
Structural Characterization of Receptor–Receptor Interactions in the Allosteric Modulation of G Protein-Coupled Receptor (GPCR) Dimers



Abstract
1. Introduction
2. Role of Receptor–Receptor Interactions in the Allosteric Modulation of GPCR Activation
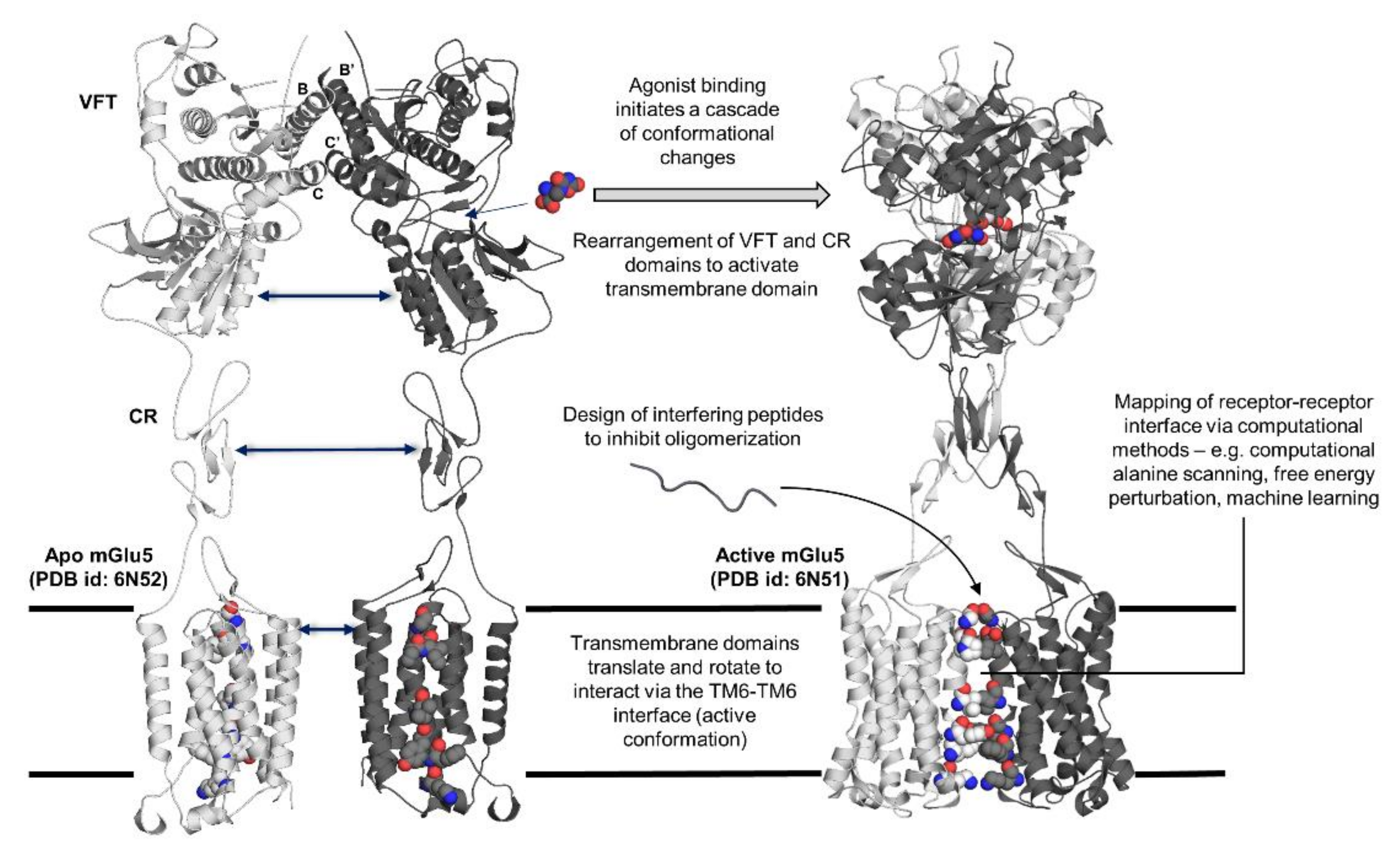
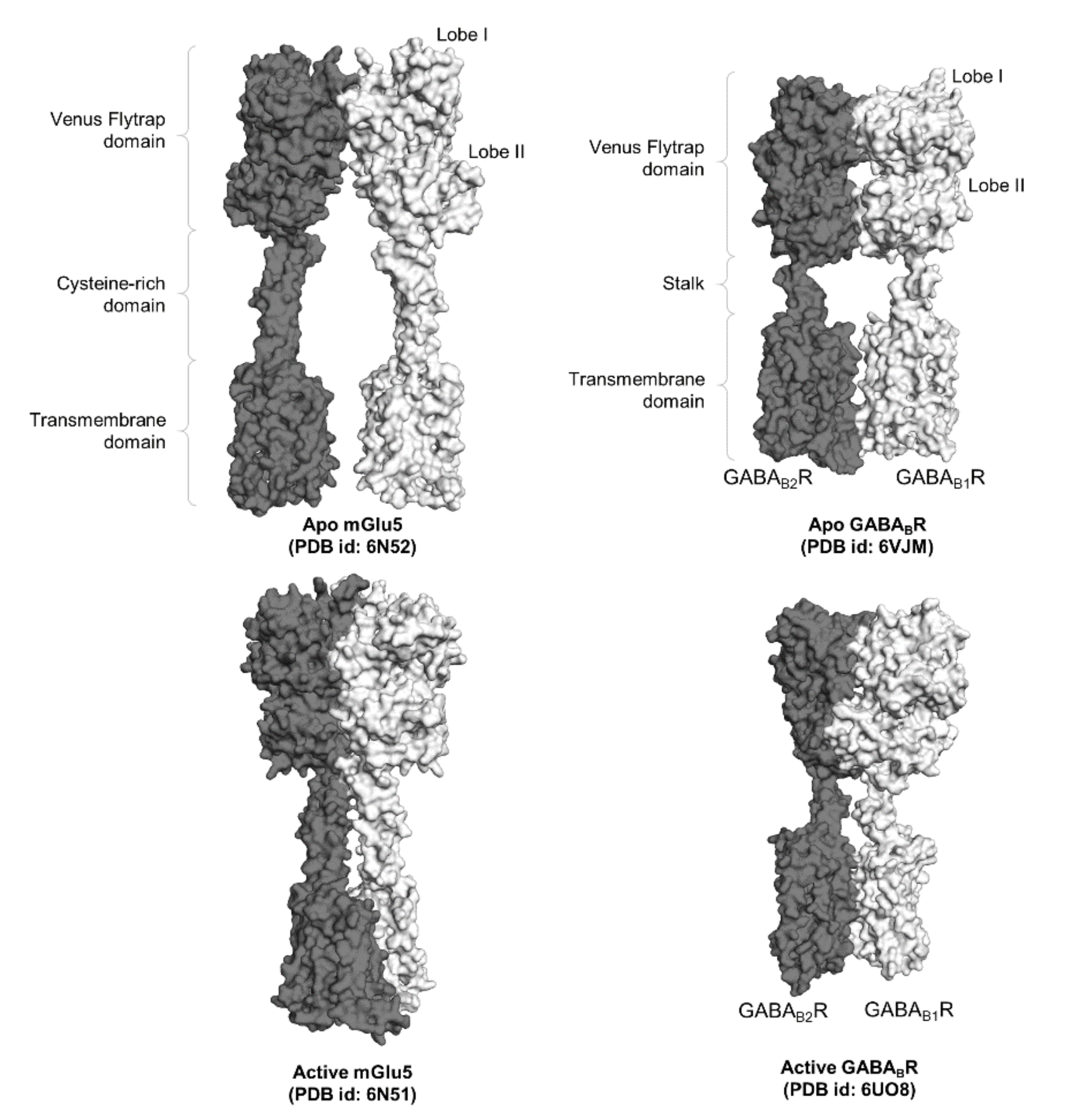
2.1. Class C GPCRs: A Potential Model for GPCR Trans-Activation
2.2. Elucidation of Allosteric Modulation via Full-Length Structures of Class C GPCR Dimers
2.3. Altered GPCR Activities Induced through Heterodimerization
3. Computational Methods Utilized for the Understanding of Receptor–Receptor Interactions in GPCR Dimers
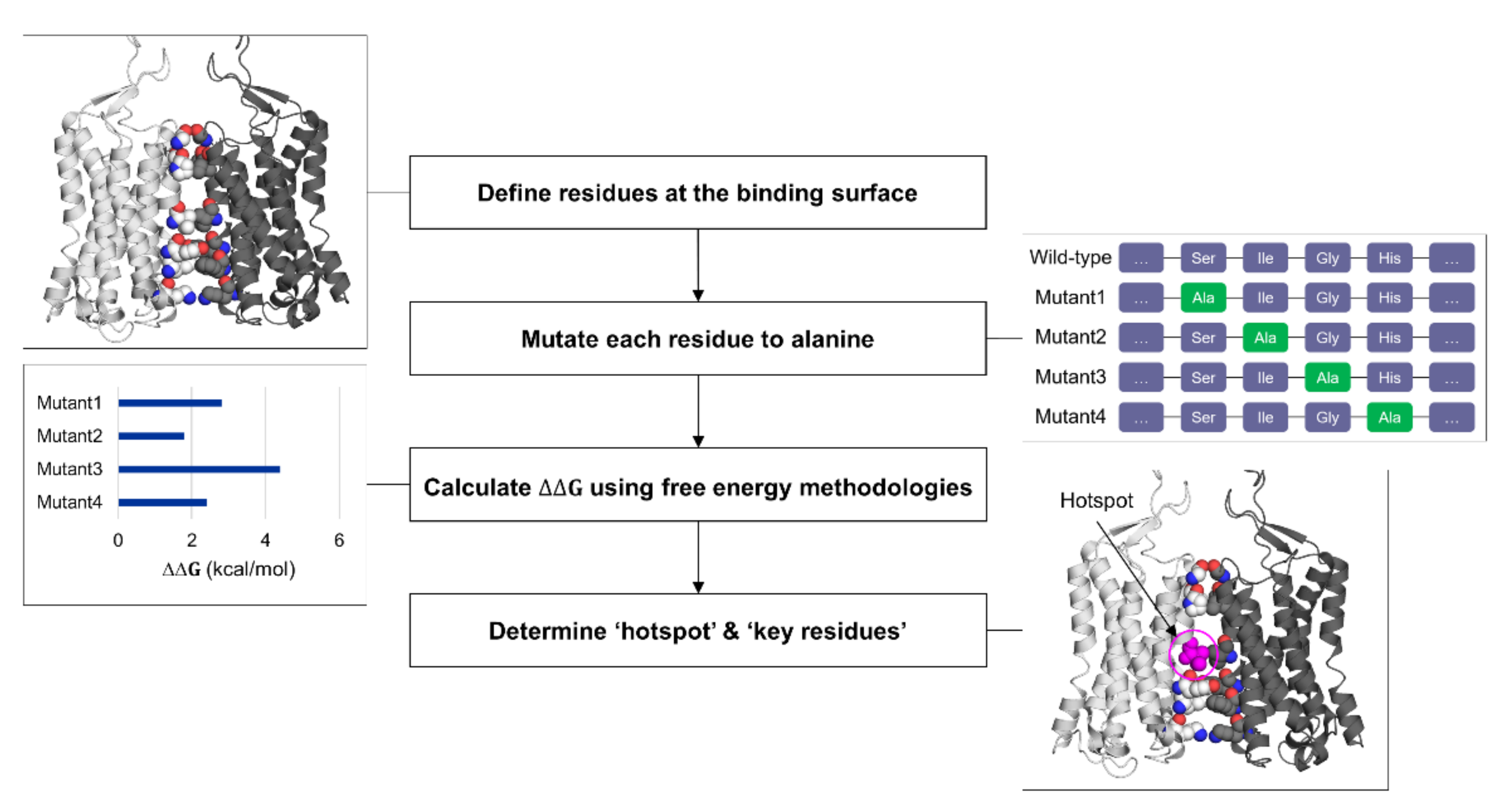
3.1. Hot-Spot and Interface Interaction Discovery Using Computational Methods
3.2. Application of Artificial Intelligence to Predict PPIs
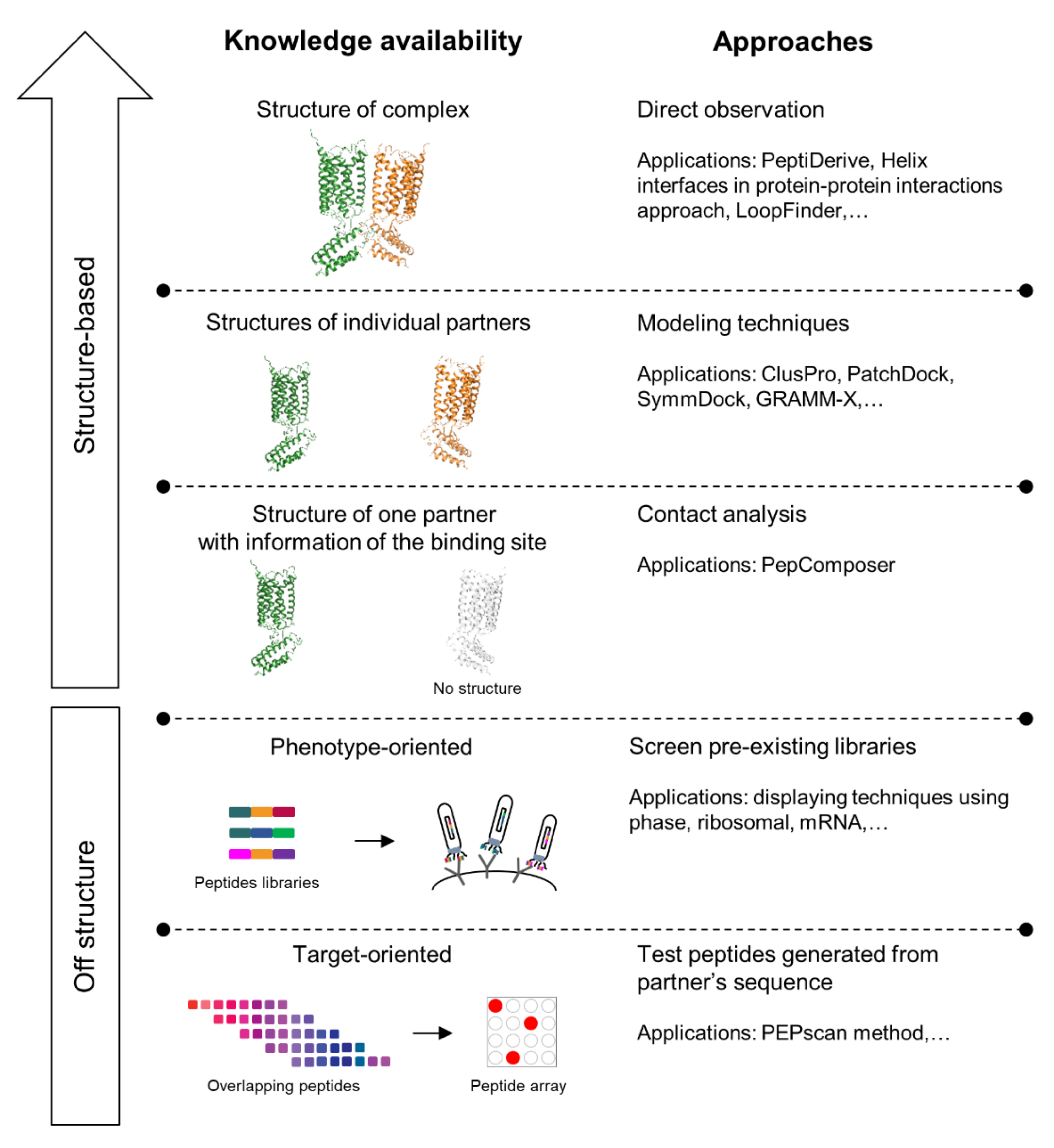
4. Design of Interface Interfering Peptides (IPs) to Prevent GPCR Dimerization
4.1. Interfering Peptide (IP) Identification
4.2. IP Optimization
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Retamal, J.S.; Ramírez-García, P.D.; Shenoy, P.A.; Poole, D.P.; Veldhuis, N.A. Internalized GPCRs as Potential Therapeutic Targets for the Management of Pain. Front. Mol. Neurosci. 2019, 12, 273. [Google Scholar] [CrossRef] [PubMed]
- Bagnato, A.; Rosanò, L. New Routes in GPCR/β-Arrestin-Driven Signaling in Cancer Progression and Metastasis. Front. Pharmacol. 2019, 10, 114. [Google Scholar] [CrossRef]
- Sloop, K.W.; Emmerson, P.J.; Statnick, M.A.; Willard, F.S. The current state of GPCR-based drug discovery to treat metabolic disease. Br. J. Pharmacol. 2018, 175, 4060–4071. [Google Scholar] [CrossRef] [PubMed]
- Gendaszewska-Darmach, E.; Drzazga, A.; Koziołkiewicz, M. Targeting GPCRs Activated by Fatty Acid-Derived Lipids in Type 2 Diabetes. Trends Mol. Med. 2019, 25, 915–929. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Todd, N.; Thathiah, A. The role of GPCRs in neurodegenerative diseases: Avenues for therapeutic intervention. Curr. Opin. Pharmacol. 2017, 32, 96–110. [Google Scholar] [CrossRef] [PubMed]
- May, L.T.; Leach, K.; Sexton, P.M.; Christopoulos, A. Allosteric Modulation of G Protein–Coupled Receptors. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 1–51. [Google Scholar] [CrossRef]
- Lee, Y.; Lazim, R.; Macalino, S.J.Y.; Choi, S. Importance of protein dynamics in the structure-based drug discovery of class A G protein-coupled receptors (GPCRs). Curr. Opin. Struct. Biol. 2019, 55, 147–153. [Google Scholar] [CrossRef]
- Alfonso-Prieto, M.; Navarini, L.; Carloni, P. Understanding Ligand Binding to G-Protein Coupled Receptors Using Multiscale Simulations. Front. Mol. Biosci. 2019, 6, 29. [Google Scholar] [CrossRef]
- Van Eps, N.; Altenbach, C.; Caro, L.N.; Latorraca, N.R.; Hollingsworth, S.A.; Dror, R.O.; Ernst, O.P.; Hubbell, W.L. Gi and Gs-coupled GPCRs show different modes of G-protein binding. Proc. Natl. Acad. Sci. USA 2018, 115, 2383–2388. [Google Scholar] [CrossRef]
- Ge, B.; Lao, J.; Li, J.; Chen, Y.; Song, Y.; Huang, F. Single-molecule imaging reveals dimerization/oligomerization of CXCR4 on plasma membrane closely related to its function. Sci. Rep. 2017, 7, 16873. [Google Scholar] [CrossRef]
- Møller, T.C.; Hottin, J.; Clerté, C.; Zwier, J.M.; Durroux, T.; Rondard, P.; Prézeau, L.; Royer, C.A.; Pin, J.-P.; Margeat, E.; et al. Oligomerization of a G protein-coupled receptor in neurons controlled by its structural dynamics. Sci. Rep. 2018, 8, 10414. [Google Scholar] [CrossRef]
- Townsend-Nicholson, A.; Altwaijry, N.; Potterton, A.; Morao, I.; Heifetz, A. Computational prediction of GPCR oligomerization. Curr. Opin. Struct. Biol. 2019, 55, 178–184. [Google Scholar] [CrossRef]
- Milligan, G.; Ward, R.J.; Marsango, S. GPCR homo-oligomerization. Curr. Opin. Cell Biol. 2019, 57, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Kasai, R.S.; Ito, S.V.; Awane, R.M.; Fujiwara, T.K.; Kusumi, A. The Class-A GPCR Dopamine D2 Receptor Forms Transient Dimers Stabilized by Agonists: Detection by Single-Molecule Tracking. Cell Biochem. Biophys. 2018, 76, 29–37. [Google Scholar] [CrossRef]
- Möller, J.; Isbilir, A.; Sungkaworn, T.; Osberg, B.; Karathanasis, C.; Sunkara, V.; Grushevskyi, E.O.; Bock, A.; Annibale, P.; Heilemann, M.; et al. Single-molecule analysis reveals agonist-specific dimer formation of µ-opioid receptors. Nat. Chem. Biol. 2020, 16, 946–954. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Gong, Z.; Lu, Y.-B.; Xu, C.-J.; Wei, T.-F.; Yang, M.-S.; Zhan, T.-W.; Yang, Y.-H.; Lin, L.; Liu, J.; et al. FLIM–FRET-Based Structural Characterization of a Class-A GPCR Dimer in the Cell Membrane. J. Mol. Biol. 2020, 432, 4596–4611. [Google Scholar] [CrossRef]
- Møller, T.C.; Moreno-Delgado, D.; Pin, J.-P.; Kniazeff, J. Class C G protein-coupled receptors: Reviving old couples with new partners. Biophys. Rep. 2017, 3, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Pin, J.-P.; Bettler, B. Organization and functions of mGlu and GABAB receptor complexes. Nature 2016, 540, 60–68. [Google Scholar] [CrossRef]
- Angers, S.; Salahpour, A.; Joly, E.; Hilairet, S.; Chelsky, D.; Dennis, M.; Bouvier, M. Detection of beta 2-adrenergic receptor dimerization in living cells using bioluminescence resonance energy transfer (BRET). Proc. Natl. Acad. Sci. USA 2000, 97, 3684–3689. [Google Scholar] [CrossRef] [PubMed]
- Angers, S.; Salahpour, A.; Bouvier, M. Dimerization: An emerging concept for G protein-coupled receptor ontogeny and function. Annu. Rev. Pharmacol. Toxicol. 2002, 42, 409–435. [Google Scholar] [CrossRef]
- Limbird, L.E.; Meyts, P.D.; Lefkowitz, R.J. β-Adrenergic receptors: Evidence for negative cooperativity. Biochem. Biophys. Res. Commun. 1975, 64, 1160–1168. [Google Scholar] [CrossRef]
- Ng, S.; Lee, L.; Chow, B. Receptor oligomerization: From early evidence to current understanding in class B GPCRs. Front. Endocrinol. 2013, 3, 175. [Google Scholar] [CrossRef]
- Freudenberg, J.M.; Dunham, I.; Sanseau, P.; Rajpal, D.K. Uncovering new disease indications for G-protein coupled receptors and their endogenous ligands. BMC Bioinform. 2018, 19, 345. [Google Scholar] [CrossRef] [PubMed]
- Szymańska, K.; Kałafut, J.; Przybyszewska, A.; Paziewska, B.; Adamczuk, G.; Kiełbus, M.; Rivero-Müller, A. FSHR Trans-Activation and Oligomerization. Front. Endocrinol. 2018, 9, 760. [Google Scholar] [CrossRef]
- Pin, J.-P.; Kniazeff, J.; Prézeau, L.; Liu, J.-F.; Rondard, P. GPCR interaction as a possible way for allosteric control between receptors. Mol. Cell. Endocrinol. 2019, 486, 89–95. [Google Scholar] [CrossRef]
- Ellaithy, A.; Gonzalez-Maeso, J.; Logothetis, D.A.; Levitz, J. Structural and Biophysical Mechanisms of Class C G Protein-Coupled Receptor Function. Trends Biochem. Sci. 2020, 45, 1049–1064. [Google Scholar] [CrossRef] [PubMed]
- Stewart, G.D.; Comps-Agrar, L.; Nørskov-Lauritsen, L.B.; Pin, J.-P.; Kniazeff, J. Allosteric interactions between GABAB1 subunits control orthosteric binding sites occupancy within GABAB oligomers. Neuropharmacology 2018, 136, 92–101. [Google Scholar] [CrossRef]
- Koehl, A.; Hu, H.; Feng, D.; Sun, B.; Zhang, Y.; Robertson, M.J.; Chu, M.; Kobilka, T.S.; Laeremans, T.; Steyaert, J.; et al. Structural insights into the activation of metabotropic glutamate receptors. Nature 2019, 566, 79–84. [Google Scholar] [CrossRef]
- Wu, H.; Wang, C.; Gregory, K.J.; Han, G.W.; Cho, H.P.; Xia, Y.; Niswender, C.M.; Katritch, V.; Meiler, J.; Cherezov, V.; et al. Structure of a Class C GPCR Metabotropic Glutamate Receptor 1 Bound to an Allosteric Modulator. Science 2014, 344, 58. [Google Scholar] [CrossRef] [PubMed]
- Bruno, A.; Costantino, G.; de Fabritiis, G.; Pastor, M.; Selent, J. Membrane-Sensitive Conformational States of Helix 8 in the Metabotropic Glu2 Receptor, a Class C GPCR. PLoS ONE 2012, 7, e42023. [Google Scholar] [CrossRef]
- Binet, V.; Duthey, B.; Lecaillon, J.; Vol, C.; Quoyer, J.; Labesse, G.; Pin, J.-P.; Prézeau, L. Common Structural Requirements for Heptahelical Domain Function in Class A and Class C G Protein-coupled Receptors. J. Biol. Chem. 2007, 282, 12154–12163. [Google Scholar] [CrossRef] [PubMed]
- Doré, A.S.; Okrasa, K.; Patel, J.C.; Serrano-Vega, M.; Bennett, K.; Cooke, R.M.; Errey, J.C.; Jazayeri, A.; Khan, S.; Tehan, B.; et al. Structure of class C GPCR metabotropic glutamate receptor 5 transmembrane domain. Nature 2014, 511, 557–562. [Google Scholar] [CrossRef]
- Ballesteros, J.A.; Weinstein, H. [19] Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. In Methods in Neurosciences; Sealfon, S.C., Ed.; Academic Press: Cambridge, MA, USA, 1995; Volume 25, pp. 366–428. [Google Scholar]
- Chang, W.; Chen, T.-H.; Pratt, S.; Shoback, D. Amino Acids in the Second and Third Intracellular Loops of the Parathyroid Ca2+-sensing Receptor Mediate Efficient Coupling to Phospholipase C*. J. Biol. Chem. 2000, 275, 19955–19963. [Google Scholar] [CrossRef]
- Beqollari, D.; Betzenhauser, M.J.; Kammermeier, P.J. Altered G-Protein Coupling in an mGluR6 Point Mutant Associated with Congenital Stationary Night Blindness. Mol. Pharmacol. 2009, 76, 992. [Google Scholar] [CrossRef] [PubMed]
- Lundström, L.; Bissantz, C.; Beck, J.; Wettstein, J.G.; Woltering, T.J.; Wichmann, J.; Gatti, S. Structural determinants of allosteric antagonism at metabotropic glutamate receptor 2: Mechanistic studies with new potent negative allosteric modulators. Br. J. Pharmacol. 2011, 164, 521–537. [Google Scholar] [CrossRef] [PubMed]
- Malherbe, P.; Kratochwil, N.; Zenner, M.-T.; Piussi, J.; Diener, C.; Kratzeisen, C.; Fischer, C.; Porter, R.H.P. Mutational Analysis and Molecular Modeling of the Binding Pocket of the Metabotropic Glutamate 5 Receptor Negative Modulator 2-Methyl-6-(phenylethynyl)-pyridine. Mol. Pharmacol. 2003, 64, 823. [Google Scholar] [CrossRef]
- Bu, L.; Michino, M.; Wolf, R.M.; Brooks Iii, C.L. Improved model building and assessment of the Calcium-sensing receptor transmembrane domain. Proteins: Struct. Funct. Bioinform. 2008, 71, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Goudet, C.; Gaven, F.; Kniazeff, J.; Vol, C.; Liu, J.; Cohen-Gonsaud, M.; Acher, F.; Prézeau, L.; Pin, J.P. Heptahelical domain of metabotropic glutamate receptor 5 behaves like rhodopsin-like receptors. Proc. Natl. Acad. Sci. USA 2004, 101, 378. [Google Scholar] [CrossRef]
- Brien, J.A.; Lemaire, W.; Chen, T.-B.; Chang, R.S.L.; Jacobson, M.A.; Ha, S.N.; Lindsley, C.W.; Schaffhauser, H.J.; Sur, C.; Pettibone, D.J.; et al. A Family of Highly Selective Allosteric Modulators of the Metabotropic Glutamate Receptor Subtype 5. Mol. Pharmacol. 2003, 64, 731. [Google Scholar] [CrossRef] [PubMed]
- Orgován, Z.; Ferenczy, G.G.; Keserű, G.M. The role of water and protein flexibility in the structure-based virtual screening of allosteric GPCR modulators: An mGlu5 receptor case study. J. Comput. Aided Mol. Des. 2019, 33, 787–797. [Google Scholar] [CrossRef]
- Evenseth, L.S.M.; Ocello, R.; Gabrielsen, M.; Masetti, M.; Recanatini, M.; Sylte, I.; Cavalli, A. Exploring Conformational Dynamics of the Extracellular Venus flytrap Domain of the GABAB Receptor: A Path-Metadynamics Study. J. Chem. Inf. Model. 2020, 60, 2294–2303. [Google Scholar] [CrossRef]
- Mao, C.; Shen, C.; Li, C.; Shen, D.-D.; Xu, C.; Zhang, S.; Zhou, R.; Shen, Q.; Chen, L.-N.; Jiang, Z.; et al. Cryo-EM structures of inactive and active GABAB receptor. Cell Res. 2020, 30, 564–573. [Google Scholar] [CrossRef]
- Park, J.; Fu, Z.; Frangaj, A.; Liu, J.; Mosyak, L.; Shen, T.; Slavkovich, V.N.; Ray, K.M.; Taura, J.; Cao, B.; et al. Structure of human GABAB receptor in an inactive state. Nature 2020, 584, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Shaye, H.; Ishchenko, A.; Lam, J.H.; Han, G.W.; Xue, L.; Rondard, P.; Pin, J.-P.; Katritch, V.; Gati, C.; Cherezov, V. Structural basis of the activation of a metabotropic GABA receptor. Nature 2020, 584, 298–303. [Google Scholar] [CrossRef]
- Papasergi-Scott, M.M.; Robertson, M.J.; Seven, A.B.; Panova, O.; Mathiesen, J.M.; Skiniotis, G. Structures of metabotropic GABAB receptor. Nature 2020, 584, 310–314. [Google Scholar] [CrossRef]
- Kniazeff, J.; Bessis, A.-S.; Maurel, D.; Ansanay, H.; Prézeau, L.; Pin, J.-P. Closed state of both binding domains of homodimeric mGlu receptors is required for full activity. Nat. Struct. Mol. Biol. 2004, 11, 706–713. [Google Scholar] [CrossRef]
- Huang, S.; Cao, J.; Jiang, M.; Labesse, G.; Liu, J.; Pin, J.-P.; Rondard, P. Interdomain movements in metabotropic glutamate receptor activation. Proc. Natl. Acad. Sci. USA 2011, 108, 15480–15485. [Google Scholar] [CrossRef]
- Hlavackova, V.; Zabel, U.; Frankova, D.; Bätz, J.; Hoffmann, C.; Prezeau, L.; Pin, J.-P.; Blahos, J.; Lohse, M.J. Sequential Inter- and Intrasubunit Rearrangements During Activation of Dimeric Metabotropic Glutamate Receptor 1. Sci. Signal. 2012, 5, ra59. [Google Scholar] [CrossRef] [PubMed]
- Brock, C.; Oueslati, N.; Soler, S.; Boudier, L.; Rondard, P.; Pin, J.-P. Activation of a Dimeric Metabotropic Glutamate Receptor by Intersubunit Rearrangement. J. Biol. Chem. 2007, 282, 33000–33008. [Google Scholar] [CrossRef]
- Werthmann, R.C.; Tzouros, M.; Lamerz, J.; Augustin, A.; Fritzius, T.; Trovo, L.; Stawarski, M.; Raveh, A.; Diener, C.; Fischer, C.; et al. Symmetric signal transduction and negative allosteric modulation of heterodimeric mGlu1/5 receptors. Neuropharmacology 2020, 108426. [Google Scholar] [CrossRef] [PubMed]
- Galvez, T.; Duthey, B.; Kniazeff, J.; Blahos, J.; Rovelli, G.; Bettler, B.; Prézeau, L.; Pin, J.-P. Allosteric interactions between GB1 and GB2 subunits are required for optimal GABAB receptor function. EMBO 2001, 20, 2152–2159. [Google Scholar] [CrossRef] [PubMed]
- Kniazeff, J.; Galvez, T.; Labesse, G.; Pin, J.-P. No Ligand Binding in the GB2 Subunit of the GABABReceptor Is Required for Activation and Allosteric Interaction between the Subunits. J. Neurosci. 2002, 22, 7352. [Google Scholar] [CrossRef]
- Robbins, M.J.; Calver, A.R.; Filippov, A.K.; Hirst, W.D.; Russell, R.B.; Wood, M.D.; Nasir, S.; Couve, A.; Brown, D.A.; Moss, S.J.; et al. GABAB2 Is Essential for G-Protein Coupling of the GABAB Receptor Heterodimer. J. Neurosci. 2001, 21, 8043. [Google Scholar] [CrossRef]
- Binet, V.; Brajon, C.; Le Corre, L.; Acher, F.; Pin, J.-P.; Prézeau, L. The Heptahelical Domain of GABAB2 Is Activated Directly by CGP7930, a Positive Allosteric Modulator of the GABAB Receptor. J. Biol. Chem. 2004, 279, 29085–29091. [Google Scholar] [CrossRef] [PubMed]
- Lecat-Guillet, N.; Monnier, C.; Rovira, X.; Kniazeff, J.; Lamarque, L.; Zwier, J.M.; Trinquet, E.; Pin, J.-P.; Rondard, P. FRET-Based Sensors Unravel Activation and Allosteric Modulation of the GABAB Receptor. Cell Chem. Biol. 2017, 24, 360–370. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Huang, S.; Qian, J.; Huang, J.; Jin, L.; Su, Z.; Yang, J.; Liu, J. Evolution of the class C GPCR Venus flytrap modules involved positive selected functional divergence. BMC Evol. Biol. 2009, 9, 67. [Google Scholar] [CrossRef]
- Xue, L.; Sun, Q.; Zhao, H.; Rovira, X.; Gai, S.; He, Q.; Pin, J.-P.; Liu, J.; Rondard, P. Rearrangement of the transmembrane domain interfaces associated with the activation of a GPCR hetero-oligomer. Nat. Commun. 2019, 10, 2765. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, Z.; Moreno-Delgado, D.; Dalton, J.A.R.; Rovira, X.; Trapero, A.; Goudet, C.; Llebaria, A.; Giraldo, J.; Yuan, Q.; et al. Allosteric control of an asymmetric transduction in a G protein-coupled receptor heterodimer. eLife 2017, 6, e26985. [Google Scholar] [CrossRef]
- Levitz, J.; Habrian, C.; Bharill, S.; Fu, Z.; Vafabakhsh, R.; Isacoff, E.Y. Mechanism of Assembly and Cooperativity of Homomeric and Heteromeric Metabotropic Glutamate Receptors. Neuron 2016, 92, 143–159. [Google Scholar] [CrossRef]
- Lee, J.; Munguba, H.; Gutzeit, V.A.; Singh, D.R.; Kristt, M.; Dittman, J.S.; Levitz, J. Defining the Homo- and Heterodimerization Propensities of Metabotropic Glutamate Receptors. Cell Rep. 2020, 31, 107605. [Google Scholar] [CrossRef] [PubMed]
- Pandya, N.J.; Klaassen, R.V.; van der Schors, R.C.; Slotman, J.A.; Houtsmuller, A.; Smit, A.B.; Li, K.W. Group 1 metabotropic glutamate receptors 1 and 5 form a protein complex in mouse hippocampus and cortex. Proteomics 2016, 16, 2698–2705. [Google Scholar] [CrossRef]
- Hlavackova, V.; Goudet, C.; Kniazeff, J.; Zikova, A.; Maurel, D.; Vol, C.; Trojanova, J.; Prézeau, L.; Pin, J.-P.; Blahos, J. Evidence for a single heptahelical domain being turned on upon activation of a dimeric GPCR. EMBO 2005, 24, 499–509. [Google Scholar] [CrossRef]
- Moreno Delgado, D.; Møller, T.C.; Ster, J.; Giraldo, J.; Maurel, D.; Rovira, X.; Scholler, P.; Zwier, J.M.; Perroy, J.; Durroux, T.; et al. Pharmacological evidence for a metabotropic glutamate receptor heterodimer in neuronal cells. eLife 2017, 6, e25233. [Google Scholar] [CrossRef] [PubMed]
- Goudet, C.; Kniazeff, J.; Hlavackova, V.; Malhaire, F.; Maurel, D.; Acher, F.; Blahos, J.; Prézeau, L.; Pin, J.-P. Asymmetric Functioning of Dimeric Metabotropic Glutamate Receptors Disclosed by Positive Allosteric Modulators*. J. Biol. Chem. 2005, 280, 24380–24385. [Google Scholar] [CrossRef] [PubMed]
- Toneatti, R.; Shin, J.M.; Shah, U.H.; Mayer, C.R.; Saunders, J.M.; Fribourg, M.; Arsenovic, P.T.; Janssen, W.G.; Sealfon, S.C.; López-Giménez, J.F.; et al. Interclass GPCR heteromerization affects localization and trafficking. Sci. Signal. 2020, 13, eaaw3122. [Google Scholar] [CrossRef]
- Ferré, S.; Karcz-Kubicha, M.; Hope, B.T.; Popoli, P.; Burgueño, J.; Gutiérrez, M.A.; Casadó, V.; Fuxe, K.; Goldberg, S.R.; Lluis, C.; et al. Synergistic interaction between adenosine A2A and glutamate mGlu5 receptors: Implications for striatal neuronal function. Proc. Natl. Acad. Sci. USA 2002, 99, 11940. [Google Scholar] [CrossRef] [PubMed]
- Hideshima, K.S.; Hojati, A.; Saunders, J.M.; On, D.M.; de la Fuente Revenga, M.; Shin, J.M.; Sánchez-González, A.; Dunn, C.M.; Pais, A.B.; Pais, A.C.; et al. Role of mGlu2 in the 5-HT 2A receptor-dependent antipsychotic activity of clozapine in mice. Psychopharmacology 2018, 235, 3149–3165. [Google Scholar] [CrossRef] [PubMed]
- Moreno, J.L.; Miranda-Azpiazu, P.; García-Bea, A.; Younkin, J.; Cui, M.; Kozlenkov, A.; Ben-Ezra, A.; Voloudakis, G.; Fakira, A.K.; Baki, L.; et al. Allosteric signaling through an mGlu2 and 5-HT2A; heteromeric receptor complex and its potential contribution to schizophrenia. Sci. Signal. 2016, 9, ra5. [Google Scholar] [CrossRef] [PubMed]
- González-Maeso, J.; Ang, R.L.; Yuen, T.; Chan, P.; Weisstaub, N.V.; López-Giménez, J.F.; Zhou, M.; Okawa, Y.; Callado, L.F.; Milligan, G.; et al. Identification of a serotonin/glutamate receptor complex implicated in psychosis. Nature 2008, 452, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Sebastianutto, I.; Goyet, E.; Andreoli, L.; Font-Ingles, J.; Moreno-Delgado, D.; Bouquier, N.; Jahannault-Talignani, C.; Moutin, E.; Di Menna, L.; Maslava, N.; et al. D1-mGlu5 heteromers mediate noncanonical dopamine signaling in Parkinson’s disease. J. Clin. Investig. 2020, 130, 1168–1184. [Google Scholar] [CrossRef] [PubMed]
- Akgün, E.; Javed, M.I.; Lunzer, M.M.; Smeester, B.A.; Beitz, A.J.; Portoghese, P.S. Ligands that interact with putative MOR-mGluR5 heteromer in mice with inflammatory pain produce potent antinociception. Proc. Natl. Acad. Sci. USA 2013, 110, 11595. [Google Scholar] [CrossRef]
- Kwan, C.; Frouni, I.; Nuara, S.G.; Belliveau, S.; Kang, W.; Hamadjida, A.; Bédard, D.; Beaudry, F.; Panisset, M.; Gourdon, J.C.; et al. Combined 5-HT2A and mGlu2 modulation for the treatment of dyskinesia and psychosis in Parkinson’s disease. Neuropharmacology 2021, 186, 108465. [Google Scholar] [CrossRef] [PubMed]
- Gahbauer, S.; Bockmann, R.A. Membrane-Mediated Oligomerization of G Protein Coupled Receptors and Its Implications for GPCR Function. Front. Physiol. 2016, 7, 494. [Google Scholar] [CrossRef]
- Ghosh, E.; Kumari, P.; Jaiman, D.; Shukla, A.K. Methodological advances: The unsung heroes of the GPCR structural revolution. Nat. Rev. Mol. Cell Biol. 2015, 16, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Xiang, J.; Chun, E.; Liu, C.; Jing, L.; Al-Sahouri, Z.; Zhu, L.; Liu, W. Successful Strategies to Determine High-Resolution Structures of GPCRs. Trends Pharmacol. Sci. 2016, 37, 1055–1069. [Google Scholar] [CrossRef]
- Bolla, J.R.; Agasid, M.T.; Mehmood, S.; Robinson, C.V. Membrane Protein–Lipid Interactions Probed Using Mass Spectrometry. Annu. Rev. Biochem. 2019, 88, 85–111. [Google Scholar] [CrossRef] [PubMed]
- Bogan, A.A.; Thorn, K.S. Anatomy of hot spots in protein interfaces11Edited by J. Wells. J. Mol. Biol. 1998, 280, 1–9. [Google Scholar] [CrossRef]
- Morrison, K.L.; Weiss, G.A. Combinatorial alanine-scanning. Curr. Opin. Chem. Biol. 2001, 5, 302–307. [Google Scholar] [CrossRef]
- Massova, I.; Kollman, P.A. Computational Alanine Scanning to Probe Protein−Protein Interactions: A Novel Approach to Evaluate Binding Free Energies. J. Am. Chem. Soc. 1999, 121, 8133–8143. [Google Scholar] [CrossRef]
- Moreira, I.S.; Fernandes, P.A.; Ramos, M.J. Computational alanine scanning mutagenesis—An improved methodological approach. J. Comput. Chem. 2007, 28, 644–654. [Google Scholar] [CrossRef]
- Lazim, R.; Suh, D.; Choi, S. Advances in Molecular Dynamics Simulations and Enhanced Sampling Methods for the Study of Protein Systems. Int. J. Mol. Sci. 2020, 21, 6339. [Google Scholar] [CrossRef] [PubMed]
- Bowman, G.R.; Pande, V.S.; Noé, F.A. Introduction to Markov State Models and their Application to Long Timescale Molecular Simulation; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2013; Volume 797. [Google Scholar]
- Chipot, C.; Kollman, P.A.; Pearlman, D.A. Alternative approaches to potential of mean force calculations: Free energy perturbation versus thermodynamic integration. Case study of some representative nonpolar interactions. J. Comput. Chem. 1996, 17, 1112–1131. [Google Scholar] [CrossRef]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.; Hou, T. End-point binding free energy calculation with MM/PBSA and MM/GBSA: Strategies and applications in drug design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef] [PubMed]
- Schütte, C.; Huisinga, W.; Deuflhard, P. Transfer operator approach to conformational dynamics in biomolecular systems. In Ergodic Theory, Analysis, and Efficient Simulation of Dynamical Systems; Springer: Berlin/Heidelberg, Germany, 2001; pp. 191–223. [Google Scholar]
- Bitencourt-Ferreira, G.; de Azevedo, W.F. Development of a machine-learning model to predict Gibbs free energy of binding for protein-ligand complexes. Biophys. Chem. 2018, 240, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Barlow, K.A.; Ó Conchúir, S.; Thompson, S.; Suresh, P.; Lucas, J.E.; Heinonen, M.; Kortemme, T. Flex ddG: Rosetta Ensemble-Based Estimation of Changes in Protein–Protein Binding Affinity upon Mutation. J. Phys. Chem. B 2018, 122, 5389–5399. [Google Scholar] [CrossRef] [PubMed]
- Ollikainen, N.; Smith, C.A.; Fraser, J.S.; Kortemme, T. Flexible backbone sampling methods to model and design protein alternative conformations. Methods Enzymol. 2013, 523, 61–85. [Google Scholar] [PubMed]
- Ibarra, A.A.; Bartlett, G.J.; Hegedüs, Z.; Dutt, S.; Hobor, F.; Horner, K.A.; Hetherington, K.; Spence, K.; Nelson, A.; Edwards, T.A.; et al. Predicting and Experimentally Validating Hot-Spot Residues at Protein–Protein Interfaces. ACS Chem. Biol. 2019, 14, 2252–2263. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Liu, C.; Deng, L. Machine learning approaches for protein–protein interaction hot spot prediction: Progress and comparative assessment. Molecules 2018, 23, 2535. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Ascher, D.B.; Blundell, T.L. mCSM: Predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics 2014, 30, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Shan, J.; Khelashvili, G.; Mondal, S.; Mehler, E.L.; Weinstein, H. Ligand-dependent conformations and dynamics of the serotonin 5-HT 2A receptor determine its activation and membrane-driven oligomerization properties. PLoS Comput. Biol. 2012, 8, e1002473. [Google Scholar] [CrossRef]
- Baltoumas, F.A.; Theodoropoulou, M.C.; Hamodrakas, S.J. Molecular dynamics simulations and structure-based network analysis reveal structural and functional aspects of G-protein coupled receptor dimer interactions. J. Comput. Aided Mol. Des. 2016, 30, 489–512. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.M.; Wang, H.; Provasi, D.; Filizola, M. Assessing the relative stability of dimer interfaces in g protein-coupled receptors. PLoS Comput. Biol. 2012, 8, e1002649. [Google Scholar] [CrossRef]
- Giorgi, F.; Bruni, L.E.; Maggio, R. Receptor Oligomerization as a Process Modulating Cellular Semiotics. Biosemiotics 2010, 3, 157–176. [Google Scholar] [CrossRef]
- Ma, B.; Elkayam, T.; Wolfson, H.; Nussinov, R. Protein–protein interactions: Structurally conserved residues distinguish between binding sites and exposed protein surfaces. Proc. Natl. Acad. Sci. USA 2003, 100, 5772–5777. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Panchenko, A.R. Mechanisms of protein oligomerization, the critical role of insertions and deletions in maintaining different oligomeric states. Proc. Natl. Acad. Sci. USA 2010, 107, 20352–20357. [Google Scholar] [CrossRef] [PubMed]
- Tarakanov, A.O.; Fuxe, K.G. Triplet puzzle: Homologies of receptor heteromers. J. Mol. Neurosci. 2010, 41, 294–303. [Google Scholar] [CrossRef]
- Borroto-Escuela, D.O.; Tarakanov, A.O.; Brito, I.; Fuxe, K. Glutamate heteroreceptor complexes in the brain. Pharmacol. Rep. 2018, 70, 936–950. [Google Scholar] [CrossRef]
- Lu, H.; Zhou, Q.; He, J.; Jiang, Z.; Peng, C.; Tong, R.; Shi, J. Recent advances in the development of protein–protein interactions modulators: Mechanisms and clinical trials. Signal. Transduct. Target. Ther. 2020, 5, 1–23. [Google Scholar] [CrossRef]
- Borroto-Escuela, D.O.; Pérez-Alea, M.; Narvaez, M.; Tarakanov, A.O.; Mudó, G.; Jiménez-Beristain, A.; Agnati, L.F.; Ciruela, F.; Belluardo, N.; Fuxe, K. Enhancement of the FGFR1 signaling in the FGFR1-5-HT1A heteroreceptor complex in midbrain raphe 5-HT neuron systems. Relevance for neuroplasticity and depression. Biochem. Biophys. Res. Commun. 2015, 463, 180–186. [Google Scholar] [CrossRef]
- Fuxe, K.; Borroto-Escuela, D.O.; Romero-Fernandez, W.; Palkovits, M.; Tarakanov, A.O.; Ciruela, F.; Agnati, L.F. Moonlighting proteins and protein–protein interactions as neurotherapeutic targets in the G protein-coupled receptor field. Neuropsychopharmacology 2014, 39, 131–155. [Google Scholar] [CrossRef] [PubMed]
- Schiedel, A.C.; Kose, M.; Barreto, C.; Bueschbell, B.; Morra, G.; Sensoy, O.; Moreira, I.S. Prediction and targeting of interaction interfaces in G-protein coupled receptor oligomers. Curr. Top. Med. Chem. 2018, 18, 714–746. [Google Scholar] [CrossRef]
- Romero-Molina, S.; Ruiz-Blanco, Y.B.; Harms, M.; Münch, J.; Sanchez-Garcia, E. PPI-Detect: A support vector machine model for sequence-based prediction of protein–protein interactions. J. Comput. Chem. 2019, 40, 1233–1242. [Google Scholar] [CrossRef] [PubMed]
- Baspinar, A.; Cukuroglu, E.; Nussinov, R.; Keskin, O.; Gursoy, A. PRISM: A web server and repository for prediction of protein-protein interactions and modeling their 3D complexes. Nucleic Acids Res. 2014, 42, W285–W289. [Google Scholar] [CrossRef]
- Qin, S.; Zhou, H.X. meta-PPISP: A meta web server for protein-protein interaction site prediction. Bioinformatics 2007, 23, 3386–3387. [Google Scholar] [CrossRef]
- Hosur, R.; Peng, J.; Vinayagam, A.; Stelzl, U.; Xu, J.; Perrimon, N.; Bienkowska, J.; Berger, B. A computational framework for boosting confidence in high-throughput protein-protein interaction datasets. Genome Biol. 2012, 13, R76. [Google Scholar] [CrossRef]
- Li, Y.; Ilie, L. SPRINT: Ultrafast protein-protein interaction prediction of the entire human interactome. BMC Bioinform. 2017, 18, 485. [Google Scholar] [CrossRef] [PubMed]
- Bakail, M.; Ochsenbein, F. Targeting protein–protein interactions, a wide open field for drug design. C. R. Chim. 2016, 19, 19–27. [Google Scholar] [CrossRef]
- Fuxe, K.; Marcellino, D.; Borroto-Escuela, D.O.; Frankowska, M.; Ferraro, L.; Guidolin, D.; Ciruela, F.; Agnati, L.F. The changing world of G protein-coupled receptors: From monomers to dimers and receptor mosaics with allosteric receptor–receptor interactions. J. Recept. Signal. Transduct. 2010, 30, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Casadó, V.; Cortés, A.; Mallol, J.; Pérez-Capote, K.; Ferré, S.; Lluis, C.; Franco, R.; Canela, E.I. GPCR homomers and heteromers: A better choice as targets for drug development than GPCR monomers? Pharmacol. Ther. 2009, 124, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Borroto-Escuela, D.O.; Fuxe, K. Oligomeric receptor complexes and their allosteric receptor-receptor interactions in the plasma membrane represent a new biological principle for integration of signals in the CNS. Front. Mol. Neurosci. 2019, 12, 230. [Google Scholar] [CrossRef] [PubMed]
- Pelay-Gimeno, M.; Glas, A.; Koch, O.; Grossmann, T.N. Structure-based design of inhibitors of protein–protein interactions: Mimicking peptide binding epitopes. Angew. Chem. Int. Ed. 2015, 54, 8896–8927. [Google Scholar] [CrossRef]
- Guidolin, D.; Marcoli, M.; Tortorella, C.; Maura, G.; Agnati, L.F. Receptor-receptor interactions as a widespread phenomenon: Novel targets for drug development? Front. Endocrinol. 2019, 10, 53. [Google Scholar] [CrossRef]
- Sedan, Y.; Marcu, O.; Lyskov, S.; Schueler-Furman, O. Peptiderive server: Derive peptide inhibitors from protein–protein interactions. Nucleic Acids Res. 2016, 44, W536–W541. [Google Scholar] [CrossRef]
- Bullock, B.N.; Jochim, A.L.; Arora, P.S. Assessing helical protein interfaces for inhibitor design. J. Am. Chem. Soc. 2011, 133, 14220–14223. [Google Scholar] [CrossRef]
- Gavenonis, J.; Sheneman, B.A.; Siegert, T.R.; Eshelman, M.R.; Kritzer, J.A. Comprehensive analysis of loops at protein-protein interfaces for macrocycle design. Nat. Chem. Biol. 2014, 10, 716–722. [Google Scholar] [CrossRef] [PubMed]
- Guerler, A.; Govindarajoo, B.; Zhang, Y. Mapping monomeric threading to protein–protein structure prediction. J. Chem. Inf. Model. 2013, 53, 717–725. [Google Scholar] [CrossRef]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein–protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for rigid and symmetric docking. Nucleic Acids Res. 2005, 33 (Suppl. 2), W363–W367. [Google Scholar] [CrossRef] [PubMed]
- Tovchigrechko, A.; Vakser, I.A. GRAMM-X public web server for protein–protein docking. Nucleic Acids Res. 2006, 34 (Suppl. 2), W310–W314. [Google Scholar] [CrossRef]
- Bruzzoni-Giovanelli, H.; Alezra, V.; Wolff, N.; Dong, C.-Z.; Tuffery, P.; Rebollo, A. Interfering peptides targeting protein–protein interactions: The next generation of drugs? Drug Discov. Today 2018, 23, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Obarska-Kosinska, A.; Iacoangeli, A.; Lepore, R.; Tramontano, A. PepComposer: Computational design of peptides binding to a given protein surface. Nucleic Acids Res. 2016, 44, W522–W528. [Google Scholar] [CrossRef]
- Henninot, A.; Collins, J.C.; Nuss, J.M. The current state of peptide drug discovery: Back to the future? J. Med. Chem. 2018, 61, 1382–1414. [Google Scholar] [CrossRef]
- Aghebati-Maleki, L.; Bakhshinejad, B.; Baradaran, B.; Motallebnezhad, M.; Aghebati-Maleki, A.; Nickho, H.; Yousefi, M.; Majidi, J. Phage display as a promising approach for vaccine development. J. Biomed. Sci. 2016, 23, 1–18. [Google Scholar] [CrossRef]
- Lau, Y.H.; De Andrade, P.; Wu, Y.; Spring, D.R. Peptide stapling techniques based on different macrocyclisation chemistries. Chem. Soc. Rev. 2015, 44, 91–102. [Google Scholar] [CrossRef]
- Zhai, L.; Otani, Y.; Hori, Y.; Tomita, T.; Ohwada, T. Peptide-based short single β-strand mimics without hydrogen bonding or aggregation. Chem. Commun. 2020, 56, 1573–1576. [Google Scholar] [CrossRef]
- Zheng, J.; Liu, C.; Sawaya, M.R.; Vadla, B.; Khan, S.; Woods, R.J.; Eisenberg, D.; Goux, W.J.; Nowick, J.S. Macrocyclic β-sheet peptides that inhibit the aggregation of a tau-protein-derived hexapeptide. J. Am. Chem. Soc. 2011, 133, 3144–3157. [Google Scholar] [CrossRef] [PubMed]
- Kalafatovic, D.; Giralt, E. Cell-penetrating peptides: Design strategies beyond primary structure and amphipathicity. Molecules 2017, 22, 1929. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, K.; Milech, N.; Juraja, S.M.; Cunningham, P.T.; Stone, S.R.; Francis, R.W.; Anastasas, M.; Hall, C.M.; Heinrich, T.; Bogdawa, H.M. A platform for discovery of functional cell-penetrating peptides for efficient multi-cargo intracellular delivery. Sci. Rep. 2018, 8, 12538. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Peng, H.; Kang, J.; Sun, D. Cell-penetrating peptides: Possible transduction mechanisms and therapeutic applications. Biomed. Rep. 2016, 4, 528–534. [Google Scholar] [CrossRef]
- Pei, L.; Li, S.; Wang, M.; Diwan, M.; Anisman, H.; Fletcher, P.J.; Nobrega, J.N.; Liu, F. Uncoupling the dopamine D1–D2 receptor complex exerts antidepressant-like effects. Nat. Med. 2010, 16, 1393–1395. [Google Scholar] [CrossRef]
- Lee, A.C.-L.; Harris, J.L.; Khanna, K.K.; Hong, J.-H. A comprehensive review on current advances in peptide drug development and design. Int. J. Mol. Sci. 2019, 20, 2383. [Google Scholar] [CrossRef] [PubMed]
- Botta, J.; Bibic, L.; Killoran, P.; McCormick, P.J.; Howell, L.A. Design and development of stapled transmembrane peptides that disrupt the activity of G-protein–coupled receptor oligomers. J. Biol. Chem. 2019, 294, 16587–16603. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Description | Website |
|---|---|---|
| PPI-Detect | Sequence-based prediction. Based on a support vector machine model trained using pairwise descriptors derived via numerical encoding of the primary sequences of protein pairs embedded as vectors. | https://ppi-detect.zmb.uni-due.de/ (accessed on 22 March 2021) |
| SPRINT | Sequence-based prediction. Developed based on the assumption that a target protein pair is likely to interact if their subsequence pairs exhibit high degree of similarity with a known interacting protein pair. | https://github.com/lucian-ilie/SPRINT/ (accessed on 22 March 2021) |
| Coev2Net | Structure-based prediction. Prediction and assessment of individual interactions from a high-throughput experiment. Uses protein threading to generate a homology model of the target, from which extent of co-evolution is calculated. | http://cb.csail.mit.edu/cb/coev2net/ (accessed on 22 March 2021) |
| PRISM | Structure-based prediction. Uses evolutionary conservation of hotspot PPI residues and considers shape complementarities of protein pairs. | http://cosbi.ku.edu.tr/prism/ (accessed on 22 March 2021) |
| meta-PPISP | Structure-based prediction of PPI interface residues. Uses scores from three webservers—cons-PPISP, Promate, and PINUP—train a linear regression to predict residues located at protein–protein interface. | https://pipe.rcc.fsu.edu/meta-ppisp.html (accessed on 22 March 2021) |
| Cons-PPISP | Consensus-based neural network approach for the prediction of residues making up the binding site at the protein interface. Features used to train the neural network include sequence profile and solvent accessibility of neighboring residues. | https://pipe.rcc.fsu.edu/ppisp.html (accessed on 22 March 2021) |
| Promate | Structure-based prediction of PPI binding sites. Constructed based on quantitative comparison between the PPI interface and other parts of the protein surface in terms of amino acid composition, type of secondary structure, evolutionary conservation, atomic fluctuation, and crystallographic waters. | http://bioportal.weizmann.ac.il/promate/ (accessed on 22 March 2021) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lazim, R.; Suh, D.; Lee, J.W.; Vu, T.N.L.; Yoon, S.; Choi, S. Structural Characterization of Receptor–Receptor Interactions in the Allosteric Modulation of G Protein-Coupled Receptor (GPCR) Dimers. Int. J. Mol. Sci. 2021, 22, 3241. https://doi.org/10.3390/ijms22063241
Lazim R, Suh D, Lee JW, Vu TNL, Yoon S, Choi S. Structural Characterization of Receptor–Receptor Interactions in the Allosteric Modulation of G Protein-Coupled Receptor (GPCR) Dimers. International Journal of Molecular Sciences. 2021; 22(6):3241. https://doi.org/10.3390/ijms22063241
Chicago/Turabian StyleLazim, Raudah, Donghyuk Suh, Jai Woo Lee, Thi Ngoc Lan Vu, Sanghee Yoon, and Sun Choi. 2021. "Structural Characterization of Receptor–Receptor Interactions in the Allosteric Modulation of G Protein-Coupled Receptor (GPCR) Dimers" International Journal of Molecular Sciences 22, no. 6: 3241. https://doi.org/10.3390/ijms22063241
APA StyleLazim, R., Suh, D., Lee, J. W., Vu, T. N. L., Yoon, S., & Choi, S. (2021). Structural Characterization of Receptor–Receptor Interactions in the Allosteric Modulation of G Protein-Coupled Receptor (GPCR) Dimers. International Journal of Molecular Sciences, 22(6), 3241. https://doi.org/10.3390/ijms22063241