA) Contributes to the Adhesion of Neisseria meningitidis to Human Epithelial Cells. PLoS ONE 2016, 11, e0162878. [Google Scholar] [CrossRef] [PubMed]
- Stockmann, C.; Ampofo, K.; Byington, C.L.; Filloux, F.; Hersh, A.L.; Blaschke, A.J.; Cowan, P.; Korgenski, K.; Mason, E.O.; Pavia, A.T. Pneumococcal meningitis in children: Epidemiology, serotypes, and outcomes from 1997–2010 in Utah. Pediatrics 2013, 132, 421–428. [Google Scholar] [CrossRef] [PubMed]
- INSST. Streptococcus Pneumoniae 2018. Available online: https://www.insst.es/documents/94886/353165/Streptococcus+pneumoniae+-+A%C3%B1o+2019.pdf/93020441–818d-4981-b2a6-a38336430e49 (accessed on 20 May 2020).
- Saez-Llorens, X.; McCracken, G.H., Jr. Bacterial meningitis in children. Lancet 2003, 361, 2139–2148. [Google Scholar] [CrossRef]
- Correa, J.; Gómez, J.; Posada, R. Streptococcus Pneumoniae (Serotipos 6, 9, 14, 18 y 23) Niños: Fundamentos de. Available online: https://books.google.es/books?id=cpfGDwAAQBAJ&pg=PT1083&lpg=PT1083&dq=Streptococcus+pneumoniae+(serotipos+6,+9,+14,+18+y+23)+ni%C3%B1os&source=bl&ots=EcgOVi1KAq&sig=ACfU3U0zL43-vDONM_LjL-8pr1r75x_aSw&hl=es&sa=X&ved=2ahUKEwju6ueIhpDqAhW0D2MBHUT3AyMQ6AEwBHoECAsQAQ#v=onepage&q=Streptococcus%20pneumoniae%20(serotipos%206%2C%209%2C%2014%2C%2018%20y%2023)%20ni%C3%B1os&f=false (accessed on 20 May 2020).
- Tunkel, A.R.; Hartman, B.J.; Kaplan, S.L.; Kaufman, B.A.; Roos, K.L.; Scheld, W.M.; Whitley, R.J. Practice guidelines for the management of bacterial meningitis. Clin. Infect. Dis. 2004, 39, 1267–1284. [Google Scholar] [CrossRef]
- Jauneikaite, E.; Tocheva, A.S.; Jefferies, J.M.; Gladstone, R.A.; Faust, S.N.; Christodoulides, M.; Hibberd, M.L.; Clarke, S.C. Current methods for capsular typing of Streptococcus pneumoniae. J. Microbiol. Methods 2015, 113, 41–49. [Google Scholar] [CrossRef]
- Ahmad, N.; Plorde, J.W.D. Sherris Medical Microbiology, 5th ed.; McGraw-Hill: New York, NY, USA, 2018. [Google Scholar]
- Servicio Madrileño de Salud. Protocolo de Actuación Frente a Enfermedad Meningocócica. Red de Vigilancia Epidemiológica. 2011. Available online: http://www.madrid.org/cs/Satellite?blobcol=urldata&blobheader=application%2Fpdf&blobheadername1=Content-disposition&blobheadername2=cadena&blobheadervalue1=filename%3DEnfermedad+Meningococica.pdf&blobheadervalue2=language%3Des%26site%3DPortalSalud&blobkey=id&blobtable=MungoBlobs&blobwhere=1352862630691&ssbinary=true (accessed on 20 May 2020).
- Arditi, M.; Mason, E.O., Jr.; Bradley, J.S.; Tan, T.Q.; Barson, W.J.; Schutze, G.E.; Wald, E.R.; Givner, L.B.; Kim, K.S.; Yogev, R.; et al. Three-year multicenter surveillance of pneumococcal meningitis in children: Clinical characteristics, and outcome related to penicillin susceptibility and dexamethasone use. Pediatrics 1998, 102, 1087–1097. [Google Scholar] [CrossRef]
- Braun, J.S.; Sublett, J.E.; Freyer, D.; Mitchell, T.J.; Cleveland, J.L.; Tuomanen, E.I.; Weber, J.R. Pneumococcal pneumolysin and H(2)O(2) mediate brain cell apoptosis during meningitis. J. Clin. Investig. 2002, 109, 19–27. [Google Scholar] [CrossRef]
- Loughran, A.J.; Orihuela, C.J.; Tuomanen, E.I. Streptococcus pneumoniae: Invasion and Inflammation. Microbiol. Spectr. 2019, 7. [Google Scholar] [CrossRef]
- Vademecum. Ampicilina. 2016. Available online: https://www.vademecum.es/principios-activos-ampicilina-J01CA01 (accessed on 20 May 2020).
- Baker, C.J.; Long, S.S. 50 Years Ago in The Journal of Pediatrics: Ampicillin in the Treatment of Meningitis due to Haemophilus influenzae: An Appraisal after 6 Years of Experience. J. Pediatrics 2019, 208, 37. [Google Scholar] [CrossRef]
- Toltzis, P. 50 Years Ago in The Journal of Pediatrics: Relapse of Hemophilus influenzae Type b Meningitis during Intravenous Therapy with Ampicillin. J. Pediatrics 2019, 208, 182. [Google Scholar] [CrossRef] [PubMed]
- Vademecum. Cefotaxima. Available online: https://www.vademecum.es/principios-activos-cefotaxima-j01dd01 (accessed on 10 February 2021).
- Fang, C.C.; Yen, C.J.; Tsai, T.J.; Chen, R.H.; Lee, P.H.; Tomino, Y. Antibiotics induce apoptosis of human peritoneal mesothelial cells. Nephrol. Carlton 2003, 8, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Friedland, I.R.; Klugman, K.P. Cerebrospinal fluid bactericidal activity against cephalosporin-resistant Streptococcus pneumoniae in children with meningitis treated with high-dosage cefotaxime. Antimicrob. Agents Chemother. 1997, 41, 1888–1891. [Google Scholar] [CrossRef]
- Vademecum. Cefalosporina 2019. Available online: https://www.vademecum.es/principios-activos-ceftriaxona-J01DD04 (accessed on 10 February 2021).
- Rumbaugh, J.A.; Li, G.; Rothstein, J.; Nath, A. Ceftriaxone protects against the neurotoxicity of human immunodeficiency virus proteins. J. Neurovirol. 2007, 13, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Maltecca, F.; Baseggio, E.; Consolato, F.; Mazza, D.; Podini, P.; Young, S.M., Jr.; Casari, G. Purkinje neuron Ca2+ influx reduction rescues ataxia in SCA28 model. J. Clin. Investig. 2015, 125, 263–274. [Google Scholar] [CrossRef]
- Zeng, L.; Choonara, I.; Zhang, L.; Xue, S.; Chen, Z.; He, M. Safety of ceftriaxone in paediatrics: A systematic review protocol. BMJ Open 2017, 7, e016273. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Merino, E.; Solis, J.; Solera, J.; Mateos, F. Protocolo de Diagnóstico y Tratamiento de la Meningitis Aguda: Servicio de Medicina Interna. Available online: https://www.chospab.es/area_medica/medicinainterna/PROTOCOLOS/meningitis.htm (accessed on 20 May 2020).
- World Health Organization. Meningitis Meningocócica 2018. Available online: https://www.who.int/es/news-room/fact-sheets/detail/meningococcal-meningitis (accessed on 20 May 2020).
- World Health Organization (WHO). WHO Vaccine-Preventable Diseases: Monitoring System 2019. Available online: https://apps.who.int/immunization_monitoring/globalsummary/schedules (accessed on 20 May 2020).
- Barichello, T.F.D.; Generoso, J.; Elias, S.; Simões, L.; Teixeira, A. Pathophysiology of neonatal acute bacterial meningitis. J. Med Microbiol. 2013, 62, 1781–1789. [Google Scholar] [CrossRef]
- Murawska-Cialowicz, E.S.Z.; Trebusiewicz, B. Nitric oxide production during bacterial and viral meningitis in children. Int. J. Clin. Lab. Res. 2000, 30, 127–131. [Google Scholar] [CrossRef]
- Wang, X.; Miyazaki, Y.; Inaoka, D.K.; Hartuti, E.D.; Watanabe, Y.I.; Shiba, T.; Harada, S.; Saimoto, H.; Burrows, J.N.; Benito, F.J.G.; et al. Identification of Plasmodium falciparum Mitochondrial Malate: Quinone Oxidoreductase Inhibitors from the Pathogen Box. Genes 2019, 10, 471. [Google Scholar] [CrossRef]
- Biagini, G.A.; Viriyavejakul, P.; O’Neill, P.M.; Bray, P.G.; Ward, S.A. Functional characterization and target validation of alternative complex I of Plasmodium falciparum mitochondria. Antimicrob. Agents Chemother. 2006, 50, 1841–1851. [Google Scholar] [CrossRef]
- Becker, K.; Kirk, K. Of malaria, metabolism and membrane transport. Trends Parasitol. 2004, 20, 590–596. [Google Scholar] [CrossRef]
- Milner, D.A., Jr. Malaria Pathogenesis. Cold Spring Harb. Perspect. Med. 2018, 8, a025569. [Google Scholar] [CrossRef]
- Garcia, L.S. Malaria. Clin. Lab Med. 2010, 30, 93–129. [Google Scholar] [CrossRef] [PubMed]
- Kiatfuengfoo, R.; Suthiphongchai, T.; Prapunwattana, P.; Yuthavong, Y. Mitochondria as the site of action of tetracycline on Plasmodium falciparum. Mol. Biochem. Parasitol. 1989, 34, 109–115. [Google Scholar] [CrossRef]
- Infosalus. 10 Datos Sobre la Malaria 2015 [25/05/2020]. Available online: https://m.infosalus.com/salud-investigacion/noticia-10-datos-malaria-20170425151150.html (accessed on 20 May 2020).
- Malaria: Resumen Práctico para la Consulta del Pediatra.: AEPAP 2018. Available online: https://www.aepap.org/sites/default/files/documento/archivos-adjuntos/malaria.pdf (accessed on 20 May 2020).
- Harada, S.; Inaoka, D.K.; Ohmori, J.; Kita, K. Diversity of parasite complex II. Biochimica et Biophysica Acta (BBA)-Bioenergetics. 2013, 1827, 658–667. [Google Scholar] [CrossRef] [PubMed]
- Mogi, T.; Kita, K. Identification of mitochondrial Complex II subunits SDH3 and SDH4 and ATP synthase subunits a and b in Plasmodium spp. Mitochondrion 2009, 9, 443–453. [Google Scholar] [CrossRef]
- Hino, A.; Hirai, M.; Tanaka, T.Q.; Watanabe, Y.; Matsuoka, H.; Kita, K. Critical roles of the mitochondrial complex II in oocyst formation of rodent malaria parasite Plasmodium berghei. J. Biochem. 2012, 152, 259–268. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Paludismo 2019. Available online: https://www.who.int/es/news-room/facts-in-pictures/detail/10-datos-sobre-el-paludismo (accessed on 20 May 2020).
- Viajarseguro.org. Malaria en Niños. 2020. Available online: http://fundacionio.org/viajar/enfermedades/malaria/malaria%20kids.html (accessed on 20 May 2020).
- Fowler, C.; Cserti-Gazdewich, C.; Dhabangi, A.; Musoke, C.; Sharma, H.; Amr, S.S.; Dzik, W. Mitochondrial gene sequence variants in children with severe malaria anaemia with or without lactic acidosis: A case control study. Malar. J. 2018, 17, 467. [Google Scholar] [CrossRef]
- Boldt, A.B.W.; van Tong, H.; Grobusch, M.P.; Kalmbach, Y.; Dzeing Ella, A.; Kombila, M.; Meyer, C.G.; Kun, J.F.J.; Kremsner, P.G.; Velavan, T.P. The blood transcriptome of childhood malaria. EBioMedicine 2019, 40, 614–625. [Google Scholar] [CrossRef]
- Vademecum. Quinina [cited]. Available online: https://www.iqb.es/cbasicas/farma/farma04/q004.htm (accessed on 10 February 2021).
- Pukrittayakamee, S.; Chantra, A.; Vanijanonta, S.; Clemens, R.; Looareesuwan, S.; White, N.J. Therapeutic responses to quinine and clindamycin in multidrug-resistant falciparum malaria. Antimicrob. Agents Chemother. 2000, 44, 2395–2398. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Huang, L.; Li, J.; Fan, Q.; Long, Y.; Li, Y.; Zhou, B. Artemisinin directly targets malarial mitochondria through its specific mitochondrial activation. PLoS ONE 2010, 5, e9582. [Google Scholar] [CrossRef] [PubMed]
- Giraldo, C.; Trujillo, S. Mefloquina: Revisión de Tema Medellin: Revista Medica Universidad de Antioquia 2003. Available online: http://www.scielo.org.co/scielo.php?script=sci_arttext&pid=S0121–07932003000100003 (accessed on 10 February 2021).
- Garcia Lopez Hortelano, M.; Fumado Perez, V.; Gonzalez Tome, M.I.; Grupo de Trabajo de Enfermedades Tropicales de la Sociedad de Infectologia Pediátrica. Update in the diagnosis and treatment of malaria. An. Pediatr. Barc. 2013, 78, 124.e1–124.e8. [Google Scholar]
- Vatsveen, T.K.; Myhre, M.R.; Steen, C.B.; Walchli, S.; Lingjaerde, O.C.; Bai, B.; Dillard, P.; Theodossiou, T.A.; Holien, T.; Sundan, A.; et al. Artesunate shows potent anti-tumor activity in B-cell lymphoma. J. Hematol. Oncol. 2018, 11, 23. [Google Scholar] [CrossRef] [PubMed]
- Dowshen, S. Malaria: Kidshealth from Nemours 2017. Available online: https://kidshealth.org/es/parents/malaria-esp.html (accessed on 10 February 2021).
- Adebayo, J.O.; Tijjani, H.; Adegunloye, A.P.; Ishola, A.A.; Balogun, E.A.; Malomo, S.O. Enhancing the antimalarial activity of artesunate. Parasitol. Res. 2020, 119, 2749–2764. [Google Scholar] [CrossRef] [PubMed]
- Vademecum. Artesumato. Available online: https://www.iqb.es/cbasicas/farma/farma04/a121.htm (accessed on 20 May 2020).
- CDC. Malaria’s Impact Worldwide: Center for Disease Control 2021. Available online: https://www.cdc.gov/malaria/malaria_worldwide/impact.html (accessed on 10 February 2021).
- Ramond, E.; Jamet, A.; Coureuil, M.; Charbit, A. Pivotal role of mitochondria in macrophage response to bacterial pathogens. Front. Immunol. 2019, 10, 2461. [Google Scholar] [CrossRef]
- Claus, C.; Liebert, U.G. A renewed focus on the interplay between viruses and mitochondrial metabolism. Arch. Virol. 2014, 159, 1267–1277. [Google Scholar] [CrossRef] [PubMed]
- Talmon, G.; Cornell, L.D.; Lager, D.J. Mitochondrial changes in cidofovir therapy for BK virus nephropathy. Transplant. Proc. 2010, 42, 1713–1715. [Google Scholar] [CrossRef]
- Laukoter, S.; Rauschka, H.; Troscher, A.R.; Kock, U.; Saji, E.; Jellinger, K.; Lassmann, H.; Bauer, J. Differences in T cell cytotoxicity and cell death mechanisms between progressive multifocal leukoencephalopathy, herpes simplex virus encephalitis and cytomegalovirus encephalitis. Acta Neuropathol. 2017, 133, 613–627. [Google Scholar] [CrossRef]
- Hos, N.J.; Ganesan, R.; Gutierrez, S.; Hos, D.; Klimek, J.; Abdullah, Z.; Kronke, M.; Robinson, N. Type I interferon enhances necroptosis of Salmonella Typhimurium-infected macrophages by impairing antioxidative stress responses. J. Cell Biol. 2017, 216, 4107–4121. [Google Scholar] [CrossRef]
- Poderoso, J.J.; Helfenberger, K.; Poderoso, C. The effect of nitric oxide on mitochondrial respiration. Nitric Oxide 2019, 88, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.W. Lynn Margulis and the endosymbiont hypothesis: 50 years later. Mol. Biol. Cell 2017, 28, 1285–1287. [Google Scholar] [CrossRef] [PubMed]
- Moren, C.; Noguera-Julian, A.; Rovira, N.; Corrales, E.; Garrabou, G.; Hernandez, S.; Nicolas, M.; Tobias, E.; Cardellach, F.; Miro, O.; et al. Mitochondrial impact of human immunodeficiency virus and antiretrovirals on infected pediatric patients with or without lipodystrophy. Pediatrics Infect. Dis. J. 2011, 30, 992–995. [Google Scholar] [CrossRef] [PubMed]
- Williamson, C.D.; DeBiasi, R.L.; Colberg-Poley, A.M. Viral product trafficking to mitochondria, mechanisms and roles in pathogenesis. Infect. Disord. Drug Targets 2012, 12, 18–37. [Google Scholar] [CrossRef] [PubMed]
- Ouzzani, M.; Hammady, H.; Fedorowicz, Z.; Elmagarmid, A. Rayyan-a web and mobile app for systematic reviews. Syst. Rev. 2016, 5, 210. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TB | Children | Adults |
|---|---|---|
| Cause | A complication of the pathophysiologic events surrounding the initial infection | A reactivation of organisms that were lodged in the apices of the lungs during hematogenous dissemination at the time of primary infection |
| Incubation period | Often only weeks to months | Often long (years to decades) |
| Pulmonary and extrapulmonary TB | Children are more prone to extrapulmonary disease but rarely develop contagious pulmonary disease | Adults are more prone to developing contagious pulmonary disease |
| Pulmonary location | Anywhere (25% multilobar) | Apical |
| Adenopathy | Usual | Rare (except HIV related) |
| Cavitation | Rare (except adolescents) | Common |
| Signs and symptoms | Unspecific | Typical |
| Name | Characteristics | Mechanism of Action |
|---|---|---|
| Cpn60.2 | Mtb chaperone protein |
|
| LpqH | Mtb protein |
|
| ESAT 6 | Mtb protein |
|
| Cyclophilin D (CypD) | Mitochondrial matrix protein |
|
| Anti-TB Family | Characteristics | Mechanism of Action | Mitochondrial Dysfunction | Clinical Secondary Effects | Pediatric Studies |
|---|---|---|---|---|---|
| Isoniazid (INH) |
|
|
| ||
| Rifampin (RIF) |
|
|
|
|
|
| Ethambutol (EMB) |
|
|
| ||
| Pyrazynamide (PZA) |
|
|
|
|
|
| Drug | Antibiotic Type | Mechanism of Action | Mitochondrial Damage | Mitochondrial Interactions and Pediatric Studies |
|---|---|---|---|---|
| Gentamicin | Aminoglycoside |
|
|
|
| Linezolid | 2-oxazolidone |
|
| |
| Doxycycline | Tetracycline |
|
|
|
| Tigecycline | Tetracycline |
|
|
|
| Vancomycin | Glycopeptide |
|
|
| Meningitis Type | Microorganisms Responsible | |
|---|---|---|
| Bacterial meningitis | In newborns and young infants (<3 months) | Group B Streptococcus |
| E. coli | ||
| Listeria monocytogenes | ||
| In infants (>3 months) and children | Hemophilus influenzae type b * | |
| Neisseria meningitidis | ||
| Streptococcus pneumoniae | ||
| Others | Syphilis | |
| TB | ||
| Viral meningitis | Poliovirus * | |
| Enterovirus (e.g., coxsackie virus and echovirus) | ||
| Mumps (paramyxovirus) * | ||
| Herpes Simplex Virus (HSV) | ||
| Other organisms that can also cause meningitis | Borrelia burgdorferi (Lyme disease) | |
| Fungi such as candida, Aspergillus, or Cryptococcus neoformans in immunosuppressed patients | ||
| Drug | Antibiotic Type | Mechanism of Action | Mitochondrial Damage | Pediatric Studies |
|---|---|---|---|---|
| Ampicillin | Betalactamic |
|
| Ampicillin is a “broad-spectrum penicillin” used as therapy for suspected bacterial meningitis. Penetrates the blood–brain barrier sufficiently with an adequate dose and inflamed meninges. This drug revolutionized the treatment of bacterial meningitis in children [169,170] |
| Cefotaxime | Cephalosporin (also Betalactamic) |
|
| High-dose cefotaxime, while safe, is not reliably sufficient therapy for cephalosporin-non-susceptible pneumococcal meningitis, and combination therapy is recommended in children [173] |
| Ceftriaxone |
| Ceftriaxone, widely used in children in the treatment of sepsis, is not reliably sufficient therapy for cephalosporin-non-susceptible pneumococcal meningitis [177] |
| Type | Parasite and Resistance | Drug | Parasite’s Mitochondria Involvement |
|---|---|---|---|
| Uncomplicated malaria (no signs of severity) | P. falciparum or chloroquine-resistant strains | Atovaquone/proguanil | |
| Quinine sulfate + clindamycin or doxycycline (>8 years old) | |||
| derivatives of artemisin, in Spain dihydroartemisin + piperaquine |
| ||
| Mefloquine |
| ||
| P. vivax, ovale, malariae, or falciparum from chloroquine-sensitive area | Chloroquine |
| |
| Primaquine |
| ||
| Severe malaria | Normally caused by P. falciparum | Quinine gluconate IV diluted in glucose 5% + clindamycin IV (>8 years old) | |
| Artesunate |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romero-Cordero, S.; Kirwan, R.; Noguera-Julian, A.; Cardellach, F.; Fortuny, C.; Morén, C. A Mitocentric View of the Main Bacterial and Parasitic Infectious Diseases in the Pediatric Population. Int. J. Mol. Sci. 2021, 22, 3272. https://doi.org/10.3390/ijms22063272
Romero-Cordero S, Kirwan R, Noguera-Julian A, Cardellach F, Fortuny C, Morén C. A Mitocentric View of the Main Bacterial and Parasitic Infectious Diseases in the Pediatric Population. International Journal of Molecular Sciences. 2021; 22(6):3272. https://doi.org/10.3390/ijms22063272
Chicago/Turabian StyleRomero-Cordero, Sonia, Richard Kirwan, Antoni Noguera-Julian, Francesc Cardellach, Clàudia Fortuny, and Constanza Morén. 2021. "A Mitocentric View of the Main Bacterial and Parasitic Infectious Diseases in the Pediatric Population" International Journal of Molecular Sciences 22, no. 6: 3272. https://doi.org/10.3390/ijms22063272
APA StyleRomero-Cordero, S., Kirwan, R., Noguera-Julian, A., Cardellach, F., Fortuny, C., & Morén, C. (2021). A Mitocentric View of the Main Bacterial and Parasitic Infectious Diseases in the Pediatric Population. International Journal of Molecular Sciences, 22(6), 3272. https://doi.org/10.3390/ijms22063272