
Analysis of HPV Integrations in Mexican Pre-Tumoral Cervical Lesions Reveal Centromere-Enriched Breakpoints and Abundant Unspecific HPV Regions

 ,
,  ,
,  ,
,  ,
, 


Abstract
1. Introduction
2. Results
2.1. Patient Samples
2.2. HPV Genotyping by qPCR and Sequencing
2.3. HIVID-NGS HPV Genotype Detection
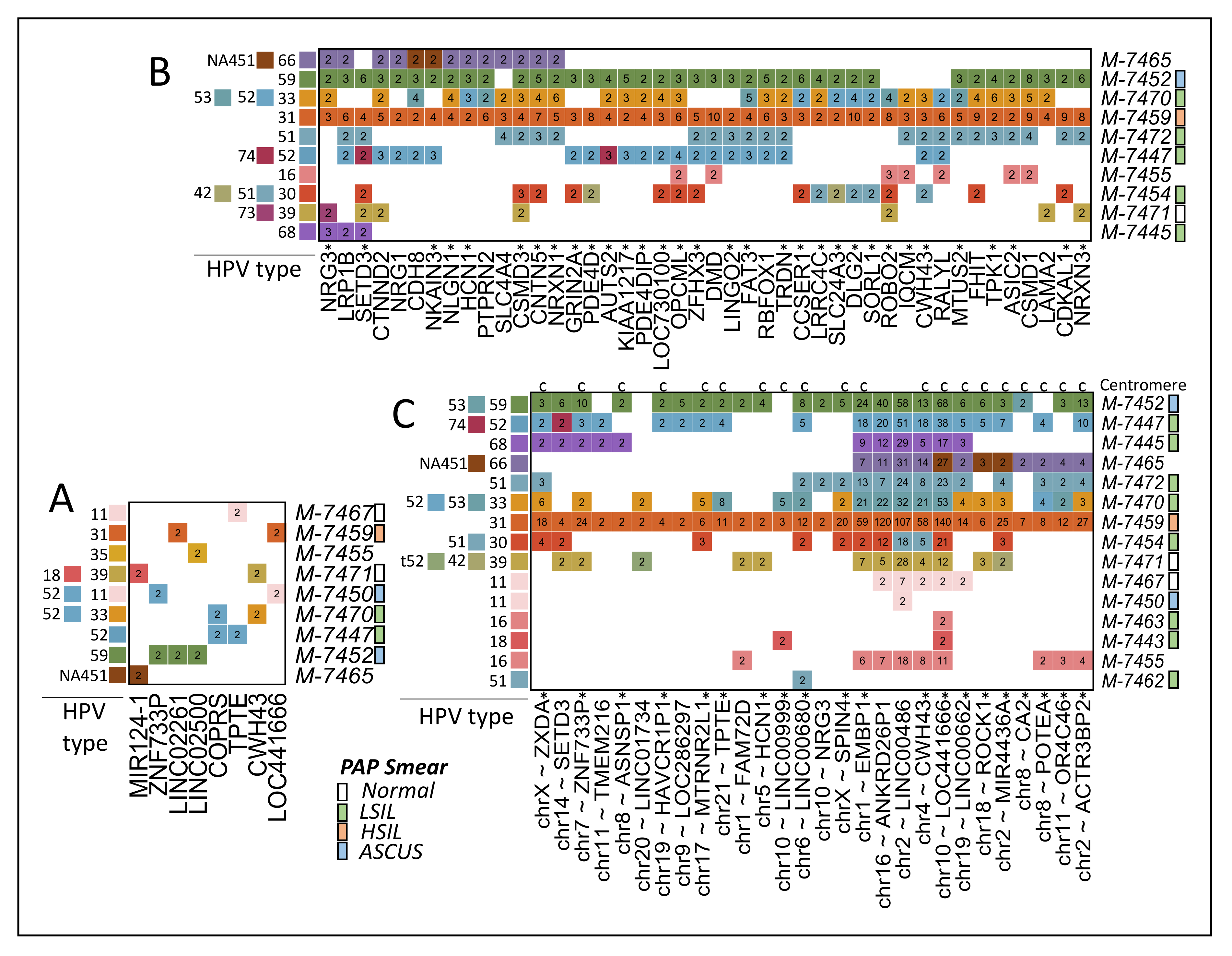
2.4. Identification of Viral Integrations
3. Discussion
3.1. HPV Genotyping
3.2. HPVint
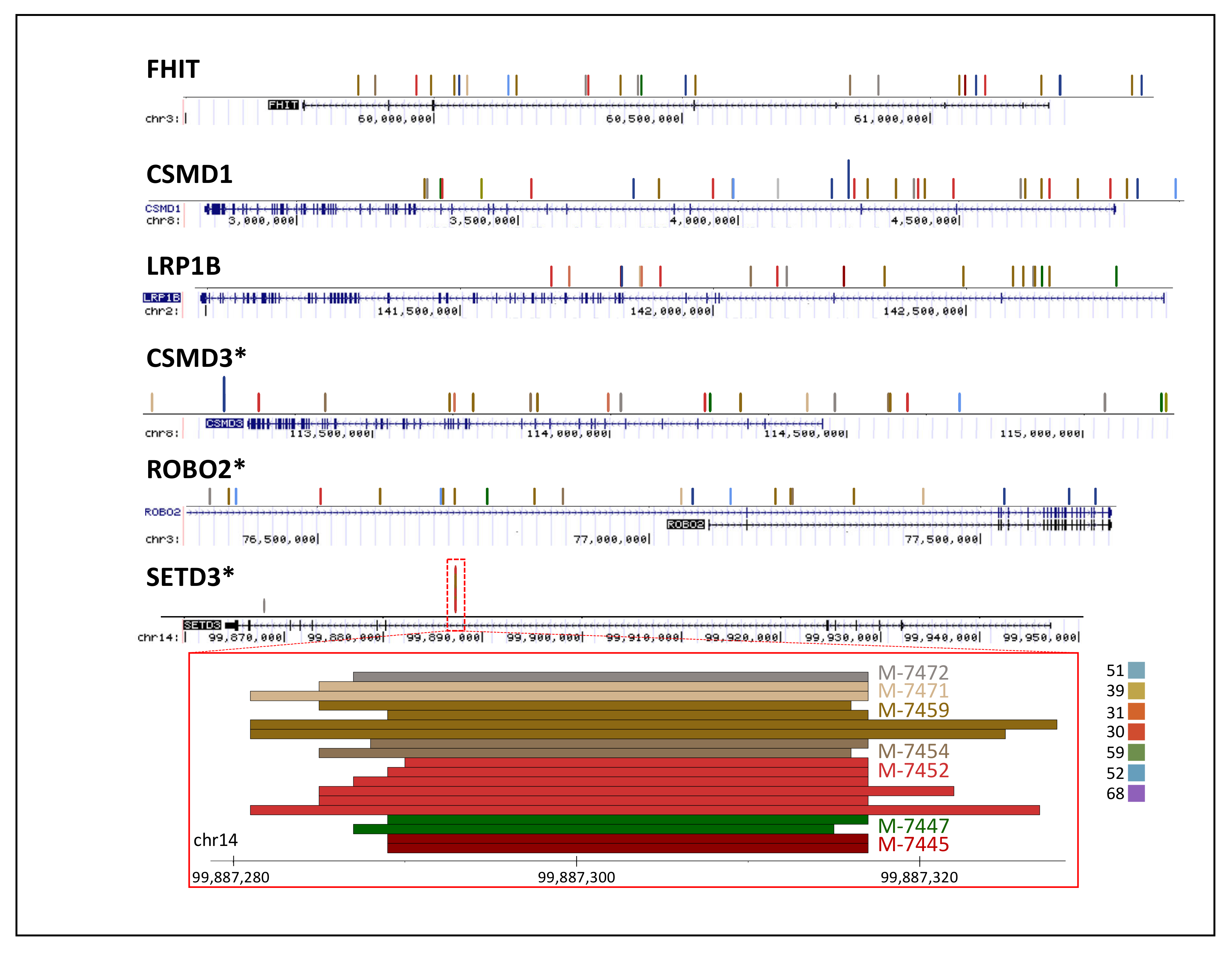
3.3. Centromere Integrations
3.4. HPVint Ratio and Breakpoint
3.5. Comparison of Reported HPV Genes vs. Observed
3.6. Comparison of Reported Human Genes
4. Materials and Methods
4.1. Study Population
4.2. Sample Collection and DNA Extraction
4.3. HPV Genotyping
4.4. Capturing and Sequencing
4.5. HPV Mapping and Typing from Sequencing
4.6. Viral Integration Pipeline
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wold Health Organization. Cervical Cancer: Overview. Available online: https://www.who.int/health-topics/cervical-cancer#tab=tab_1 (accessed on 9 February 2021).
- Human Papillomavirus and Related Diseases in Mexico. Summary Report—IARC Information Centre on HPV and Cancer (HPV Information Centre). Available online: http://www.hpvcentre.net/statistics/reports/MEX.pdf (accessed on 10 December 2020).
- Münger, K.; Baldwin, A.; Edwards, K.M.; Hayakawa, H.; Nguyen, C.L.; Owens, M.; Grace, M.; Huh, K. Mechanisms of Human Papillomavirus-Induced Oncogenesis. J. Virol. 2004, 78, 11451–11460. [Google Scholar] [CrossRef]
- Bzhalava, D.; Eklund, C.; Dillner, J. International standardization and classification of human papillomavirus types. Virology 2015, 476, 341–344. [Google Scholar] [CrossRef]
- Munger, K. The role of human papillomaviruses in human cancers. Front. Biosci. 2002, 7, d641–d649. [Google Scholar] [CrossRef]
- Bernard, H.-U.; Calleja-Macias, I.E.; Dunn, S.T. Genome variation of human papillomavirus types: Phylogenetic and medical implications. Int. J. Cancer 2005, 118, 1071–1076. [Google Scholar] [CrossRef]
- McLaughlin-Drubin, M.E.; Münger, K. Oncogenic activities of human papillomaviruses. Virus Res. 2009, 143, 195–208. [Google Scholar] [CrossRef] [PubMed]
- De Villiers, E.M. Cross-roads in the classification of papillomaviruses. Virology 2013, 445, 2–10. [Google Scholar] [CrossRef]
- Cricca, M.; Venturoli, S.; Leo, E.; Costa, S.; Musiani, M.; Zerbini, M. Disruption of HPV 16 E1 and E2 genes in precancerous cervical lesions. J. Virol. Methods 2009, 158, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Zhu, D.; Wang, W.; Li, W.; Jia, W.; Zeng, X.; Ding, W.; Yu, L.; Wang, X.; Wang, L.; et al. Genome-wide profiling of HPV integration in cervical cancer identifies clustered genomic hot spots and a potential microhomology-mediated integration mechanism. Nat. Genet. 2015, 47, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.V. The human papillomavirus replication cycle, and its links to cancer progression: A comprehensive review. Clin. Sci. 2017, 131, 2201–2221. [Google Scholar] [CrossRef] [PubMed]
- Pett, M.; Coleman, N. Integration of high-risk human papillomavirus: A key event in cervical carcinogenesis? J. Pathol. 2007, 212, 356–367. [Google Scholar] [CrossRef] [PubMed]
- Wentzensen, N.; Vinokurova, S.; Doeberitz, M.V.K. Systematic Review of Genomic Integration Sites of Human Papillomavirus Genomes in Epithelial Dysplasia and Invasive Cancer of the Female Lower Genital Tract. Cancer Res. 2004, 64, 3878–3884. [Google Scholar] [CrossRef] [PubMed]
- Oyervides-Muñoz, M.A.; Pérez-Maya, A.A.; Rodríguez-Gutiérrez, H.F.; Gómez-Macias, G.S.; Fajardo-Ramírez, O.R.; Treviño, V.; Barrera-Saldaña, H.A.; Garza-Rodríguez, M.L. Understanding the HPV integration and its progression to cervical cancer. Infect. Genet. Evol. 2018, 61, 134–144. [Google Scholar] [CrossRef]
- Zhao, J.-W.; Fang, F.; Guo, Y.; Zhu, T.-L.; Yu, Y.-Y.; Kong, F.-F.; Han, L.-F.; Chen, D.-S.; Li, F. HPV16 integration probably contributes to cervical oncogenesis through interrupting tumor suppressor genes and inducing chromosome instability. J. Exp. Clin. Cancer Res. 2016, 35, 180. [Google Scholar] [CrossRef]
- Li, H.; Yang, Y.; Zhang, R.; Cai, Y.; Yang, X.; Wang, Z.; Li, Y.; Cheng, X.; Ye, X.; Xiang, Y.; et al. Preferential sites for the integration and disruption of human papillomavirus 16 in cervical lesions. J. Clin. Virol. 2013, 56, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Liu, Y.; Dong, R.; Liu, J.; Lang, J.; Yang, J.; Wang, W.; Li, J.; Meng, B.; Tian, G. Accurate Detection of HPV Integration Sites in Cervical Cancer Samples Using the Nanopore MinION Sequencer Without Error Correction. Front. Genet. 2020, 11, 660. [Google Scholar] [CrossRef]
- Warburton, A.; Redmond, C.J.; Dooley, K.E.; Fu, H.; Gillison, M.L.; Akagi, K.; Symer, D.E.; Aladjem, M.I.; McBride, A.A. HPV integration hijacks and multimerizes a cellular enhancer to generate a viral-cellular super-enhancer that drives high viral oncogene expression. PLoS Genet. 2018, 14, e1007179. [Google Scholar] [CrossRef]
- Vinokurova, S.; Wentzensen, N.; Kraus, I.; Klaes, R.; Driesch, C.; Melsheimer, P.; Kisseljov, F.; Dürst, M.; Schneider, A.; Doeberitz, M.V.K. Type-Dependent Integration Frequency of Human Papillomavirus Genomes in Cervical Lesions. Cancer Res. 2008, 68, 307–313. [Google Scholar] [CrossRef]
- Ojesina, A.I.; Lichtenstein, L.; Freeman, S.S.; Pedamallu, C.S.; Imaz-Rosshandler, I.; Pugh, T.J.; Cherniack, A.D.; Ambrogio, L.; Cibulskis, K.; Bertelsen, B.; et al. Landscape of genomic alterations in cervical carcinomas. Nature 2014, 506, 371–375. [Google Scholar] [CrossRef]
- Nkili-Meyong, A.A.; Moussavou-Boundzanga, P.; Labouba, I.; Koumakpayi, I.H.; Jeannot, E.; Descorps-Declère, S.; Sastre-Garau, X.; Leroy, E.M.; Belembaogo, E.; Berthet, N. Genome-wide profiling of human papillomavirus DNA integration in liquid-based cytology specimens from a Gabonese female population using HPV capture technology. Sci. Rep. 2019, 9, 1504. [Google Scholar] [CrossRef] [PubMed]
- McBride, A.A.; Warburton, A. The role of integration in oncogenic progression of HPV-associated cancers. PLoS Pathog. 2017, 13, e1006211. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Dai, S.-Z.; Chu, H.-J.; Cui, H.-F.; Xu, X.-Y. Integration sites and genotype distributions of human papillomavirus in cervical intraepithelial neoplasia. Asian Pac. J. Cancer Prev. 2013, 14, 3837–3841. [Google Scholar] [CrossRef]
- Hudelist, G.; Manavi, M.; Pischinger, K.I.; Watkins-Riedel, T.; Singer, C.F.; Kubista, E.; Czerwenka, K.F. Physical state and expression of HPV DNA in benign and dysplastic cervical tissue: Different levels of viral integration are correlated with lesion grade. Gynecol. Oncol. 2004, 92, 873–880. [Google Scholar] [CrossRef] [PubMed]
- Arias-Pulido, H.; Peyton, C.L.; Joste, N.E.; Vargas, H.; Wheeler, C.M. Human Papillomavirus Type 16 Integration in Cervical Carcinoma In Situ and in Invasive Cervical Cancer. J. Clin. Microbiol. 2006, 44, 1755–1762. [Google Scholar] [CrossRef]
- Groves, I.J.; Coleman, N. Human papillomavirus genome integration in squamous carcinogenesis: What have next-generation sequencing studies taught us? J. Pathol. 2018, 245, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zeng, X.; Lee, N.P.; Liu, X.; Chen, S.; Guo, B.; Yi, S.; Zhuang, X.; Chen, F.; Wang, G.; et al. HIVID: An efficient method to detect HBV integration using low coverage sequencing. Genomics 2013, 102, 338–344. [Google Scholar] [CrossRef]
- Chandrani, P.; Kulkarni, V.; Iyer, P.K.; Upadhyay, P.K.; Chaubal, R.; Das, P.K.; Mulherkar, R.; Singh, R.; Dutt, A. NGS-based approach to determine the presence of HPV and their sites of integration in human cancer genome. Br. J. Cancer 2015, 112, 1958–1965. [Google Scholar] [CrossRef]
- Chen, X.; Kost, J.; Li, D. Comprehensive comparative analysis of methods and software for identifying viral integrations. Brief. Bioinform. 2018, 20, 2088–2097. [Google Scholar] [CrossRef]
- Tang, K.-W.; Alaei-Mahabadi, B.; Samuelsson, T.; Lindh, M.; Larsson, E. The landscape of viral expression and host gene fusion and adaptation in human cancer. Nat. Commun. 2013, 4, 2513. [Google Scholar] [CrossRef]
- Zapatka, M.; Pathogens, P.; Borozan, I.; Brewer, D.S.; Iskar, M.; Grundhoff, A.; Alawi, M.; Desai, N.; Sültmann, H.; Moch, H.; et al. The landscape of viral associations in human cancers. Nat. Genet. 2020, 52, 320–330. [Google Scholar] [CrossRef]
- Mohseni, M.; Cidado, J.; Croessmann, S.; Cravero, K.; Cimino-Mathews, A.; Wong, H.Y.; Scharpf, R.; Zabransky, D.J.; Abukhdeir, A.M.; Garay, J.P.; et al. MACROD2overexpression mediates estrogen independent growth and tamoxifen resistance in breast cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 17606–17611. [Google Scholar] [CrossRef] [PubMed]
- Kamal, M.; Lameiras, S.; Deloger, M.; Morel, A.; Vacher, S.; Lecerf, C.; Dupain, C.; Jeannot, E.; Girard, E.; Baulande, S.; et al. Human papilloma virus (HPV) integration signature in Cervical Cancer: Identification of MACROD2 gene as HPV hot spot integration site. Br. J. Cancer 2021, 124, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Shen, C.; Zhao, L.; Wang, J.; McCrae, M.A.; Chen, X.; Lu, F. Dysregulation of host cellular genes targeted by human papillomavirus (HPV) integration contributes to HPV-related cervical carcinogenesis. Int. J. Cancer 2015, 138, 1163–1174. [Google Scholar] [CrossRef]
- Gao, G.; Johnson, S.H.; Vasmatzis, G.; Pauley, C.E.; Tombers, N.M.; Kasperbauer, J.L.; Smith, D.I. Common fragile sites (CFS) and extremely large CFS genes are targets for human papillomavirus integrations and chromosome rearrangements in oropharyngeal squamous cell carcinoma. Genes Chromosom. Cancer 2016, 56, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Yaginuma, Y.; Yoshimoto, M.; Eguchi, A.; Tokuda, A.; Takahashi, S. The human papillomavirus18 E7 protein inhibits CENP-C binding to α-satellite DNA. Virus Res. 2015, 205, 27–32. [Google Scholar] [CrossRef]
- Yaginuma, Y.; Eguchi, A.; Yoshimoto, M.; Ogawa, K. The PxDLLCxE Sequence in Conserved Region 2 of Human Papilloma Virus 18 Protein E7 Is Required for E7 Binding to Centromere Protein C. Oncology 2012, 83, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Flores-Miramontes, M.G.; Torres-Reyes, L.A.; Alvarado-Ruíz, L.; Romero-Martínez, S.A.; Ramírez-Rodríguez, V.; Balderas-Peña, L.M.A.; Vallejo-Ruíz, V.; Piña-Sánchez, P.; Cortés-Gutiérrez, E.I.; Jave-Suárez, L.F.; et al. Human papillomavirus genotyping by Linear Array and Next-Generation Sequencing in cervical samples from Western Mexico. Virol. J. 2015, 12, 161. [Google Scholar] [CrossRef]
- Hausen, H.Z. Papillomaviruses in the causation of human cancers—A brief historical account. Virology 2009, 384, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Padilla-Mendoza, J.R.; De Oca, A.C.-M.; Rodríguez, M.A.; López-Casamichana, M.; Bolaños, J.; Quintas-Granados, L.I.; Reyes-Hernández, O.D.; Fragozo-Sandoval, F.; Reséndiz-Albor, A.A.; Arellano-Gutiérrez, C.V.; et al. Protein Phosphorylation in Serine Residues Correlates with Progression from Precancerous Lesions to Cervical Cancer in Mexican Patients. BioMed Res. Int. 2020, 2020, 5058928-11. [Google Scholar] [CrossRef]
- Pretet, J.L.; Jacquard, A.C.; Saunier, M.; Clavel, C.; Dachez, R.; Gondry, J.; Pradat, P.; Soubeyrand, B.; Leocmach, Y.; Mougin, C.; et al. Human papillomavirus genotype distribution in low-grade squamous intraepithelial lesions in France and comparison with CIN2/3 and invasive cervical cancer: The EDiTH III study. Gynecol. Oncol. 2008, 110, 179–184. [Google Scholar] [CrossRef]
- DelaRosa-Martínez, R.; Sánchez-Garza, M.; López-Revilla, R. HPV genotype distribution and anomalous association of HPV33 to cervical neoplastic lesions in San Luis Potosí, Mexico. Infect. Agents Cancer 2016, 11, 16. [Google Scholar] [CrossRef][Green Version]
- Arroyo, L.S.; Smelov, V.; Bzhalava, D.; Eklund, C.; Hultin, E.; Dillner, J. Next generation sequencing for human papillomavirus genotyping. J. Clin. Virol. 2013, 58, 437–442. [Google Scholar] [CrossRef]
- Barzon, L.; Militello, V.; Lavezzo, E.; Franchin, E.; Peta, E.; Squarzon, L.; Trevisan, M.; Pagni, S.; Bello, F.D.; Toppo, S.; et al. Human papillomavirus genotyping by 454 next generation sequencing technology. J. Clin. Virol. 2011, 52, 93–97. [Google Scholar] [CrossRef]
- Groves, I.J.; Coleman, N. Pathogenesis of human papillomavirus-associated mucosal disease. J. Pathol. 2015, 235, 527–538. [Google Scholar] [CrossRef]
- Bodelon, C.; Untereiner, M.E.; Machiela, M.J.; Vinokurova, S.; Wentzensen, N. Genomic characterization of viral integration sites in HPV-related cancers. Int. J. Cancer 2016, 139, 2001–2011. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Tian, S.; Wang, P.; Zang, Y.; Chen, X.; Yao, Y.; Li, W. The characteristics of HPV integration in cervical intraepithelial cells. J. Cancer 2019, 10, 2783–2787. [Google Scholar] [CrossRef]
- Bodelon, C.; Vinokurova, S.; Sampson, J.N.; Boon, J.A.D.; Walker, J.L.; Horswill, M.A.; Korthauer, K.; Schiffman, M.; Sherman, M.E.; Zuna, R.E.; et al. Chromosomal copy number alterations and HPV integration in cervical precancer and invasive cancer. Carcinogenesis 2015, 37, 188–196. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Integrated genomic and molecular characterization of cervical cancer. Nature 2017, 543, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Chung, T.K.; Van Hummelen, P.; Chan, P.K.; Cheung, T.H.; Yim, S.F.; Yu, M.Y.; Ducar, M.D.; Thorner, A.R.; MacConaill, L.E.; Doran, G.; et al. Genomic aberrations in cervical adenocarcinomas in Hong Kong Chinese women. Int. J. Cancer 2015, 137, 776–783. [Google Scholar] [CrossRef]
- Duncavage, E.J.; Magrini, V.; Becker, N.; Armstrong, J.R.; Demeter, R.T.; Wylie, T.; Abel, H.J.; Pfeifer, J.D. Hybrid Capture and Next-Generation Sequencing Identify Viral Integration Sites from Formalin-Fixed, Paraffin-Embedded Tissue. J. Mol. Diagn. 2011, 13, 325–333. [Google Scholar] [CrossRef]
- Holmes, A.; Lameiras, S.; Jeannot, E.; Marie, Y.; Castera, L.; Sastre-Garau, X.; Nicolas, A. Mechanistic signatures of HPV insertions in cervical carcinomas. NPJ Genom. Med. 2016, 1, 16004. [Google Scholar] [CrossRef] [PubMed]
- Luft, F.; Klaes, R.; Nees, M.; Durst, M.; Heilmann, V.; Melsheimer, P.; von Knebel Doeberitz, M. Detection of integrated papillomavirus sequences by ligation-mediated PCR (DIPS-PCR) and molecular characterization in cervical cancer cells. Int. J. Cancer 2001, 92, 9–17. [Google Scholar] [CrossRef]
- De Marco, L.; Gillio-Tos, A.; Bonello, L.; Ghisetti, V.; Ronco, G.; Merletti, F. Detection of human papillomavirus type 16 integration in pre-neoplastic cervical lesions and confirmation by DIPS-PCR and sequencing. J. Clin. Virol. 2007, 38, 7–13. [Google Scholar] [CrossRef]
- Xu, F.; Cao, M.; Shi, Q.; Chen, H.; Wang, Y.; Li, X. Integration of the full-length HPV16 genome in cervical cancer and Caski and Siha cell lines and the possible ways of HPV integration. Virus Genes 2015, 50, 210–220. [Google Scholar] [CrossRef]
- Romanczuk, H.; Howley, P.M. Disruption of either the E1 or the E2 regulatory gene of human papillomavirus type 16 increases viral immortalization capacity. Proc. Natl. Acad. Sci. USA 1992, 89, 3159–3163. [Google Scholar] [CrossRef]
- Sakthianandeswaren, A.; Parsons, M.J.; Mouradov, D.; Sieber, O.M. MACROD2 deletions cause impaired PARP1 activity and chromosome instability in colorectal cancer. Oncotarget 2018, 9, 33056–33058. [Google Scholar] [CrossRef]
- Shimizu, A.; Asakawa, S.; Sasaki, T.; Yamazaki, S.; Yamagata, H.; Kudoh, J.; Minoshima, S.; Kondo, I.; Shimizu, N. A novel giant gene CSMD3 encoding a protein with CUB and sushi multiple domains: A candidate gene for benign adult familial myoclonic epilepsy on human chromosome 8q23.3–q24.1. Biochem. Biophys. Res. Commun. 2003, 309, 143–154. [Google Scholar] [CrossRef]
- Gylfe, A.E.; Sirkiä, J.; Ahlsten, M.; Järvinen, H.; Mecklin, J.-P.; Karhu, A.; Aaltonen, L.A. Somatic mutations and germline sequence variants in patients with familial colorectal cancer. Int. J. Cancer 2010, 127, 2974–2980. [Google Scholar] [CrossRef]
- Yue, Y.; Grossmann, B.; Galetzka, D.; Zechner, U.; Haaf, T. Isolation and differential expression of two isoforms of the ROBO2/Robo2 axon guidance receptor gene in humans and mice. Genomics 2006, 88, 772–778. [Google Scholar] [CrossRef]
- Biankin, A.V.; Initiative, A.P.C.G.; Waddell, N.; Kassahn, K.S.; Gingras, M.-C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.-M.; et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012, 491, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Hassan, N.; Rutsch, N.; Győrffy, B.; Espinoza-Sánchez, N.A.; Götte, M. SETD3 acts as a prognostic marker in breast cancer patients and modulates the viability and invasion of breast cancer cells. Sci. Rep. 2020, 10, 2262. [Google Scholar] [CrossRef] [PubMed]
- Oyervides-Muñoz, M.A.; Pérez-Maya, A.A.; Sánchez-Domínguez, C.N.; Berlanga-Garza, A.; Antonio-Macedo, M.; Valdéz-Chapa, L.D.; Cerda-Flores, R.M.; Trevino, V.; Barrera-Saldaña, H.A.; Garza-Rodríguez, M.L. Multiple HPV Infections and Viral Load Association in Persistent Cervical Lesions in Mexican Women. Viruses 2020, 12, 380. [Google Scholar] [CrossRef] [PubMed]
- Van Doorslaer, K.; Li, Z.; Xirasagar, S.; Maes, P.; Kaminsky, D.; Liou, D.; Sun, Q.; Kaur, R.; Huyen, Y.; McBride, A.A. The Papillomavirus Episteme: A major update to the papillomavirus sequence database. Nucleic Acids Res. 2017, 45, D499–D506. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.S.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Smear | Reads | Mapped * | %Map ¶ | HPV Types + | Percent + |
|---|---|---|---|---|---|---|
| M-7440 | CC | 4,562,484 | 6474 | 0.1 | t45 | 100 |
| M-7443 | LSIL | 716,396 | 381,893 | 53.3 | 18, 74 | 99, 1 |
| M-7445 | LSIL | 15,287,436 | 8,496,237 | 55.6 | 68, NA440, NA448, t45 | 42, 36, 22, <1 |
| M-7447 | LSIL | 13,480,394 | 8,317,461 | 61.7 | 52, t52, 74, 87, 44, t45, t66 | 39, 38, 22, 1, <1, <1, <1 |
| M-7448 | ASCUS | 417,768 | 29,188 | 7.0 | 6, t45 | 98, 2 |
| M-7449 | Normal | 147,160 | 411 | 0.3 | t45 | 100 |
| M-7450 | ASCUS | 1,143,332 | 366,366 | 32.0 | 11, 52, t52, t56, 56, NA450 | 79, 8, 7, 2, 2, 1 |
| M-7452 | ASCUS | 17,740,624 | 11,991,175 | 67.6 | 59, 89, 53, t45 | 98, 1, 1, <1 |
| M-7454 | LSIL | 5,251,310 | 3,219,573 | 61.3 | 30, 51, 42, 11, t45 | 53, 38, 7, 2, <1 |
| M-7455 | NA | 6,350,088 | 2,897,763 | 45.6 | 16, 35, NA446, t45 | 85, 8, 7, <1 |
| M-7456 | HSIL | 3,879,422 | 8527 | 0.2 | t45, 16 | 86, 14 |
| M-7457 | LSIL | 367,094 | 13,371 | 3.6 | 6, t45, 51, 90 | 47, 44, 6, 3 |
| M-7458 | LSIL | 272,030 | 18,674 | 6.9 | 6, 86, t45 | 92, 5, 3 |
| M-7459 | HSIL | 28,572,654 | 21,901,105 | 76.7 | 31, t45 | 100, <1 |
| M-7460 | LSIL | 494,080 | 56,676 | 11.5 | 51, 70, t45, 34 | 74, 22, 2, 2 |
| M-7461 | LSIL | 365,174 | 2367 | 0.6 | 44, t45, 54, 53 | 49, 27, 13, 11 |
| M-7462 | LSIL | 884,470 | 238,060 | 26.9 | 51, 54 | 96, 4 |
| M-7463 | LSIL | 702,672 | 121,121 | 17.2 | 16, NA446, t45, 40 | 90, 8, 1, 1 |
| M-7464 | LSIL | 481,510 | 40,073 | 8.3 | 11, 31, 34, t45 | 61, 30, 5, 3 |
| M-7465 | NA | 9,826,324 | 6,183,616 | 62.9 | t66, NA451, 66, t45 | 49, 31, 19, <1 |
| M-7467 | Normal | 3,173,490 | 1,445,628 | 45.6 | 11, t45 | 100, <1 |
| M-7470 | LSIL | 13,217,166 | 7,266,920 | 55.0 | 53, 33, t33, 52, t52, NA436, t45 | 39, 16, 13, 11, 11, 10, <1 |
| M-7471 | Normal | 9,791,996 | 5,128,463 | 52.4 | 39, 82, t39, NA447, 73, 42, 66, NA449, 52, t52, t45 | 33, 27, 16, 10, 4, 3, 2, 2, 1, 1, <1 |
| M-7472 | LSIL | 6,241,940 | 3,953,761 | 63.3 | 51, t45 | 100, <1 |
| Type | Samples | Reads | Within Sample % ¶ |
|---|---|---|---|
| t45 | M-7440, M-7445, M-7447, M-7448, M-7449, M-7452, M-7454, M-7455, M-7456, M-7457, M-7458, M-7459, M-7460, M-7461, M-7463, M-7464, M-7465, M-7467, M-7470, M-7471, M-7472 | 177,864 | 100, <1, <1, 2, 100, <1, <1, <1, 86, 6, 3, <1, 2, 27, 1, 3, <1, <1, <1, <1, <1 |
| 51 | M-7454, M-7457, M-7460, M-7462, M-7472 | 5,433,163 | 38, 44, 74, 96, 100 |
| 52 | M-7447, M-7450, M-7470, M-7471 | 4,197,311 | 39, 8, 11, 1 |
| t52 | M-7447, M-7450, M-7470, M-7471 | 4,018,079 | 38, 7, 11, 1 |
| 11 | M-7450, M-7454, M-7464, M-7467 | 1,815,076 | 79, 2, 61, 100 |
| 53 | M-7452, M-7461, M-7470 | 2,901,407 | 1, 11, 39 |
| 16 | M-7455, M-7456, M-7463 | 2,561,367 | 85, 14, 90 |
| 6 | M-7448, M-7457, M-7458 | 45,980 | 98, 3, 92 |
| 31 | M-7459, M-7464 | 21,889,052 | 100, 30 |
| t66 | M-7447, M-7465 | 3,058,716 | <1, 49 |
| 74 | M-7443, M-7447 | 1,795,576 | 1, 22 |
| 66 | M-7465, M-7471 | 1,298,463 | 19, 2 |
| 42 | M-7454, M-7471 | 377,645 | 7, 3 |
| NA446 | M-7455, M-7463 | 219,032 | 7, 8 |
| 44 | M-7447, M-7461 | 29,574 | <1, 49 |
| 54 | M-7461, M-7462 | 9174 | 13, 4 |
| 34 | M-7460, M-7464 | 2977 | 2, 5 |
| Sample | % Integration | Integrations | After Marks * | % Chr Y | Types ¶ | Percents ¶ |
|---|---|---|---|---|---|---|
| M-7440 | 1.71% | 111 | 39 | 0.60% | t45 | 100 |
| M-7443 | 0.04% | 164 | 161 | 0.04% | 18 | 100 |
| M-7445 | 0.02% | 1508 | 1491 | 0.02% | 68, NA440, t45, NA448 | 96, 3.5, <1, <1 |
| M-7447 | 0.05% | 4554 | 4507 | 0.05% | 52, 74, t52, 87, 44, t45, t66 | 77, 19, 3, <1, <1, <1, <1 |
| M-7448 | 0.10% | 28 | 26 | 0.09% | 6 | 100 |
| M-7449 | 0.73% | 3 | 2 | 0.49% | t45 | 100 |
| M-7450 | 0.04% | 154 | 148 | 0.04% | 11, 52, 56, t52, NA450 | 70, 21, 7, <1, <1 |
| M-7452 | 0.07% | 8249 | 7803 | 0.07% | 59, 53, 89, t45 | 99, <1, <1, <1 |
| M-7454 | 0.07% | 2337 | 2292 | 0.07% | 30, 51, 42, 11, t45 | 54, 39, 6, 1, <1 |
| M-7455 | 0.06% | 1790 | 1769 | 0.06% | 16, 35, NA446, t45 | 91, 7, 2, <1 |
| M-7456 | 1.52% | 130 | 44 | 0.52% | t45 | 100 |
| M-7457 | 0.04% | 5 | 3 | 0.02% | 90, 51 | 67, 33 |
| M-7458 | 0.06% | 11 | 7 | 0.04% | 6, t45, 86 | 71, 14, 14 |
| M-7459 | 0.08% | 18,080 | 17,476 | 0.08% | 31, t45 | 100, <1 |
| M-7460 | 0.07% | 38 | 34 | 0.06% | 51, 70 | 74, 26 |
| M-7461 | 0.17% | 4 | 0 | 0.00% | - | - |
| M-7462 | 0.06% | 137 | 129 | 0.05% | 51, 54 | 98, 2 |
| M-7463 | 0.10% | 118 | 115 | 0.09% | 16, 40, NA446 | 92, 6, 2 |
| M-7464 | 0.05% | 20 | 18 | 0.04% | 11, 31 | 56, 34 |
| M-7465 | 0.04% | 2573 | 2539 | 0.04% | 66, NA451, t66, t45 | 53, 36, 11, <1 |
| M-7467 | 0.05% | 737 | 691 | 0.05% | 11, t45 | 99, <1 |
| M-7470 | 0.08% | 5952 | 5805 | 0.08% | 33, 53, 52, NA436, t52, t45, t33 | 39, 39, 20, 1, <1, <1, <1 |
| M-7471 | 0.03% | 1754 | 1695 | 0.03% | 39, 82, 73, 66, 42, 52, NA447, t39, t52, t45, NA449 | 67, 11, 10, 3, 3, 3, 1, <1, <1, <1, <1 |
| M-7472 | 0.12% | 4647 | 4418 | 0.11% | 51, t45 | 100, <1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garza-Rodríguez, M.L.; Oyervides-Muñoz, M.A.; Pérez-Maya, A.A.; Sánchez-Domínguez, C.N.; Berlanga-Garza, A.; Antonio-Macedo, M.; Valdés-Chapa, L.D.; Vidal-Torres, D.; Vidal-Gutiérrez, O.; Pérez-Ibave, D.C.; et al. Analysis of HPV Integrations in Mexican Pre-Tumoral Cervical Lesions Reveal Centromere-Enriched Breakpoints and Abundant Unspecific HPV Regions. Int. J. Mol. Sci. 2021, 22, 3242. https://doi.org/10.3390/ijms22063242
Garza-Rodríguez ML, Oyervides-Muñoz MA, Pérez-Maya AA, Sánchez-Domínguez CN, Berlanga-Garza A, Antonio-Macedo M, Valdés-Chapa LD, Vidal-Torres D, Vidal-Gutiérrez O, Pérez-Ibave DC, et al. Analysis of HPV Integrations in Mexican Pre-Tumoral Cervical Lesions Reveal Centromere-Enriched Breakpoints and Abundant Unspecific HPV Regions. International Journal of Molecular Sciences. 2021; 22(6):3242. https://doi.org/10.3390/ijms22063242
Chicago/Turabian StyleGarza-Rodríguez, María Lourdes, Mariel Araceli Oyervides-Muñoz, Antonio Alí Pérez-Maya, Celia Nohemí Sánchez-Domínguez, Anais Berlanga-Garza, Mauro Antonio-Macedo, Lezmes Dionicio Valdés-Chapa, Diego Vidal-Torres, Oscar Vidal-Gutiérrez, Diana Cristina Pérez-Ibave, and et al. 2021. "Analysis of HPV Integrations in Mexican Pre-Tumoral Cervical Lesions Reveal Centromere-Enriched Breakpoints and Abundant Unspecific HPV Regions" International Journal of Molecular Sciences 22, no. 6: 3242. https://doi.org/10.3390/ijms22063242
APA StyleGarza-Rodríguez, M. L., Oyervides-Muñoz, M. A., Pérez-Maya, A. A., Sánchez-Domínguez, C. N., Berlanga-Garza, A., Antonio-Macedo, M., Valdés-Chapa, L. D., Vidal-Torres, D., Vidal-Gutiérrez, O., Pérez-Ibave, D. C., & Treviño, V. (2021). Analysis of HPV Integrations in Mexican Pre-Tumoral Cervical Lesions Reveal Centromere-Enriched Breakpoints and Abundant Unspecific HPV Regions. International Journal of Molecular Sciences, 22(6), 3242. https://doi.org/10.3390/ijms22063242