
Pneumoviral Phosphoprotein, a Multidomain Adaptor-Like Protein of Apparent Low Structural Complexity and High Conformational Versatility


Abstract
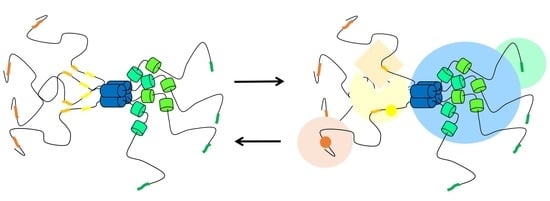
{kind=link}
{kind=link}
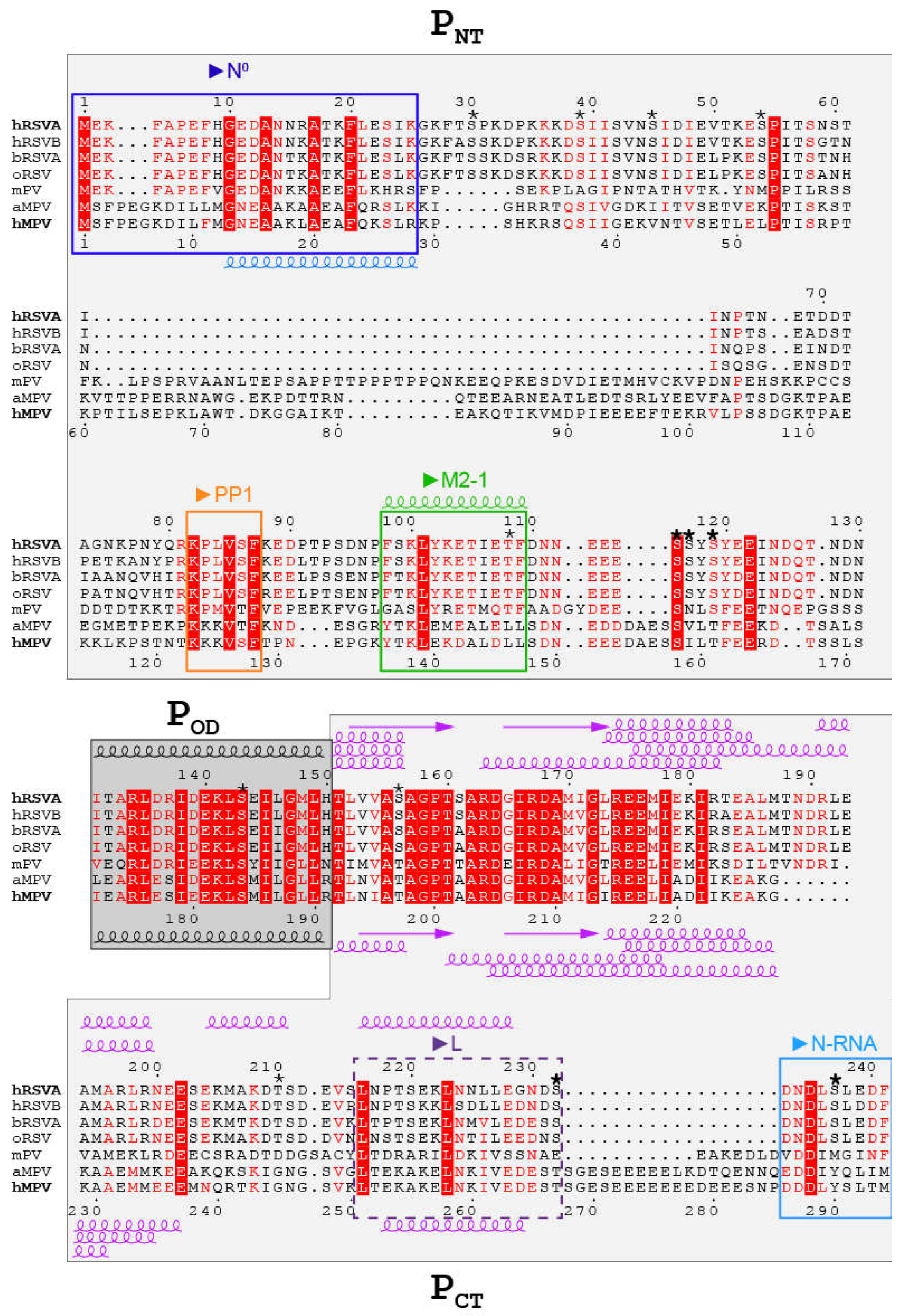{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
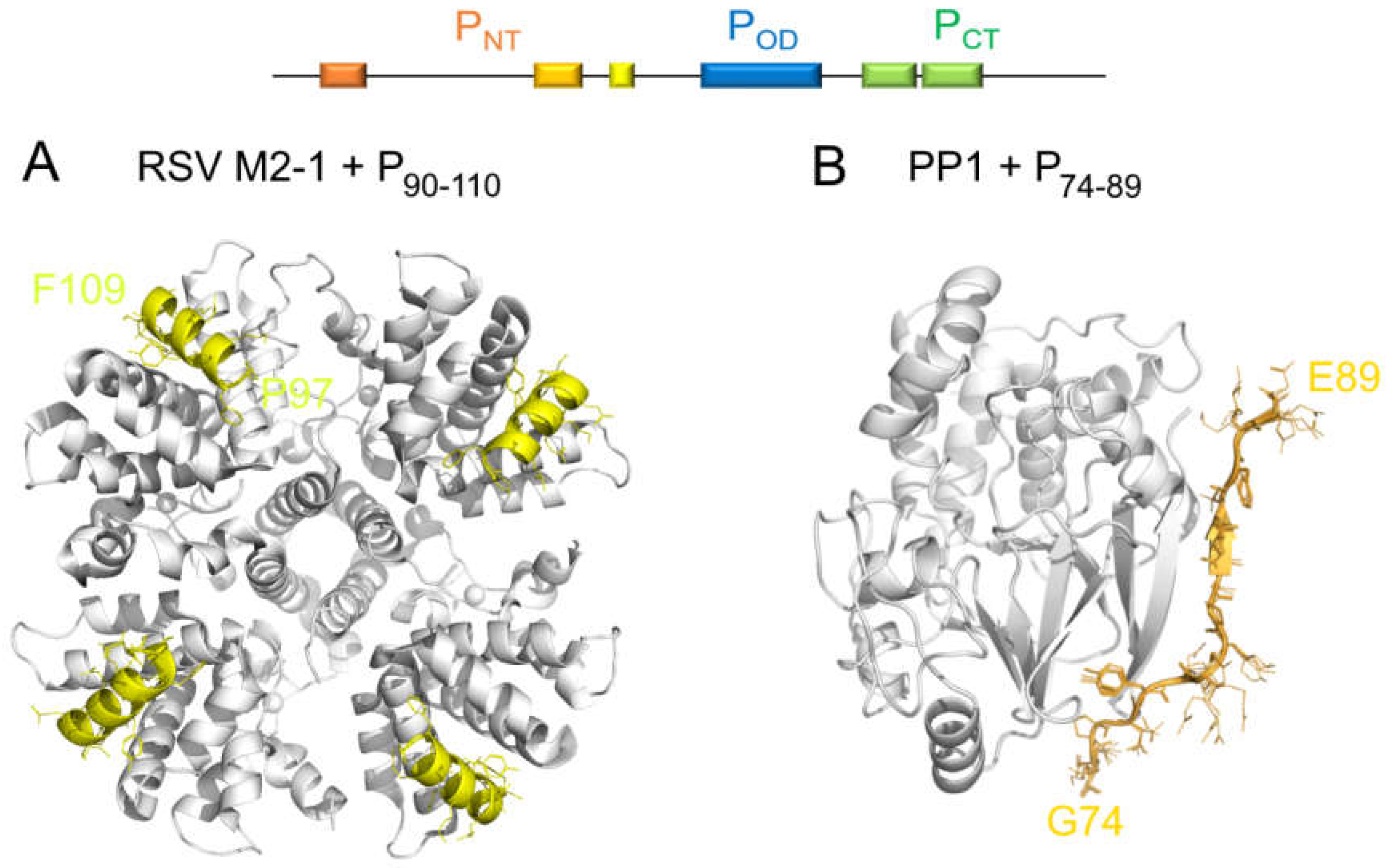{kind=link}
1. Introduction
2. Structural Features of Mononegavirales Phosphoproteins
3. The Three Domains in Pneumoviridae Phosphoproteins Display Multiple Binding Sites Associated to Multiple Functions
4. Pneumoviridae Phosphoproteins Display a High Degree of Intrinsic Disorder
4.1. The Oligomerisation Domain Is the Only Stably Folded Domain in RSV/hMPV Phosphoproteins
4.2. Relative Disorder in the N- and C-terminal Regions of RSV/hMPV Phosphoproteins
4.3. Characterization of Disorder at the Residue-Level of RSV Phosphoprotein
4.4. Disorder Correlates with Fuzzy Binding of the Phosphoprotein to the Nucleoprotein
5. Folding Upon Binding in the Intrinsically Disordered Domains of Pneumoviridae Phosphoprotein
5.1. The Disordered N-Terminus of P Folds in the N0–P Complex
5.2. Pneumoviridae P Protomers Each Adopt a Unique Fold in the L–P Complex
6. Pneumoviridae P Protein Serves as a Docking Platform for a Tertiary Complex Involved in Transcription Regulation
6.1. A Disordered Short Linear Motif in PNT Folds Into an α-Helix in the RSV M2-1–P Complex
6.2. A Short Linear Motif in RSV PNT Binds to the PP1 Phosphatase That Dephosphorylates M2-1
7. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| IB | Inclusion body |
| L | Large subunit of polymerase |
| hMPV | Human Metapneumovirus |
| HSQC | Heteronuclear Single Quantum Correlation |
| N | Nucleoprotein |
| NMR | Nuclear magnetic resonance |
| NOE | Nuclear Overhauser Effect |
| P | P protein, phosphoprotein |
| PCT | P protein C-terminal domain |
| PNT | P protein N-terminal domain |
| POD | P protein oligomerization domain |
| RNP | Ribonucleoprotein complex |
| hRSV | Human Respiratory syncytial virus |
| SAXS | Small angle X-ray scattering |
| SLiM | Short linear motif |
| SSP | Secondary Structure Propensity |
References
- Uversky, V.N. Unusual biophysics of intrinsically disordered proteins. Biochim. Biophys. Acta 2013, 1834, 932–951. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Santambrogio, C.; Brocca, S.; Grandori, R. Length-dependent compaction of intrinsically disordered proteins. FEBS Lett. 2012, 586, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Amarasinghe, G.K.; Ayllon, M.A.; Bao, Y.; Basler, C.F.; Bavari, S.; Blasdell, K.R.; Briese, T.; Brown, P.A.; Bukreyev, A.; Balkema-Buschmann, A.; et al. Taxonomy of the order Mononegavirales: Update 2019. Arch. Virol. 2019, 164, 1967–1980. [Google Scholar] [CrossRef] [PubMed]
- Collins, P.L.; Karron, R.A. Respiratory Syncytial Virus and Metapneumovirus. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippinscot Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 1086–1123. [Google Scholar]
- Shi, T.; McAllister, D.A.; O’Brien, K.L.; Simoes, E.A.F.; Madhi, S.A.; Gessner, B.D.; Polack, F.P.; Balsells, E.; Acacio, S.; Aguayo, C.; et al. Global, regional, and national disease burden estimates of acute lower respiratory infections due to respiratory syncytial virus in young children in 2015: A systematic review and modelling study. Lancet 2017, 390, 946–958. [Google Scholar] [CrossRef]
- Coultas, J.A.; Smyth, R.; Openshaw, P.J. Respiratory syncytial virus (RSV): A scourge from infancy to old age. Thorax 2019, 74, 986–993. [Google Scholar] [CrossRef]
- Sacco, R.E.; McGill, J.L.; Pillatzki, A.E.; Palmer, M.V.; Ackermann, M.R. Respiratory syncytial virus infection in cattle. Vet. Pathol. 2014, 51, 427–436. [Google Scholar] [CrossRef]
- Caracciolo, S.; Minini, C.; Colombrita, D.; Rossi, D.; Miglietti, N.; Vettore, E.; Caruso, A.; Fiorentini, S. Human metapneumovirus infection in young children hospitalized with acute respiratory tract disease: Virologic and clinical features. Pediatr. Infect. Dis. J. 2008, 27, 406–412. [Google Scholar] [CrossRef]
- Edwards, K.M.; Zhu, Y.; Griffin, M.R.; Weinberg, G.A.; Hall, C.B.; Szilagyi, P.G.; Staat, M.A.; Iwane, M.; Prill, M.M.; Williams, J.V. Burden of human metapneumovirus infection in young children. N. Engl. J. Med. 2013, 368, 633–643. [Google Scholar] [CrossRef]
- Ruigrok, R.W.; Crepin, T.; Kolakofsky, D. Nucleoproteins and nucleocapsids of negative-strand RNA viruses. Curr. Opin. Microbiol. 2011, 14, 504–510. [Google Scholar] [CrossRef]
- Jamin, M.; Yabukarski, F. Nonsegmented Negative-Sense RNA Viruses-Structural Data Bring New Insights Into Nucleocapsid Assembly. Adv. Virus Res. 2017, 97, 143–185. [Google Scholar] [CrossRef]
- Morin, B.; Kranzusch, P.J.; Rahmeh, A.A.; Whelan, S.P. The polymerase of negative-stranded RNA viruses. Curr. Opin. Virol. 2013, 3, 103–110. [Google Scholar] [CrossRef]
- Collins, P.L.; Melero, J.A. Progress in understanding and controlling respiratory syncytial virus: Still crazy after all these years. Virus Res. 2011, 162, 80–99. [Google Scholar] [CrossRef]
- Fearns, R.; Plemper, R.K. Polymerases of paramyxoviruses and pneumoviruses. Virus Res. 2017, 234, 87–102. [Google Scholar] [CrossRef]
- Liang, B. Structures of the Mononegavirales Polymerases. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Cox, R.; Pickar, A.; Qiu, S.; Tsao, J.; Rodenburg, C.; Dokland, T.; Elson, A.; He, B.; Luo, M. Structural studies on the authentic mumps virus nucleocapsid showing uncoiling by the phosphoprotein. Proc. Natl. Acad. Sci. USA 2014, 111, 15208–15213. [Google Scholar] [CrossRef]
- Ding, H.; Green, T.J.; Lu, S.; Luo, M. Crystal structure of the oligomerization domain of the phosphoprotein of vesicular stomatitis virus. J. Virol. 2006, 80, 2808–2814. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.; Crepin, T.; Jamin, M.; Ruigrok, R.W. Structure of the dimerization domain of the rabies virus phosphoprotein. J. Virol. 2010, 84, 3707–3710. [Google Scholar] [CrossRef]
- Communie, G.; Crepin, T.; Maurin, D.; Jensen, M.R.; Blackledge, M.; Ruigrok, R.W. Structure of the tetramerization domain of measles virus phosphoprotein. J. Virol. 2013, 87, 7166–7169. [Google Scholar] [CrossRef] [PubMed]
- Leyrat, C.; Renner, M.; Harlos, K.; Grimes, J.M. Solution and crystallographic structures of the central region of the phosphoprotein from human metapneumovirus. PLoS ONE 2013, 8, e80371. [Google Scholar] [CrossRef]
- Bruhn, J.F.; Kirchdoerfer, R.N.; Urata, S.M.; Li, S.; Tickle, I.J.; Bricogne, G.; Saphire, E.O. Crystal Structure of the Marburg Virus VP35 Oligomerization Domain. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Zinzula, L.; Nagy, I.; Orsini, M.; Weyher-Stingl, E.; Bracher, A.; Baumeister, W. Structures of Ebola and Reston Virus VP35 Oligomerization Domains and Comparative Biophysical Characterization in All Ebolavirus Species. Structure 2019, 27, 39–54. [Google Scholar] [CrossRef]
- Karlin, D.; Belshaw, R. Detecting Remote Sequence Homology in Disordered Proteins: Discovery of Conserved Motifs in the N-Termini of Mononegavirales phosphoproteins. PLoS ONE 2012, 7, e31719. [Google Scholar] [CrossRef]
- Longhi, S. Structural disorder within paramyxoviral nucleoproteins. FEBS Lett. 2015, 589, 2649–2659. [Google Scholar] [CrossRef]
- Whelan, J.N.; Reddy, K.D.; Uversky, V.N.; Teng, M.N. Functional correlations of respiratory syncytial virus proteins to intrinsic disorder. Mol. Biosyst. 2016, 12, 1507–1526. [Google Scholar] [CrossRef]
- Habchi, J.; Longhi, S. Structural Disorder within Paramyxoviral Nucleoproteins and Phosphoproteins in Their Free and Bound Forms: From Predictions to Experimental Assessment. Int. J. Mol. Sci. 2015, 16, 15688–15726. [Google Scholar] [CrossRef]
- Mavrakis, M.; McCarthy, A.A.; Roche, S.; Blondel, D.; Ruigrok, R.W. Structure and function of the C-terminal domain of the polymerase cofactor of rabies virus. J. Mol. Biol. 2004, 343, 819–831. [Google Scholar] [CrossRef]
- Ivanov, I.; Yabukarski, F.; Ruigrok, R.W.; Jamin, M. Structural insights into the rhabdovirus transcription/replication complex. Virus Res. 2011, 162, 126–137. [Google Scholar] [CrossRef]
- Leung, D.W.; Ginder, N.D.; Fulton, D.B.; Nix, J.; Basler, C.F.; Honzatko, R.B.; Amarasinghe, G.K. Structure of the Ebola VP35 interferon inhibitory domain. Proc. Natl. Acad. Sci. USA 2009, 106, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Castagne, N.; Barbier, A.; Bernard, J.; Rezaei, H.; Huet, J.C.; Henry, C.; Da Costa, B.; Eleouet, J.F. Biochemical characterization of the respiratory syncytial virus P-P and P-N protein complexes and localization of the P protein oligomerization domain. J. Gen. Virol. 2004, 85, 1643–1653. [Google Scholar] [CrossRef] [PubMed]
- Llorente, M.T.; Taylor, I.A.; Lopez-Vinas, E.; Gomez-Puertas, P.; Calder, L.J.; Garcia-Barreno, B.; Melero, J.A. Structural properties of the human respiratory syncytial virus P protein: Evidence for an elongated homotetrameric molecule that is the smallest orthologue within the family of paramyxovirus polymerase cofactors. Proteins Struct. Funct. Bioinform. 2008, 72, 946–958. [Google Scholar] [CrossRef] [PubMed]
- Gilman, M.S.A.; Liu, C.; Fung, A.; Behera, I.; Jordan, P.; Rigaux, P.; Ysebaert, N.; Tcherniuk, S.; Sourimant, J.; Eleouet, J.F.; et al. Structure of the Respiratory Syncytial Virus Polymerase Complex. Cell 2019, 179, 193–204. [Google Scholar] [CrossRef]
- Cao, D.; Gao, Y.; Roesler, C.; Rice, S.; D’Cunha, P.; Zhuang, L.; Slack, J.; Domke, M.; Antonova, A.; Romanelli, S.; et al. Cryo-EM structure of the respiratory syncytial virus RNA polymerase. Nat. Commun. 2020, 11, 368. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Qian, X.; Lattmann, S.; El Sahili, A.; Yeo, T.H.; Jia, H.; Cressey, T.; Ludeke, B.; Noton, S.; Kalocsay, M.; et al. Structure of the human metapneumovirus polymerase phosphoprotein complex. Nature 2020, 577, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Gouet, P.; Robert, X.; Courcelle, E. ESPript/ENDscript: Extracting and rendering sequence and 3D information from atomic structures of proteins. Nucleic Acids Res. 2003, 31, 3320–3323. [Google Scholar] [CrossRef]
- Sourimant, J.; Rameix-Welti, M.A.; Gaillard, A.L.; Chevret, D.; Galloux, M.; Gault, E.; Eleouet, J.F. Fine mapping and characterization of the L-polymerase-binding domain of the respiratory syncytial virus phosphoprotein. J. Virol. 2015, 89, 4421–4433. [Google Scholar] [CrossRef]
- Khattar, S.K.; Yunus, A.S.; Samal, S.K. Mapping the domains on the phosphoprotein of bovine respiratory syncytial virus required for N-P and P-L interactions using a minigenome system. J. Gen. Virol. 2001, 82, 775–779. [Google Scholar] [CrossRef] [PubMed]
- Galloux, M.; Tarus, B.; Blazevic, I.; Fix, J.; Duquerroy, S.; Eleouet, J.F. Characterization of a viral phosphoprotein binding site on the surface of the respiratory syncytial nucleoprotein. J. Virol. 2012, 86, 8375–8387. [Google Scholar] [CrossRef]
- Tran, T.L.; Castagne, N.; Bhella, D.; Varela, P.F.; Bernard, J.; Chilmonczyk, S.; Berkenkamp, S.; Benhamo, V.; Grznarova, K.; Grosclaude, J.; et al. The nine C-terminal amino acids of the respiratory syncytial virus protein P are necessary and sufficient for binding to ribonucleoprotein complexes in which six ribonucleotides are contacted per N protein protomer. J. Gen. Virol. 2007, 88, 196–206. [Google Scholar] [CrossRef]
- Bakker, S.E.; Duquerroy, S.; Galloux, M.; Loney, C.; Conner, E.; Eleouet, J.F.; Rey, F.A.; Bhella, D. The respiratory syncytial virus nucleoprotein-RNA complex forms a left-handed helical nucleocapsid. J. Gen. Virol. 2013, 94, 1734–1738. [Google Scholar] [CrossRef] [PubMed]
- Galloux, M.; Risso-Ballester, J.; Richard, C.A.; Fix, J.; Rameix-Welti, M.A.; Eleouet, J.F. Minimal Elements Required for the Formation of Respiratory Syncytial Virus Cytoplasmic Inclusion Bodies In Vivo and In Vitro. MBio 2020, 11. [Google Scholar] [CrossRef]
- Garcia-Barreno, B.; Delgado, T.; Melero, J.A. Identification of protein regions involved in the interaction of human respiratory syncytial virus phosphoprotein and nucleoprotein: Significance for nucleocapsid assembly and formation of cytoplasmic inclusions. J. Virol. 1996, 70, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Rincheval, V.; Lelek, M.; Gault, E.; Bouillier, C.; Sitterlin, D.; Blouquit-Laye, S.; Galloux, M.; Zimmer, C.; Eleouet, J.F.; Rameix-Welti, M.A. Functional organization of cytoplasmic inclusion bodies in cells infected by respiratory syncytial virus. Nat. Commun. 2017, 8, 563. [Google Scholar] [CrossRef]
- Cifuentes-Munoz, N.; Ellis Dutch, R. To assemble or not to assemble: The changing rules of pneumovirus transmission. Virus Res. 2019, 265, 68–73. [Google Scholar] [CrossRef]
- Derdowski, A.; Peters, T.R.; Glover, N.; Qian, R.; Utley, T.J.; Burnett, A.; Williams, J.V.; Spearman, P.; Crowe, J.E. Human metapneumovirus nucleoprotein and phosphoprotein interact and provide the minimal requirements for inclusion body formation. J. Gen. Virol. 2008, 89, 2698–2708. [Google Scholar] [CrossRef]
- Hardy, R.W.; Wertz, G.W. The product of the respiratory syncytial virus M2 gene ORF1 enhances readthrough of intergenic junctions during viral transcription. J. Virol. 1998, 72, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Collins, P.L.; Hill, M.G.; Cristina, J.; Grosfeld, H. Transcription elongation factor of respiratory syncytial virus, a nonsegmented negative-strand RNA virus. Proc. Natl. Acad. Sci. USA 1996, 93, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Cowton, V.M.; McGivern, D.R.; Fearns, R. Unravelling the complexities of respiratory syncytial virus RNA synthesis. J. Gen. Virol. 2006, 87, 1805–1821. [Google Scholar] [CrossRef]
- Buchholz, U.J.; Biacchesi, S.; Pham, Q.N.; Tran, K.C.; Yang, L.; Luongo, C.L.; Skiadopoulos, M.H.; Murphy, B.R.; Collins, P.L. Deletion of M2 gene open reading frames 1 and 2 of human metapneumovirus: Effects on RNA synthesis, attenuation, and immunogenicity. J. Virol. 2005, 79, 6588–6597. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.; Garcia-Barreno, B.; Vivo, A.; Melero, J.A. Cytoplasmic inclusions of respiratory syncytial virus-infected cells: Formation of inclusion bodies in transfected cells that coexpress the nucleoprotein, the phosphoprotein, and the 22K protein. Virology 1993, 195, 243–247. [Google Scholar] [CrossRef]
- Blondot, M.L.; Dubosclard, V.; Fix, J.; Lassoued, S.; Aumont-Nicaise, M.; Bontems, F.; Eleouet, J.F.; Sizun, C. Structure and Functional Analysis of the RNA and Viral Phosphoprotein Binding Domain of Respiratory Syncytial Virus M2-1 Protein. PLoS Pathog. 2012, 8, e1002734. [Google Scholar] [CrossRef]
- Richard, C.A.; Rincheval, V.; Lassoued, S.; Fix, J.; Cardone, C.; Esneau, C.; Nekhai, S.; Galloux, M.; Rameix-Welti, M.A.; Sizun, C.; et al. RSV hijacks cellular protein phosphatase 1 to regulate M2-1 phosphorylation and viral transcription. PLoS Pathog. 2018, 14, e1006920. [Google Scholar] [CrossRef]
- Navarro, J.; Lopez-Otin, C.; Villanueva, N. Location of phosphorylated residues in human respiratory syncytial virus phosphoprotein. J. Gen. Virol. 1991, 72 Pt 6, 1455–1459. [Google Scholar] [CrossRef]
- Asenjo, A.; Rodriguez, L.; Villanueva, N. Determination of phosphorylated residues from human respiratory syncytial virus P protein that are dynamically dephosphorylated by cellular phosphatases: A possible role for serine 54. J. Gen. Virol. 2005, 86, 1109–1120. [Google Scholar] [CrossRef]
- Lu, B.; Ma, C.H.; Brazas, R.; Jin, H. The major phosphorylation sites of the respiratory syncytial virus phosphoprotein are dispensable for virus replication in vitro. J. Virol. 2002, 76, 10776–10784. [Google Scholar] [CrossRef] [PubMed]
- Mazumder, B.; Barik, S. Requirement of casein kinase II-mediated phosphorylation for the transcriptional activity of human respiratory syncytial viral phosphoprotein P: Transdominant negative phenotype of phosphorylation-defective P mutants. Virology 1994, 205, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, N.; Hardy, R.; Asenjo, A.; Yu, Q.; Wertz, G. The bulk of the phosphorylation of human respiratory syncytial virus phosphoprotein is not essential but modulates viral RNA transcription and replication. Microbiology 2000, 81, 129–133. [Google Scholar] [CrossRef]
- Asenjo, A.; Mendieta, J.; Gomez-Puertas, P.; Villanueva, N. Residues in human respiratory syncytial virus P protein that are essential for its activity on RNA viral synthesis. Virus Res. 2008, 132, 160–173. [Google Scholar] [CrossRef]
- Asenjo, A.; Calvo, E.; Villanueva, N. Phosphorylation of human respiratory syncytial virus P protein at threonine 108 controls its interaction with the M2-1 protein in the viral RNA polymerase complex. J. Gen. Virol. 2006, 87, 3637–3642. [Google Scholar] [CrossRef] [PubMed]
- Renner, M.; Bertinelli, M.; Leyrat, C.; Paesen, G.C.; de Oliveira, L.F.S.; Huiskonen, J.T.; Grimes, J.M. Nucleocapsid assembly in pneumoviruses is regulated by conformational switching of the N protein. Elife 2016, 5, e12627. [Google Scholar] [CrossRef] [PubMed]
- Tawar, R.G.; Duquerroy, S.; Vonrhein, C.; Varela, P.F.; Damier-Piolle, L.; Castagne, N.; MacLellan, K.; Bedouelle, H.; Bricogne, G.; Bhella, D.; et al. Crystal structure of a nucleocapsid-like nucleoprotein-RNA complex of respiratory syncytial virus. Science 2009, 326, 1279–1283. [Google Scholar] [CrossRef] [PubMed]
- Simabuco, F.M.; Asara, J.M.; Guerrero, M.C.; Libermann, T.A.; Zerbini, L.F.; Ventura, A.M. Structural analysis of human respiratory syncytial virus P protein: Identification of intrinsically disordered domains. Braz. J. Microbiol. 2011, 42, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Esperante, S.A.; Paris, G.; de Prat-Gay, G. Modular unfolding and dissociation of the human respiratory syncytial virus phosphoprotein p and its interaction with the M2-1 antiterminator: A singular tetramer-tetramer interface arrangement. Biochemistry 2012, 51, 8100–8110. [Google Scholar] [CrossRef]
- Noval, M.G.; Esperante, S.A.; Molina, I.G.; Chemes, L.B.; de Prat-Gay, G. Intrinsic disorder to order transitions in the scaffold phosphoprotein P from the respiratory syncytial virus RNA-polymerase complex. Biochemistry 2016, 55, 1441–1454. [Google Scholar] [CrossRef] [PubMed]
- Llorente, M.T.; Garcia-Barreno, B.; Calero, M.; Camafeita, E.; Lopez, J.A.; Longhi, S.; Ferron, F.; Varela, P.F.; Melero, J.A. Structural analysis of the human respiratory syncytial virus phosphoprotein: Characterization of an alpha-helical domain involved in oligomerization. J. Gen. Virol. 2006, 87, 159–169. [Google Scholar] [CrossRef]
- Renner, M.; Paesen, G.C.; Grison, C.M.; Granier, S.; Grimes, J.M.; Leyrat, C. Structural dissection of human metapneumovirus phosphoprotein using small angle x-ray scattering. Sci. Rep. 2017, 7, 14865. [Google Scholar] [CrossRef]
- Tompa, P. Intrinsically disordered proteins: A 10-year recap. Trends Biochem. Sci. 2012, 37, 509–516. [Google Scholar] [CrossRef]
- Pereira, N.; Cardone, C.; Lassoued, S.; Galloux, M.; Fix, J.; Assrir, N.; Lescop, E.; Bontems, F.; Eleouet, J.F.; Sizun, C. New Insights into Structural Disorder in Human Respiratory Syncytial Virus Phosphoprotein and Implications for Binding of Protein Partners. J. Biol. Chem. 2017, 292, 2120–2131. [Google Scholar] [CrossRef]
- Shen, Y.; Delaglio, F.; Cornilescu, G.; Bax, A. TALOS+: A hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 2009, 44, 213–223. [Google Scholar] [CrossRef]
- Camilloni, C.; De Simone, A.; Vranken, W.F.; Vendruscolo, M. Determination of secondary structure populations in disordered states of proteins using nuclear magnetic resonance chemical shifts. Biochemistry 2012, 51, 2224–2231. [Google Scholar] [CrossRef] [PubMed]
- Guntert, P. Automated NMR Structure Calculation With CYANA. In Protein NMR Techniques; Downing, A.K., Ed.; Humana Press: Totowa, NJ, USA, 2004; pp. 353–378. [Google Scholar] [CrossRef]
- Kay, L.E.; Torchia, D.A.; Bax, A. Backbone dynamics of proteins as studied by 15N inverse detected heteronuclear NMR spectroscopy: Application to staphylococcal nuclease. Biochemistry 1989, 28, 8972–8979. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.G., III. NMR probes of molecular dynamics: Overview and comparison with other techniques. Annu. Rev. Biophys. Biomol. Struct. 2001, 30, 129–155. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.W.F.; Majumdar, A.; Zuiderweg, E.R.P. Protein NMR relaxation: Theory, applications and outlook. Prog. Nucl. Magn. Reson. Spectrosc. 1998, 33, 207–272. [Google Scholar] [CrossRef]
- Clore, G.M.; Iwahara, J. Theory, practice, and applications of paramagnetic relaxation enhancement for the characterization of transient low-population states of biological macromolecules and their complexes. Chem. Rev. 2009, 109, 4108–4139. [Google Scholar] [CrossRef]
- Ouizougun-Oubari, M.; Pereira, N.; Tarus, B.; Galloux, M.; Lassoued, S.; Fix, J.; Tortorici, M.A.; Hoos, S.; Baron, B.; England, P.; et al. A Druggable Pocket at the Nucleocapsid/Phosphoprotein Interaction Site of Human Respiratory Syncytial Virus. J. Virol. 2015, 89, 11129–11143. [Google Scholar] [CrossRef]
- Sharma, R.; Raduly, Z.; Miskei, M.; Fuxreiter, M. Fuzzy complexes: Specific binding without complete folding. FEBS Lett. 2015, 589, 2533–2542. [Google Scholar] [CrossRef]
- Tompa, P.; Fuxreiter, M. Fuzzy complexes: Polymorphism and structural disorder in protein-protein interactions. Trends Biochem. Sci. 2008, 33, 2–8. [Google Scholar] [CrossRef]
- Galloux, M.; Gabiane, G.; Sourimant, J.; Richard, C.A.; England, P.; Moudjou, M.; Aumont-Nicaise, M.; Fix, J.; Rameix-Welti, M.A.; Eleouet, J.F. Identification and characterization of the binding site of the respiratory syncytial virus phosphoprotein to RNA-free nucleoprotein. J. Virol. 2015, 89, 3484–3496. [Google Scholar] [CrossRef]
- Esneau, C.; Raynal, B.; Roblin, P.; Brule, S.; Richard, C.A.; Fix, J.; Eleouet, J.F.; Galloux, M. Biochemical characterization of the respiratory syncytial virus N0-P complex in solution. J. Biol. Chem. 2019, 294, 3647–3660. [Google Scholar] [CrossRef]
- Galloux, M.; Gsponer, N.; Gaillard, V.; Fenner, B.; Larcher, T.; Vilotte, M.; Riviere, J.; Richard, C.A.; Eleouet, J.F.; Le Goffic, R.; et al. Targeting the Respiratory Syncytial Virus N0-P complex with constrained alpha-helical peptides in cells and in mice. Antimicrob. Agents Chemother. 2020, 64. [Google Scholar] [CrossRef] [PubMed]
- Hraber, P.; O’Maille, P.E.; Silberfarb, A.; Davis-Anderson, K.; Generous, N.; McMahon, B.H.; Fair, J.M. Resources to Discover and Use Short Linear Motifs in Viral Proteins. Trends Biotechnol. 2020, 38, 113–127. [Google Scholar] [CrossRef]
- Van Roey, K.; Uyar, B.; Weatheritt, R.J.; Dinkel, H.; Seiler, M.; Budd, A.; Gibson, T.J.; Davey, N.E. Short linear motifs: Ubiquitous and functionally diverse protein interaction modules directing cell regulation. Chem. Rev. 2014, 114, 6733–6778. [Google Scholar] [CrossRef]
- Cuesta, I.; Geng, X.; Asenjo, A.; Villanueva, N. Structural phosphoprotein M2-1 of the human respiratory syncytial virus is an RNA binding protein. J. Virol. 2000, 74, 9858–9867. [Google Scholar] [CrossRef][Green Version]
- Cartee, T.L.; Wertz, G.W. Respiratory syncytial virus M2-1 protein requires phosphorylation for efficient function and binds viral RNA during infection. J. Virol. 2001, 75, 12188–12197. [Google Scholar] [CrossRef]
- Tanner, S.J.; Ariza, A.; Richard, C.A.; Kyle, H.F.; Dods, R.L.; Blondot, M.L.; Wu, W.; Trincao, J.; Trinh, C.H.; Hiscox, J.A.; et al. Crystal structure of the essential transcription antiterminator M2-1 protein of human respiratory syncytial virus and implications of its phosphorylation. Proc. Natl. Acad. Sci. USA 2014, 111, 1580–1585. [Google Scholar] [CrossRef]
- Leyrat, C.; Renner, M.; Harlos, K.; Huiskonen, J.T.; Grimes, J.M. Drastic changes in conformational dynamics of the antiterminator M2-1 regulate transcription efficiency in Pneumovirinae. Elife 2014, 3, e02674. [Google Scholar] [CrossRef]
- Mason, S.W.; Aberg, E.; Lawetz, C.; DeLong, R.; Whitehead, P.; Liuzzi, M. Interaction between human respiratory syncytial virus (RSV) M2-1 and P proteins is required for reconstitution of M2-1-dependent RSV minigenome activity. J. Virol. 2003, 77, 10670–10676. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tran, T.L.; Castagne, N.; Dubosclard, V.; Noinville, S.; Koch, E.; Moudjou, M.; Henry, C.; Bernard, J.; Yeo, R.P.; Eleouet, J.F. The respiratory syncytial virus M2-1 protein forms tetramers and interacts with RNA and P in a competitive manner. J. Virol. 2009, 83, 6363–6374. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, M.; Yegambaram, K.; Todd, E.; Richard, C.A.; Dods, R.L.; Pangratiou, G.M.; Trinh, C.H.; Moul, S.L.; Murphy, J.C.; Mankouri, J.; et al. The Structure of the Human Respiratory Syncytial Virus M2-1 Protein Bound to the Interaction Domain of the Phosphoprotein P Defines the Orientation of the Complex. MBio 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Marti-Renom, M.A.; Stuart, A.C.; Fiser, A.; Sanchez, R.; Melo, F.; Sali, A. Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 291–325. [Google Scholar] [CrossRef]
- Cai, H.; Zhang, Y.; Lu, M.; Liang, X.; Jennings, R.; Niewiesk, S.; Li, J. Phosphorylation of Human Metapneumovirus M2-1 Protein Upregulates Viral Replication and Pathogenesis. J. Virol. 2016, 90, 7323–7338. [Google Scholar] [CrossRef] [PubMed]
- Bollen, M.; Peti, W.; Ragusa, M.J.; Beullens, M. The extended PP1 toolkit: Designed to create specificity. Trends Biochem. Sci. 2010, 35, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, A.P.; Simabuco, F.M.; Tamura, R.E.; Guerrero, M.C.; Ribeiro, P.G.; Libermann, T.A.; Zerbini, L.F.; Ventura, A.M. Human respiratory syncytial virus N, P and M protein interactions in HEK-293T cells. Virus Res. 2013, 177, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Munday, D.C.; Wu, W.; Smith, N.; Fix, J.; Noton, S.L.; Galloux, M.; Touzelet, O.; Armstrong, S.D.; Dawson, J.M.; Aljabr, W.; et al. Interactome analysis of the human respiratory syncytial virus RNA polymerase complex identifies protein chaperones as important cofactors that promote L-protein stability and RNA synthesis. J. Virol. 2015, 89, 917–930. [Google Scholar] [CrossRef] [PubMed]
- Milles, S.; Jensen, M.R.; Lazert, C.; Guseva, S.; Ivashchenko, S.; Communie, G.; Maurin, D.; Gerlier, D.; Ruigrok, R.W.H.; Blackledge, M. An ultraweak interaction in the intrinsically disordered replication machinery is essential for measles virus function. Sci. Adv. 2018, 4, eaat7778. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cardone, C.; Caseau, C.-M.; Pereira, N.; Sizun, C. Pneumoviral Phosphoprotein, a Multidomain Adaptor-Like Protein of Apparent Low Structural Complexity and High Conformational Versatility. Int. J. Mol. Sci. 2021, 22, 1537. https://doi.org/10.3390/ijms22041537
Cardone C, Caseau C-M, Pereira N, Sizun C. Pneumoviral Phosphoprotein, a Multidomain Adaptor-Like Protein of Apparent Low Structural Complexity and High Conformational Versatility. International Journal of Molecular Sciences. 2021; 22(4):1537. https://doi.org/10.3390/ijms22041537
Chicago/Turabian StyleCardone, Christophe, Claire-Marie Caseau, Nelson Pereira, and Christina Sizun. 2021. "Pneumoviral Phosphoprotein, a Multidomain Adaptor-Like Protein of Apparent Low Structural Complexity and High Conformational Versatility" International Journal of Molecular Sciences 22, no. 4: 1537. https://doi.org/10.3390/ijms22041537
APA StyleCardone, C., Caseau, C.-M., Pereira, N., & Sizun, C. (2021). Pneumoviral Phosphoprotein, a Multidomain Adaptor-Like Protein of Apparent Low Structural Complexity and High Conformational Versatility. International Journal of Molecular Sciences, 22(4), 1537. https://doi.org/10.3390/ijms22041537