
Exploring the Genetic Conception of Obesity via the Dual Role of FoxO




 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. FoxO1: An Effective Tactic to Combat Obesity
2.1. FoxO1 Role in Normal Physiology and Pathogenesis of Metabolic Disorders
2.2. FoxO1: Activity and Expression Regulation
3. General Pathophysiology of Obesity
4. FoxO1 in General Metabolic Functions and Associated Metabolic Syndromes
4.1. FoxO1 in Liver
4.2. FoxO1 in Skeletal Muscles
4.3. FoxO1 in Obesity
4.4. FoxO1 in Non-Alcoholic Fatty-Liver Disease (NAFLD)
4.5. FoxO1 and Type 2 Diabetes Mellitus (T2DM)
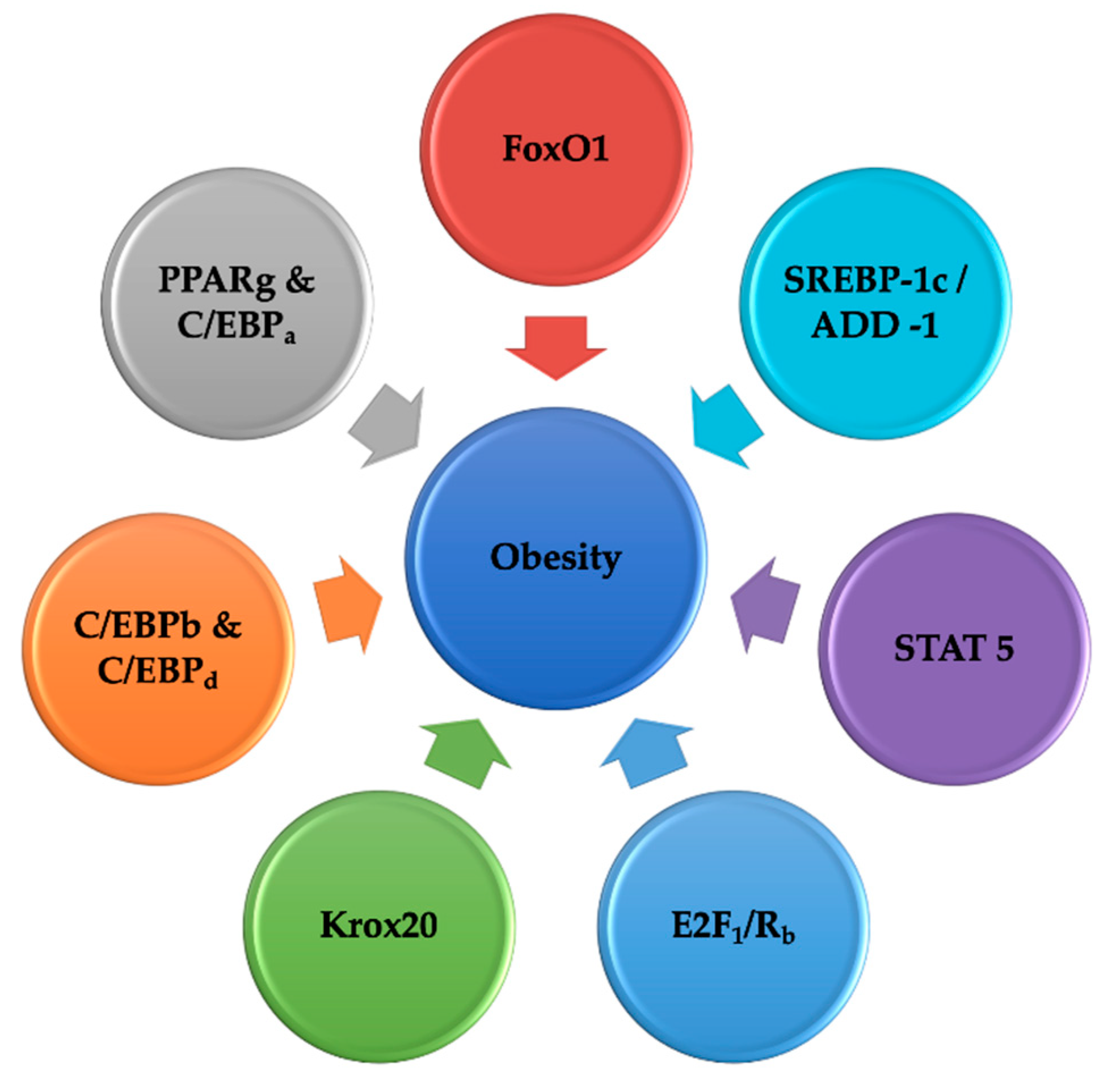
5. Interpretation of the Network of Transcription Elements Controlling Adipogenesis
5.1. Fork Head-Box O Transcription Elements
5.2. PPARg and C/EBPa: Master Controller of Fat Synthesis
5.3. C/EBPb and C/EBPd
5.4. Krox20
5.5. KLF5 and SREBP1C
5.6. E2F Family
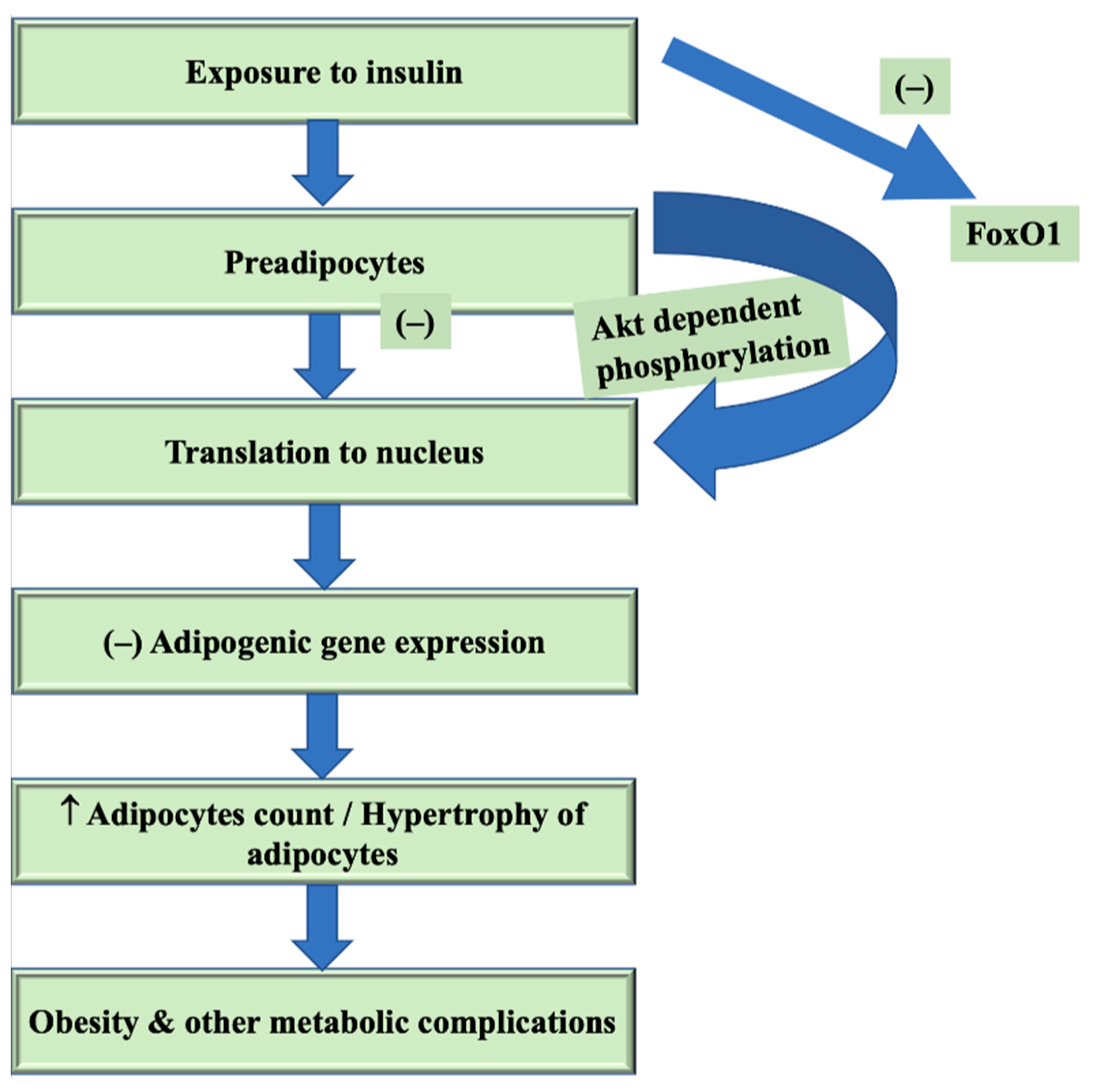
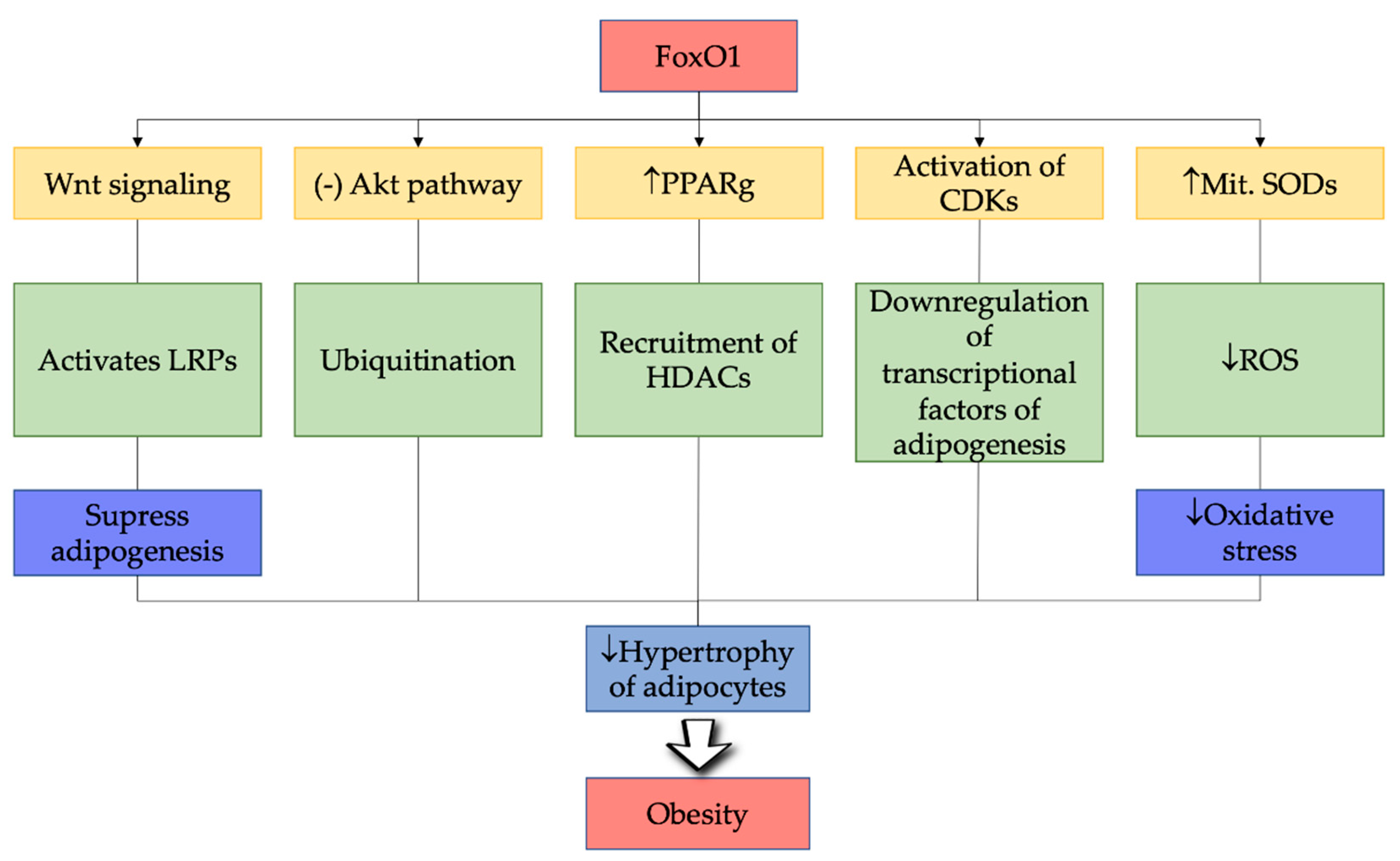
6. FoxO1 in Pathogenesis of Obesity
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Borkowski, K.; Newman, J.W.; Aghaeepour, N.; Mayo, J.A.; Blazenović, I.; Fiehn, O.; Stevenson, D.K.; Shaw, G.M.; Carmichael, S.L. Mid-gestation serum lipidomic profile associations with spontaneous preterm birth are influenced by body mass index. PLoS ONE 2020, 15, e0239115. [Google Scholar] [CrossRef] [PubMed]
- Behl, T.; Kaur, I.; Sehgal, A.; Kumar, A.; Uddin, M.S.; Bungau, S. The Interplay of ABC Transporters in Aβ Translocation and Cholesterol Metabolism: Implicating Their Roles in Alzheimer’s Disease. Mol. Neurobiol. 2020, 58, 1–19. [Google Scholar]
- Kivimäki, M.; Luukkonen, R.; Batty, G.D.; Ferrie, J.E.; Pentti, J.; Nyberg, S.T.; Shipley, M.J.; Alfredsson, L.; Fransson, E.I.; Goldberg, M. Body mass index and risk of dementia: Analysis of individual-level data from 1.3 million individuals. Alzheimer Dement. 2018, 14, 601–609. [Google Scholar] [CrossRef]
- Janssen, R.; Walk, J. Vitamin K epoxide reductase complex subunit 1 (VKORC1) gene polymorphism as determinant of differences in Covid-19-related disease severity. Med. Hypotheses 2020, 144, 110218. [Google Scholar] [CrossRef]
- Roh, E.; Hwang, S.Y.; Kim, J.A.; Lee, Y.-B.; Hong, S.; Kim, N.H.; Seo, J.A.; Kim, S.G.; Kim, N.H.; Choi, K.M. Body weight variability increases dementia risk among older adults: A nationwide population-based cohort study. Front. Endocrinol. 2020, 11, 291. [Google Scholar] [CrossRef] [PubMed]
- Karki, R.; Madan, S.; Gadiya, Y.; Domingo-Fernández, D.; Hofmann-Apitius, M. Data-Driven Modeling of Knowledge Assemblies in Understanding Comorbidity Between Type 2 Diabetes Mellitus and Alzheimer’s Disease. J. Alzheimer Dis. 2020, 78, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Shinjyo, N.; Parkinson, J.; Bell, J.; Katsuno, T.; Bligh, A. Berberine for prevention of dementia associated with diabetes and its comorbidities: A systematic review. J. Integr. Med. 2020, 18, 125–151. [Google Scholar] [CrossRef] [PubMed]
- Zaha, D.; Vesa, C.; Uivarosan, D.; Bratu, O.; Fratila, O.; Tit, D.; Pantis, C.; Diaconu, C.; Bungau, S. Influence of inflammation and adipocyte biochemical markers on the components of metabolic syndrome. Exp. Ther. Med. 2020, 20, 121–128. [Google Scholar] [CrossRef]
- Choe, S.S.; Huh, J.Y.; Hwang, I.J.; Kim, J.I.; Kim, J.B. Adipose tissue remodeling: Its role in energy metabolism and metabolic disorders. Front. Endocrinol. 2016, 7, 30. [Google Scholar] [CrossRef]
- Tong, X. Role and Regulation of microRNAs in Adipose Tissue during Aging. Ph.D. Thesis, Université Paul Sabatier-Toulouse III, Toulouse, France, 2018. [Google Scholar]
- Bär, L.; Stangl, G. Studies on the regulation of the phosphaturic hormone fibroblast growth factor 23 (FGF23). FEBS Lett. 2019, 593, 1879–1900. [Google Scholar] [CrossRef]
- Yan, K.; Da, T.-T.; Bian, Z.-H.; He, Y.; Liu, M.-C.; Liu, Q.-Z.; Long, J.; Li, L.; Gao, C.-Y.; Yang, S.-H. Multi-omics analysis identifies FoxO1 as a regulator of macrophage function through metabolic reprogramming. Cell Death Dis. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Moll, L.; Schubert, M. The role of insulin and insulin-like growth factor-1/FoxO-mediated transcription for the pathogenesis of obesity-associated dementia. Curr. Gerontol. Geriatr. Res. 2012, 2012, 38904. [Google Scholar] [CrossRef]
- Sergi, C.; Shen, F.; Liu, S.-M. Insulin/IGF-1R, SIRT1, and FOXOs pathways—an intriguing interaction platform for bone and osteosarcoma. Front. Endocrinol. 2019, 10, 93. [Google Scholar] [CrossRef] [PubMed]
- Castells-Nobau, A.; Eidhof, I.; Fenckova, M.; Brenman-Suttner, D.B.; Scheffer-de Gooyert, J.M.; Christine, S.; Schellevis, R.L.; Van der Laan, K.; Quentin, C.; Van Ninhuijs, L. Conserved regulation of neurodevelopmental processes and behavior by FoxP in Drosophila. PLoS ONE 2019, 14, e0211652. [Google Scholar] [CrossRef] [PubMed]
- Spierer, A.; Mossman, J.A.; Smith, S.P.; Crawford, L.; Ramachandran, S.; Rand, D.M. Natural variation in the regulation of neurodevelopmental genes modifies flight performance in Drosophila. bioRxiv 2020. Available online: https://www.biorxiv.org/content/10.1101/2020.05.27.118604v1 (accessed on 26 January 2021). [CrossRef]
- Laissue, P. The forkhead-box family of transcription factors: Key molecular players in colorectal cancer pathogenesis. Mol. Cancer 2019, 18, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Schmitt-Ney, M. The FOXO’s Advantages of Being a Family: Considerations on Function and Evolution. Cells 2020, 9, 787. [Google Scholar] [CrossRef]
- Jiramongkol, Y.; Lam, E.W.-F. FOXO transcription factor family in cancer and metastasis. Cancer Metastasis Rev. 2020, 39, 1–29. [Google Scholar] [CrossRef]
- Farhan, M.; Wang, H.; Gaur, U.; Little, P.J.; Xu, J.; Zheng, W. FOXO signaling pathways as therapeutic targets in cancer. Int. J. Biol. Sci. 2017, 13, 815. [Google Scholar] [CrossRef]
- Minard, A.Y.; Wong, M.K.L.; Chaudhuri, R.; Tan, S.-X.; Humphrey, S.J.; Parker, B.L.; Yang, J.Y.; Laybutt, D.R.; Cooney, G.J.; Coster, A.C.F. Hyperactivation of the insulin signaling pathway improves intracellular proteostasis by coordinately up-regulating the proteostatic machinery in adipocytes. J. Biol. Chem. 2016, 291, 25629–25640. [Google Scholar] [CrossRef] [PubMed]
- Link, W.; Fernandez-Marcos, P.J. FOXO transcription factors at the interface of metabolism and cancer. Int. J. Cancer 2017, 141, 2379–2391. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.-H.; Kar, S.; Dore, S.; Quirion, R. Insulin-like growth factor-1 (IGF-1): A neuroprotective trophic factor acting via the Akt kinase pathway. In Advances in Research on Neurodegeneration; Springer: Berlin/Heidelberg, Germany, 2000; pp. 261–272. [Google Scholar]
- Lewitt, M.S.; Boyd, G.W. The Role of Insulin-Like Growth Factors and Insulin-Like Growth Factor–Binding Proteins in the Nervous System. Biochem. Insights 2019, 12, 1178626419842176. [Google Scholar] [CrossRef]
- Zhang, C.; Lim, J.; Jeon, H.H.; Xu, F.; Tian, C.; Miao, F.; Hameedaldeen, A.; Graves, D.T. FOXO1 deletion in keratinocytes improves diabetic wound healing through MMP9 regulation. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef]
- Abuzenadah, A.; Alsaedi, S.; Karim, S.; Al-Qahtani, M. Role of Overexpressed Transcription Factor FOXO1 in. Transcr. Regul. Mol. Involv. Mech. Misregul. 2019, 19, 176. [Google Scholar]
- Weigelt, J.; Climent, I.; Dahlman-Wright, K.; Wikström, M. Solution structure of the DNA binding domain of the human forkhead transcription factor AFX (FOXO4). Biochemistry 2001, 40, 5861–5869. [Google Scholar] [CrossRef]
- Gajiwala, K.S.; Burley, S.K. Winged helix proteins. Curr. Opin. Struct. Biol. 2000, 10, 110–116. [Google Scholar] [CrossRef]
- Boura, E.; Rezabkova, L.; Brynda, J.; Obsilova, V.; Obsil, T. Structure of the human FOXO4-DBD–DNA complex at 1.9 Å resolution reveals new details of FOXO binding to the DNA. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 1351–1357. [Google Scholar] [CrossRef]
- Murtaza, G.; Khan, A.K.; Rashid, R.; Muneer, S.; Hasan, S.M.F.; Chen, J. FOXO transcriptional factors and long-term living. Oxid. Med. Cell. Longev. 2017, 2017, 3494289. [Google Scholar] [CrossRef]
- Tia, N.; Singh, A.K.; Pandey, P.; Azad, C.S.; Chaudhary, P.; Gambhir, I.S. Role of Forkhead Box O (FOXO) transcription factor in aging and diseases. Gene 2018, 648, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Melnik, B.C. The role of transcription factor FoxO1 in the pathogenesis of acne vulgaris and the mode of isotretinoin action. G. Ital. Di Dermatol. E Venereol. Organo Uff. Soc. Ital. Di Dermatol. E Sifilogr. 2010, 145, 559. [Google Scholar]
- Ma, X.; Su, P.; Yin, C.; Lin, X.; Wang, X.; Gao, Y.; Patil, S.; War, A.R.; Qadir, A.; Tian, Y. The roles of FoxO transcription factors in regulation of bone cells function. Int. J. Mol. Sci. 2020, 21, 692. [Google Scholar] [CrossRef]
- Kurutas, E.B. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: Current state. Nutr. J. 2015, 15, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.K.; Webb, A.E. Regulation of FOXO factors in mammalian cells. In Current Topics in Developmental Biology; Elsevier: Amsterdam, The Netherlands, 2018; Volume 127, pp. 165–192. ISBN 0070-2153. [Google Scholar]
- Deng, Y.; Scherer, P.E. Adipokines as novel biomarkers and regulators of the metabolic syndrome. Ann. N. Y. Acad. Sci. 2010, 1212, E1. [Google Scholar] [CrossRef]
- Balistreri, C.R.; Caruso, C.; Candore, G. The role of adipose tissue and adipokines in obesity-related inflammatory diseases. Mediat. Inflamm. 2010, 2010, 802078. [Google Scholar] [CrossRef]
- Kim, M.-S.; Pak, Y.K.; Jang, P.-G.; Namkoong, C.; Choi, Y.-S.; Won, J.-C.; Kim, K.-S.; Kim, S.-W.; Kim, H.-S.; Park, J.-Y. Role of hypothalamic Foxo1 in the regulation of food intake and energy homeostasis. Nat. Neurosci. 2006, 9, 901–906. [Google Scholar] [CrossRef]
- Kintscher, U.; Law, R.E. PPARγ-mediated insulin sensitization: The importance of fat versus muscle. Am. J. Physiol. Metab. 2005, 288, E287–E291. [Google Scholar] [CrossRef] [PubMed]
- Kousteni, S. FoxO1, the transcriptional chief of staff of energy metabolism. Bone 2012, 50, 437–443. [Google Scholar] [CrossRef]
- Zou, P.; Liu, L.; Zheng, L.; Liu, L.; Stoneman, R.E.; Cho, A.; Emery, A.; Gilbert, E.R.; Cheng, Z. Targeting FoxO1 with AS1842856 suppresses adipogenesis. Cell Cycle 2014, 13, 3759–3767. [Google Scholar] [CrossRef]
- Klotz, L.-O.; Sánchez-Ramos, C.; Prieto-Arroyo, I.; Urbánek, P.; Steinbrenner, H.; Monsalve, M. Redox regulation of FoxO transcription factors. Redox Biol. 2015, 6, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.; Wang, L.; Zhou, L.; Fu, Z. The function of FOXO1 in the late phases of the cell cycle is suppressed by PLK1-mediated phosphorylation. Cell Cycle 2014, 13, 807–819. [Google Scholar] [CrossRef] [PubMed]
- Fasano, C.; Disciglio, V.; Bertora, S.; Lepore Signorile, M.; Simone, C. FOXO3a from the nucleus to the mitochondria: A round trip in cellular stress response. Cells 2019, 8, 1110. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Tsai, L.T.Y.; Rosen, E.D. Nuclear mechanisms of insulin resistance. Trends Cell Biol. 2016, 26, 341–351. [Google Scholar] [CrossRef]
- Lei, H.; Quelle, F.W. FOXO transcription factors enforce cell cycle checkpoints and promote survival of hematopoietic cells after DNA damage. Mol. Cancer Res. 2009, 7, 1294–1303. [Google Scholar] [CrossRef] [PubMed]
- Hinman, R.M. The Role of Foxo Transcription Factors in B Cell Development and Activation. Ph.D. Thesis, University of Texas Southwestern Medical Center at Dallas, Dallas, TX, USA, 2010. [Google Scholar]
- Zhao, Y.; Wang, Y.; Zhu, W.G. Applications of post-translational modifications of FoxO family proteins in biological functions. J. Mol. Cell Biol. 2011, 3, 276–282. [Google Scholar] [CrossRef]
- Park, J.-M.; Jo, S.-H.; Kim, M.-Y.; Kim, T.-H.; Ahn, Y.-H. Role of transcription factor acetylation in the regulation of metabolic homeostasis. Protein Cell 2015, 6, 804–813. [Google Scholar] [CrossRef] [PubMed]
- Sadoul, K.; Wang, J.; Diagouraga, B.; Khochbin, S. The tale of protein lysine acetylation in the cytoplasm. J. Biomed. Biotechnol. 2010, 2011, 970382. [Google Scholar] [CrossRef]
- Hariharan, N.; Maejima, Y.; Nakae, J.; Paik, J.; DePinho, R.A.; Sadoshima, J. Deacetylation of FoxO by Sirt1 plays an essential role in mediating starvation-induced autophagy in cardiac myocytes. Circ. Res. 2010, 107, 1470–1482. [Google Scholar] [CrossRef]
- Wondisford, A.R.; Xiong, L.; Chang, E.; Meng, S.; Meyers, D.J.; Li, M.; Cole, P.A.; He, L. Control of foxo1 gene expression by co-activator p300. J. Biol. Chem. 2014, 289, 4326–4333. [Google Scholar] [CrossRef]
- Qiang, L.; Banks, A.S.; Accili, D. Uncoupling of acetylation from phosphorylation regulates FoxO1 function independent of its subcellular localization. J. Biol. Chem. 2010, 285, 27396–27401. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, Y.I.; Kitamura, T.; Kruse, J.-P.; Raum, J.C.; Stein, R.; Gu, W.; Accili, D. FoxO1 protects against pancreatic β cell failure through NeuroD and MafA induction. Cell Metab. 2005, 2, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Ioannilli, L.; Ciccarone, F.; Ciriolo, M.R. Adipose Tissue and FoxO1: Bridging Physiology and Mechanisms. Cells 2020, 9, 849. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.G.; Lee, H.; Gupta, N.; Ramachandran, S.; Kaushik, I.; Srivastava, S.; Kim, S.-H.; Srivastava, S.K. Role of Forkhead Box Class O proteins in cancer progression and metastasis. In Proceedings of the Seminars in Cancer Biology; Elsevier: Amsterdam, The Netherlands, 2018; Volume 50, pp. 142–151. [Google Scholar]
- Zhang, K.; Guo, X.; Yan, H.; Wu, Y.; Pan, Q.; Shen, J.Z.; Li, X.; Chen, Y.; Li, L.; Qi, Y.; et al. Phosphorylation of Forkhead Protein FoxO1 at S253 Regulates Glucose Homeostasis in Mice. Endocrinology 2019, 160, 1333–1347. [Google Scholar] [CrossRef]
- Yang, Y.; Li, W.; Liu, Y.; Sun, Y.; Li, Y.; Yao, Q.; Li, J.; Zhang, Q.; Gao, Y.; Gao, L. Alpha-lipoic acid improves high-fat diet-induced hepatic steatosis by modulating the transcription factors SREBP-1, FoxO1 and Nrf2 via the SIRT1/LKB1/AMPK pathway. J. Nutr. Biochem. 2014, 25, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.K.; Sinha, R.A.; Zhou, J.; Xie, S.Y.; You, S.-H.; Gauthier, K.; Yen, P.M. FoxO1 deacetylation regulates thyroid hormone-induced transcription of key hepatic gluconeogenic genes. J. Biol. Chem. 2013, 288, 30365–30372. [Google Scholar] [CrossRef] [PubMed]
- Nita, M.; Grzybowski, A. The role of the reactive oxygen species and oxidative stress in the pathomechanism of the age-related ocular diseases and other pathologies of the anterior and posterior eye segments in adults. Oxid. Med. Cell. Longev. 2016, 2016, 3164734. [Google Scholar] [CrossRef]
- Greer, E.L.; Brunet, A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene 2005, 24, 7410–7425. [Google Scholar] [CrossRef]
- Peng, S.; Li, W.; Hou, N.; Huang, N. A Review of FoxO1-Regulated Metabolic Diseases and Related Drug Discoveries. Cells 2020, 9, 184. [Google Scholar] [CrossRef]
- Fardini, Y.; Perez-Cervera, Y.; Camoin, L.; Pagesy, P.; Lefebvre, T.; Issad, T. Regulatory O-GlcNAcylation sites on FoxO1 are yet to be identified. Biochem. Biophys. Res. Commun. 2015, 462, 151–158. [Google Scholar] [CrossRef]
- Coelho, M.; Oliveira, T.; Fernandes, R. Biochemistry of adipose tissue: An endocrine organ. Arch. Med. Sci. AMS 2013, 9, 191. [Google Scholar] [CrossRef]
- Ouchi, N.; Parker, J.L.; Lugus, J.J.; Walsh, K. Adipokines in inflammation and metabolic disease. Nat. Rev. Immunol. 2011, 11, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Karpe, F.; Dickmann, J.R.; Frayn, K.N. Fatty acids, obesity, and insulin resistance: Time for a reevaluation. Diabetes 2011, 60, 2441–2449. [Google Scholar] [CrossRef]
- Björntorp, P.; Sjöström, L. Number and size of adipose tissue fat cells in relation to metabolism in human obesity. Metabolism 1971, 20, 703–713. [Google Scholar] [CrossRef]
- Walther, T.C.; Farese, R.V., Jr. Lipid droplets and cellular lipid metabolism. Annu. Rev. Biochem. 2012, 81, 687–714. [Google Scholar] [CrossRef]
- Benedict, M.; Zhang, X. Non-alcoholic fatty liver disease: An expanded review. World J. Hepatol. 2017, 9, 715. [Google Scholar] [CrossRef]
- Opazo-Ríos, L.; Mas, S.; Marín-Royo, G.; Mezzano, S.; Gómez-Guerrero, C.; Moreno, J.A.; Egido, J. Lipotoxicity and Diabetic Nephropathy: Novel Mechanistic Insights and Therapeutic Opportunities. Int. J. Mol. Sci. 2020, 21, 2632. [Google Scholar] [CrossRef] [PubMed]
- Sears, B.; Perry, M. The role of fatty acids in insulin resistance. Lipids Health Dis. 2015, 14, 1–9. [Google Scholar] [CrossRef]
- Hocking, S.; Samocha-Bonet, D.; Milner, K.-L.; Greenfield, J.R.; Chisholm, D.J. Adiposity and insulin resistance in humans: The role of the different tissue and cellular lipid depots. Endocr. Rev. 2013, 34, 463–500. [Google Scholar] [CrossRef]
- Behl, T.; Chadha, S.; Sachdeva, M.; Sehgal, A.; Kumar, A.; Dhruv; Venkatachalam, T.; Hafeez, A.; Aleya, L.; Arora, S.; et al. Understanding the possible role of endocannabinoid system in obesity. Prostaglandins Other Lipid Mediat. 2021, 152, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Majka, S.M.; Barak, Y.; Klemm, D.J. Concise review: Adipocyte origins: Weighing the possibilities. Stem Cells 2011, 29, 1034–1040. [Google Scholar] [CrossRef]
- Wang, W.; Seale, P. Control of brown and beige fat development. Nat. Rev. Mol. Cell Biol. 2016, 17, 691. [Google Scholar] [CrossRef]
- Boutens, L.; Stienstra, R. Adipose tissue macrophages: Going off track during obesity. Diabetologia 2016, 59, 879–894. [Google Scholar] [CrossRef]
- Mittal, B. Subcutaneous adipose tissue & visceral adipose tissue. Indian J. Med. Res. 2019, 149, 571. [Google Scholar]
- Hall, J.E.; do Carmo, J.M.; da Silva, A.A.; Wang, Z.; Hall, M.E. Obesity-induced hypertension: Interaction of neurohumoral and renal mechanisms. Circ. Res. 2015, 116, 991–1006. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, A.R.; Patil, S.P.; Laffan, A.M.; Polotsky, V.; Schneider, H.; Smith, P.L. Obesity and obstructive sleep apnea: Pathogenic mechanisms and therapeutic approaches. Proc. Am. Thorac. Soc. 2008, 5, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Bliddal, H.; Leeds, A.R.; Christensen, R. Osteoarthritis, obesity and weight loss: Evidence, hypotheses and horizons—A scoping review. Obes. Rev. 2014, 15, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Popa, A.R.; Fratila, O.; Rus, M.; Aron, R.; Vesa, C.; Pantis, C.; Diaconu, C.; Bratu, O.; Bungau, S.; Nemeth, S. Risk factors for adiposity in the urban population and influence on the prevalence of overweight and obesity. Exp. Ther. Med. 2020, 20, 129–133. [Google Scholar]
- Chang, J.T.; Katzka, D.A. Gastroesophageal reflux disease, Barrett esophagus, and esophageal adenocarcinoma. Arch. Intern. Med. 2004, 164, 1482–1488. [Google Scholar] [CrossRef]
- Rui, L. Energy metabolism in the liver. Compr. Physiol. 2011, 4, 177–197. [Google Scholar]
- He, L. Alterations of gut microbiota by overnutrition impact gluconeogenic gene expression and insulin signaling. Int. J. Mol. Sci. 2021, 22, 2121. [Google Scholar] [CrossRef]
- Oh, K.-J.; Han, H.-S.; Kim, M.-J.; Koo, S.-H. CREB and FoxO1: Two transcription factors for the regulation of hepatic gluconeogenesis. BMB Rep. 2013, 46, 567. [Google Scholar] [CrossRef]
- Röder, P.V.; Wu, B.; Liu, Y.; Han, W. Pancreatic regulation of glucose homeostasis. Exp. Mol. Med. 2016, 48, e219. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Pan, Q.; Yan, H.; Zhang, K.; Guo, X.; Xu, Z.; Yang, W.; Qi, Y.; Guo, C.A.; Hornsby, C.; et al. Novel mechanism of FOXO1 phosphorylation in glucagon signaling in control of glucose homeostasis. Diabetes 2018, 67, 2167–2182. [Google Scholar] [CrossRef]
- Ido-Kitamura, Y.; Sasaki, T.; Kobayashi, M.; Kim, H.-J.; Lee, Y.-S.; Kikuchi, O.; Yokota-Hashimoto, H.; Iizuka, K.; Accili, D.; Kitamura, T. Hepatic FoxO1 integrates glucose utilization and lipid synthesis through regulation of Chrebp O-glycosylation. PLoS ONE 2012, 7, e47231. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, C.M.; Ueki, K.; Kahn, C.R. Complementary roles of IRS-1 and IRS-2 in the hepatic regulation of metabolism. J. Clin. Investig. 2005, 115, 718–727. [Google Scholar] [CrossRef] [PubMed]
- Oberkofler, H.; Klein, K.; Felder, T.K.; Krempler, F.; Patsch, W. Role of peroxisome proliferator-activated receptor-γ coactivator-1α in the transcriptional regulation of the human uncoupling protein 2 gene in INS-1E cells. Endocrinology 2006, 147, 966–976. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Guo, S.; Copps, K.; Dong, X.; Kollipara, R.; Rodgers, J.T.; Depinho, R.A.; Puigserver, P.; White, M.F. Foxo1 integrates insulin signaling with mitochondrial function in the liver. Nat. Med. 2009, 15, 1307–1311. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.C.; Copps, K.D.; Guo, S.; Li, Y.; Kollipara, R.; DePinho, R.A.; White, M.F. Inactivation of hepatic Foxo1 by insulin signaling is required for adaptive nutrient homeostasis and endocrine growth regulation. Cell Metab. 2008, 8, 65–76. [Google Scholar] [CrossRef]
- Matsumoto, M.; Pocai, A.; Rossetti, L.; DePinho, R.A.; Accili, D. Impaired regulation of hepatic glucose production in mice lacking the forkhead transcription factor Foxo1 in liver. Cell Metab. 2007, 6, 208–216. [Google Scholar] [CrossRef]
- Goldstein, I.; Hager, G.L. Transcriptional and chromatin regulation during fasting–the genomic era. Trends Endocrinol. Metab. 2015, 26, 699–710. [Google Scholar] [CrossRef]
- Santoleri, D.; Titchenell, P.M. Resolving the paradox of hepatic insulin resistance. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 447–456. [Google Scholar] [CrossRef]
- Kumar, S.; Behl, T.; Sachdeva, M.; Sehgal, A.; Kumari, S.; Kumar, A.; Kaur, G.; Yadav, H.N.; Bungau, S. Implicating the effect of ketogenic diet as a preventive measure to obesity and diabetes mellitus. Life Sci. 2021, 264, 1–35. [Google Scholar] [CrossRef]
- Samuel, V.T.; Choi, C.S.; Phillips, T.G.; Romanelli, A.J.; Geisler, J.G.; Bhanot, S.; McKay, R.; Monia, B.; Shutter, J.R.; Lindberg, R.A. Targeting foxo1 in mice using antisense oligonucleotide improves hepatic and peripheral insulin action. Diabetes 2006, 55, 2042–2050. [Google Scholar] [CrossRef]
- Taniguchi, C.M.; Kondo, T.; Sajan, M.; Luo, J.; Bronson, R.; Asano, T.; Farese, R.; Cantley, L.C.; Kahn, C.R. Divergent regulation of hepatic glucose and lipid metabolism by phosphoinositide 3-kinase via Akt and PKCλ/ζ. Cell Metab. 2006, 3, 343–353. [Google Scholar] [CrossRef]
- Leavens, K.F.; Easton, R.M.; Shulman, G.I.; Previs, S.F.; Birnbaum, M.J. Akt2 is required for hepatic lipid accumulation in models of insulin resistance. Cell Metab. 2009, 10, 405–418. [Google Scholar] [CrossRef]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef] [PubMed]
- Yecies, J.L.; Zhang, H.H.; Menon, S.; Liu, S.; Yecies, D.; Lipovsky, A.I.; Gorgun, C.; Kwiatkowski, D.J.; Hotamisligil, G.S.; Lee, C.-H. Akt stimulates hepatic SREBP1c and lipogenesis through parallel mTORC1-dependent and independent pathways. Cell Metab. 2011, 14, 21–32. [Google Scholar] [CrossRef]
- Zhang, W.; Patil, S.; Chauhan, B.; Guo, S.; Powell, D.R.; Le, J.; Klotsas, A.; Matika, R.; Xiao, X.; Franks, R. FoxO1 regulates multiple metabolic pathways in the liver effects on gluconeogenic, glycolytic, and lipogenic gene expression. J. Biol. Chem. 2006, 281, 10105–10117. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; White, M.F. Targeting Forkhead box O1 from the concept to metabolic diseases: Lessons from mouse models. Antioxid. Redox Signal. 2011, 14, 649–661. [Google Scholar] [CrossRef]
- Munekata, K.; Sakamoto, K. Forkhead transcription factor Foxo1 is essential for adipocyte differentiation. Vitr. Cell. Dev. Biol. 2009, 45, 642. [Google Scholar] [CrossRef] [PubMed]
- Sparks, J.D.; Dong, H.H. FoxO1 and hepatic lipid metabolism. Curr. Opin. Lipidol. 2009, 20, 217. [Google Scholar] [CrossRef]
- Kamagate, A.; Dong, H.H. FoxO1 integrates insulin signaling to VLDL production. Cell Cycle 2008, 7, 3162–3170. [Google Scholar] [CrossRef] [PubMed]
- Pehar, M.; Harlan, B.A.; Killoy, K.M.; Vargas, M.R. Nicotinamide adenine dinucleotide metabolism and neurodegeneration. Antioxid. Redox Signal. 2018, 28, 1652–1668. [Google Scholar] [CrossRef] [PubMed]
- Rosenblat, M.; Coleman, R.; Reddy, S.T.; Aviram, M. Paraoxonase 2 attenuates macrophage triglyceride accumulation via inhibition of diacylglycerol acyltransferase 1. J. Lipid Res. 2009, 50, 870–879. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.H.; Kim, D.W.; Jo, E.J.; Kim, Y.K.; Jo, Y.S.; Park, J.H.; Yoo, S.K.; Park, M.K.; Kwak, T.H.; Kho, Y.L. Pharmacological stimulation of NADH oxidation ameliorates obesity and related phenotypes in mice. Diabetes 2009, 58, 965–974. [Google Scholar] [CrossRef]
- Liu, L. FoxO1 in the Regulation of Adipocyte Autophagy and Biology. Ph.D. Thesis, Virginia Tech, Blacksburg, VA, USA, 2016. [Google Scholar]
- Cook, J.R. Toward a Mechanistic Understanding of Hepatic Insulin Action and Resistance. Ph.D. Thesis, Columbia University, New York, NY, USA, 2014. [Google Scholar]
- Martinez, S.C.; Cras-Méneur, C.; Bernal-Mizrachi, E.; Permutt, M.A. Glucose regulates Foxo1 through insulin receptor signaling in the pancreatic islet β-cell. Diabetes 2006, 55, 1581–1591. [Google Scholar] [CrossRef] [PubMed]
- Hatting, M.; Tavares, C.D.J.; Sharabi, K.; Rines, A.K.; Puigserver, P. Insulin regulation of gluconeogenesis. Ann. N. Y. Acad. Sci. 2018, 1411, 21. [Google Scholar] [CrossRef]
- Ponugoti, B.; Dong, G.; Graves, D.T. Role of forkhead transcription factors in diabetes-induced oxidative stress. Exp. Diabetes Res. 2012, 2012. [Google Scholar] [CrossRef]
- Gross, D.N.; Van den Heuvel, A.P.J.; Birnbaum, M.J. The role of FoxO in the regulation of metabolism. Oncogene 2008, 27, 2320–2336. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Chen, W.-D.; Wang, Y.-D. DAF-16/FOXO transcription factor in aging and longevity. Front. Pharmacol. 2017, 8, 548. [Google Scholar] [CrossRef]
- Petersen, M.C.; Shulman, G.I. Mechanisms of insulin action and insulin resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef]
- Abdul-Ghani, M.A.; DeFronzo, R.A. Pathogenesis of insulin resistance in skeletal muscle. J. Biomed. Biotechnol. 2010, 2010, 476279. [Google Scholar] [CrossRef]
- Dong, X.C. FOXO transcription factors in non-alcoholic fatty liver disease. Liver Res. 2017, 1, 168–173. [Google Scholar] [CrossRef]
- Jeoung, N.H.; Harris, R.A. Role of pyruvate dehydrogenase kinase 4 in regulation of blood glucose levels. Korean Diabetes J. 2010, 34, 274–283. [Google Scholar] [CrossRef]
- Gray, L.R.; Tompkins, S.C.; Taylor, E.B. Regulation of pyruvate metabolism and human disease. Cell. Mol. life Sci. 2014, 71, 2577–2604. [Google Scholar] [CrossRef]
- Piao, L.; Sidhu, V.K.; Fang, Y.-H.; Ryan, J.J.; Parikh, K.S.; Hong, Z.; Toth, P.T.; Morrow, E.; Kutty, S.; Lopaschuk, G.D. FOXO1-mediated upregulation of pyruvate dehydrogenase kinase-4 (PDK4) decreases glucose oxidation and impairs right ventricular function in pulmonary hypertension: Therapeutic benefits of dichloroacetate. J. Mol. Med. 2013, 91, 333–346. [Google Scholar] [CrossRef] [PubMed]
- Kamei, Y.; Miura, S.; Suganami, T.; Akaike, F.; Kanai, S.; Sugita, S.; Katsumata, A.; Aburatani, H.; Unterman, T.G.; Ezaki, O. Regulation of SREBP1c gene expression in skeletal muscle: Role of retinoid X receptor/liver X receptor and forkhead-O1 transcription factor. Endocrinology 2008, 149, 2293–2305. [Google Scholar] [CrossRef] [PubMed]
- Larsson, L.; Degens, H.; Li, M.; Salviati, L.; Lee, Y.L.; Thompson, W.; Kirkland, J.L.; Sandri, M. Sarcopenia: Aging-related loss of muscle mass and function. Physiol. Rev. 2019, 99, 427–511. [Google Scholar] [CrossRef]
- O’Neill, B.T.; Bhardwaj, G.; Penniman, C.M.; Krumpoch, M.T.; Beltran, P.A.S.; Klaus, K.; Poro, K.; Li, M.; Pan, H.; Dreyfuss, J.M. FoxO transcription factors are critical regulators of diabetes-related muscle atrophy. Diabetes 2019, 68, 556–570. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M. Protein breakdown in muscle wasting: Role of autophagy-lysosome and ubiquitin-proteasome. Int. J. Biochem. Cell Biol. 2013, 45, 2121–2129. [Google Scholar] [CrossRef]
- Kyrou, I.; Randeva, H.S.; Tsigos, C.; Kaltsas, G.; Weickert, M.O. Clinical problems caused by obesity. In Endotext [Internet]; MDText. com, Inc.: South Dartmouth, MA, USA, 2018. [Google Scholar]
- Zhang, F.; Ma, D.; Zhao, W.; Wang, D.; Liu, T.; Liu, Y.; Yang, Y.; Liu, Y.; Mu, J.; Li, B. Obesity-induced overexpression of miR-802 impairs insulin transcription and secretion. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef]
- Nijhawan, P.; Behl, T.; Bungau, S.; Uddin, M.S.; Zengin, G.; Arora, S. Molecular insights into therapeutic promise of targeting of Wnt/β-catenin signaling pathway in obesity. Mol. Biol. Rep. 2020, 47, 8091–8100. [Google Scholar] [CrossRef]
- Lee, A.; Cardel, M.; Donahoo, W.T. Social and Environmental Factors Influencing Obesity. In Endotext [Internet]; MDText. com, Inc.: South Dartmouth, MA, USA, 2019. [Google Scholar]
- Rudnicki, M.; Abdifarkosh, G.; Nwadozi, E.; Ramos, S.V.; Makki, A.; Sepa-Kishi, D.M.; Ceddia, R.B.; Perry, C.G.R.; Roudier, E.; Haas, T.L. Endothelial-specific FoxO1 depletion prevents obesity-related disorders by increasing vascular metabolism and growth. eLife 2018, 7, e39780. [Google Scholar] [CrossRef]
- Nakae, J.; Cao, Y.; Oki, M.; Orba, Y.; Sawa, H.; Kiyonari, H.; Iskandar, K.; Suga, K.; Lombes, M.; Hayashi, Y. Forkhead transcription factor FoxO1 in adipose tissue regulates energy storage and expenditure. Diabetes 2008, 57, 563–576. [Google Scholar] [CrossRef]
- Cyr, N.E.; Steger, J.S.; Toorie, A.M.; Yang, J.Z.; Stuart, R.; Nillni, E.A. Central Sirt1 regulates body weight and energy expenditure along with the POMC-derived peptide α-MSH and the processing enzyme CPE production in diet-induced obese male rats. Endocrinology 2015, 156, 961–974. [Google Scholar] [CrossRef]
- Susanti, V.Y.; Sasaki, T.; Yokota-Hashimoto, H.; Matsui, S.; Lee, Y.; Kikuchi, O.; Shimpuku, M.; Kim, H.; Kobayashi, M.; Kitamura, T. Sirt1 rescues the obesity induced by insulin-resistant constitutively-nuclear FoxO1 in POMC neurons of male mice. Obesity 2014, 22, 2115–2119. [Google Scholar] [CrossRef] [PubMed]
- Marchisello, S.; Di Pino, A.; Scicali, R.; Urbano, F.; Piro, S.; Purrello, F.; Rabuazzo, A.M. Pathophysiological, molecular and therapeutic issues of nonalcoholic fatty liver disease: An overview. Int. J. Mol. Sci. 2019, 20, 1948. [Google Scholar] [CrossRef] [PubMed]
- Bungau, S.; Behl, T.; Tit, D.; Banica, F.; Bratu, O.; Diaconu, C.; Nistor-Cseppento, C.; Bustea, C.; Aron, R.; Vesa, C. Interactions between leptin and insulin resistance in patients with prediabetes, with and without NAFLD. Exp. Ther. Med. 2020, 20, 197. [Google Scholar] [CrossRef]
- Vesa, C.; Behl, T.; Nemeth, S.; Bratu, O.; Diaconu, C.; Moleriu, R.; Negrut, N.; Zaha, D.; Bustea, C.; Radu, F.; et al. Prediction of NAFLD occurrence in prediabetes patients. Exp. Ther. Med. 2020, 20, 190. [Google Scholar] [CrossRef]
- Rosato, V.; Masarone, M.; Dallio, M.; Federico, A.; Aglitti, A.; Persico, M. NAFLD and extra-hepatic comorbidities: Current evidence on a multi-organ metabolic syndrome. Int. J. Environ. Res. Public Health 2019, 16, 3415. [Google Scholar] [CrossRef] [PubMed]
- Perdomo, C.M.; Frühbeck, G.; Escalada, J. Impact of nutritional changes on nonalcoholic fatty liver disease. Nutrients 2019, 11, 677. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Li, W.; Li, J.; Li, Y.; Song, H.; Luan, Y.; Qi, H.; Ma, L.; Lu, X.; Yang, Y. Lycium barbarum polysaccharide attenuates high-fat diet-induced hepatic steatosis by up-regulating SIRT1 expression and deacetylase activity. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell. Mol. Life Sci. 2018, 75, 3313–3327. [Google Scholar] [CrossRef]
- Zatterale, F.; Longo, M.; Naderi, J.; Raciti, G.A.; Desiderio, A.; Miele, C.; Beguinot, F. Chronic adipose tissue inflammation linking obesity to insulin resistance and type 2 diabetes. Front. Physiol. 2020, 10, 1607. [Google Scholar] [CrossRef] [PubMed]
- Czech, M.P. Insulin action and resistance in obesity and type 2 diabetes. Nat. Med. 2017, 23, 804–814. [Google Scholar] [CrossRef]
- Sato, T.; Watanabe, Y.; Nishimura, Y.; Inoue, M.; Morita, A.; Miura, S. Acute fructose intake suppresses fasting-induced hepatic gluconeogenesis through the AKT-FoxO1 pathway. Biochem. Biophys. Rep. 2019, 18, 100638. [Google Scholar] [CrossRef]
- Babu, P.V.A.; Liu, D.; Gilbert, E.R. Recent advances in understanding the anti-diabetic actions of dietary flavonoids. J. Nutr. Biochem. 2013, 24, 1777–1789. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Zhu, Q.; Zhang, K.; Thomas, C.; Wu, Y.; Kumar, R.; Baker, K.M.; Xu, Z.; Chen, S.; Guo, S. Activation of Foxo1 by insulin resistance promotes cardiac dysfunction and β–myosin heavy chain gene expression. Circ. Hear. Fail. 2015, 8, 198–208. [Google Scholar] [CrossRef]
- Vesa, C.M.; Popa, L.; Popa, A.R.; Rus, M.; Zaha, A.A.; Bungau, S.; Tit, D.M.; Aron, R.A.C.; Zaha, D.C. Current data regarding the relationship between type 2 diabetes mellitus and cardiovascular risk factors. Diagnostics 2020, 10, 314. [Google Scholar] [CrossRef] [PubMed]
- Zemva, J.; Schilbach, K.; Stöhr, O.; Moll, L.; Franko, A.; Krone, W.; Wiesner, R.J.; Schubert, M. Central FoxO3a and FoxO6 expression is downregulated in obesity induced diabetes but not in aging. Exp. Clin. Endocrinol. Diabetes 2012, 120, 340–350. [Google Scholar] [PubMed]
- Salih, D.A.M.; Brunet, A. FoxO transcription factors in the maintenance of cellular homeostasis during aging. Curr. Opin. Cell Biol. 2008, 20, 126–136. [Google Scholar] [CrossRef]
- Jacobs, F.M.J.; Van der Heide, L.P.; Wijchers, P.J.E.C.; Burbach, J.P.H.; Hoekman, M.F.M.; Smidt, M.P. FoxO6, a novel member of the FoxO class of transcription factors with distinct shuttling dynamics. J. Biol. Chem. 2003, 278, 35959–35967. [Google Scholar] [CrossRef]
- Zhang, X.; Tang, N.; Hadden, T.J.; Rishi, A.K. Akt, FoxO and regulation of apoptosis. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 1978–1986. [Google Scholar] [CrossRef]
- Fu, Z.; Tindall, D.J. FOXOs, cancer and regulation of apoptosis. Oncogene 2008, 27, 2312–2319. [Google Scholar] [CrossRef]
- Luu, B.E.; Zhang, Y.; Storey, K.B. The regulation of Akt and FoxO transcription factors during dehydration in the African clawed frog (Xenopus laevis). Cell Stress Chaperones 2020, 25, 887–897. [Google Scholar] [CrossRef]
- Wang, Z.; Yu, T.; Huang, P. Post-translational modifications of FOXO family proteins. Mol. Med. Rep. 2016, 14, 4931–4941. [Google Scholar] [CrossRef]
- Huang, H.; Regan, K.M.; Wang, F.; Wang, D.; Smith, D.I.; Van Deursen, J.M.A.; Tindall, D.J. Skp2 inhibits FOXO1 in tumor suppression through ubiquitin-mediated degradation. Proc. Natl. Acad. Sci. USA 2005, 102, 1649–1654. [Google Scholar] [CrossRef] [PubMed]
- Tzivion, G.; Dobson, M.; Ramakrishnan, G. FoxO transcription factors; Regulation by AKT and 14-3-3 proteins. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 1938–1945. [Google Scholar] [CrossRef]
- Lam, E.-F.; Francis, R.E.; Petkovic, M. FOXO transcription factors: Key regulators of cell fate. Biochem. Soc. Trans. 2006, 34, 722–726. [Google Scholar] [CrossRef]
- Pramanik, K.C.; Fofaria, N.M.; Gupta, P.; Srivastava, S.K. CBP-mediated FOXO-1 acetylation inhibits pancreatic tumor growth by targeting SirT. Mol. Cancer Ther. 2014, 13, 687–698. [Google Scholar] [CrossRef]
- Satoh, A.; Stein, L.; Imai, S. The role of mammalian sirtuins in the regulation of metabolism, aging, and longevity. In Histone Deacetylases: The Biology and Clinical Implication; Springer: Berlin/Heidelberg, Germany, 2011; pp. 125–162. [Google Scholar]
- Russo, G.L.; Stampone, E.; Cervellera, C.; Borriello, A. Regulation of p27Kip1 and p57Kip2 functions by natural polyphenols. Biomolecules 2020, 10, 1316. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed]
- Akki, R.; Siracusa, R.; Cordaro, M.; Remigante, A.; Morabito, R.; Errami, M.; Marino, A. Adaptation to oxidative stress at cellular and tissue level. Arch. Physiol. Biochem. 2019, 13, 1–11. [Google Scholar] [CrossRef]
- Sengupta, S.; Yang, G.; O’Donnell, J.C.; Hinson, M.D.; McCormack, S.E.; Falk, M.J.; La, P.; Robinson, M.B.; Williams, M.L.; Yohannes, M.T.; et al. The circadian gene Rev-erbα improves cellular bioenergetics and provides preconditioning for protection against oxidative stress. Free Radic. Biol. Med. 2016, 93, 177–189. [Google Scholar] [CrossRef][Green Version]
- Wafer, R.; Tandon, P.; Minchin, J.E.N. The role of peroxisome proliferator-activated receptor gamma (PPARG) in adipogenesis: Applying knowledge from the fish aquaculture industry to biomedical research. Front. Endocrinol. 2017, 8, 102. [Google Scholar] [CrossRef] [PubMed]
- Damcott, C.M.; Moffett, S.P.; Feingold, E.; Barmada, M.M.; Marshall, J.A.; Hamman, R.F.; Ferrell, R.E. Genetic variation in fatty acid-binding protein-4 and peroxisome proliferator-activated receptor γ interactively influence insulin sensitivity and body composition in males. Metabolism 2004, 53, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Hamza, M.S.; Pott, S.; Vega, V.B.; Thomsen, J.S.; Kandhadayar, G.S.; Ng, P.W.P.; Chiu, K.P.; Pettersson, S.; Wei, C.L.; Ruan, Y. De-novo identification of PPARγ/RXR binding sites and direct targets during adipogenesis. PLoS ONE 2009, 4, e4907. [Google Scholar] [CrossRef] [PubMed]
- Eckardt, S.; McLaughlin, K.J.; Willenbring, H. Mouse chimeras as a system to investigate development, cell and tissue function, disease mechanisms and organ regeneration. Cell Cycle 2011, 10, 2091–2099. [Google Scholar] [CrossRef][Green Version]
- Aprile, M.; Ambrosio, M.R.; D’esposito, V.; Beguinot, F.; Formisano, P.; Costa, V.; Ciccodicola, A. PPARG in human adipogenesis: Differential contribution of canonical transcripts and dominant negative isoforms. PPAR Res. 2014, 2014, 537865. [Google Scholar] [CrossRef]
- Cho, Y.-W.; Hong, S.; Jin, Q.; Wang, L.; Lee, J.-E.; Gavrilova, O.; Ge, K. Histone methylation regulator PTIP is required for PPARγ and C/EBPα expression and adipogenesis. Cell Metab. 2009, 10, 27–39. [Google Scholar] [CrossRef]
- Sarjeant, K.; Stephens, J.M. Adipogenesis. Cold Spring Harb. Perspect. Biol. 2012, 4, a008417. [Google Scholar] [CrossRef]
- Rosen, E.D.; Hsu, C.-H.; Wang, X.; Sakai, S.; Freeman, M.W.; Gonzalez, F.J.; Spiegelman, B.M. C/EBPα induces adipogenesis through PPARγ: A unified pathway. Genes Dev. 2002, 16, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Farmer, S.R. Transcriptional control of adipocyte formation. Cell Metab. 2006, 4, 263–273. [Google Scholar] [CrossRef]
- Lefterova, M.I.; Zhang, Y.; Steger, D.J.; Schupp, M.; Schug, J.; Cristancho, A.; Feng, D.; Zhuo, D.; Stoeckert, C.J.; Liu, X.S. PPARγ and C/EBP factors orchestrate adipocyte biology via adjacent binding on a genome-wide scale. Genes Dev. 2008, 22, 2941–2952. [Google Scholar] [CrossRef]
- Symonds, M.E. Adipose Tissue Biology; Springer: Berlin/Heidelberg, Germany, 2012; ISBN 1461409659. [Google Scholar]
- Hishida, T.; Nishizuka, M.; Osada, S.; Imagawa, M. The role of C/EBPδ in the early stages of adipogenesis. Biochimie 2009, 91, 654–657. [Google Scholar] [CrossRef]
- Moseti, D.; Regassa, A.; Kim, W.-K. Molecular regulation of adipogenesis and potential anti-adipogenic bioactive molecules. Int. J. Mol. Sci. 2016, 17, 124. [Google Scholar] [CrossRef]
- Gormand, A.; Berggreen, C.; Amar, L.; Henriksson, E.; Lund, I.; Albinsson, S.; Goransson, O. LKB1 signalling attenuates early events of adipogenesis and responds to adipogenic cues. J. Mol. Endocrinol. 2014, 53, 117–130. [Google Scholar] [CrossRef]
- Tanaka, T.; Yoshida, N.; Kishimoto, T.; Akira, S. Defective adipocyte differentiation in mice lacking the C/EBPβ and/or C/EBPδ gene. EMBO J. 1997, 16, 7432–7443. [Google Scholar] [CrossRef] [PubMed]
- Hakim-Weber, R.; Krogsdam, A.-M.; Jørgensen, C.; Fischer, M.; Prokesch, A.; Bogner-Strauss, J.G.; Bornstein, S.R.; Hansen, J.B.; Madsen, L.; Kristiansen, K. Transcriptional regulatory program in wild-type and retinoblastoma gene-deficient mouse embryonic fibroblasts during adipocyte differentiation. BMC Res. Notes 2011, 4, 1–13. [Google Scholar] [CrossRef]
- Merrett, J.E.; Bo, T.; Psaltis, P.J.; Proud, C.G. Identification of DNA response elements regulating expression of CCAAT/enhancer-binding protein (C/EBP) β and δ during early adipogenesis. BioRxiv 2020, 9, 427–442. [Google Scholar]
- Chen, Z.; Torrens, J.I.; Anand, A.; Spiegelman, B.M.; Friedman, J.M. Krox20 stimulates adipogenesis via C/EBPβ-dependent and -independent mechanisms. Cell Metab. 2005, 1, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Topilko, P.; Schneider-Maunoury, S.; Levi, G.; Baron-Van Evercooren, A.; Chennoufi, A.B.Y.; Seitanidou, T.; Babinet, C.; Charnay, P. Krox-20 controls myelination in the peripheral nervous system. Nature 1994, 371, 796–799. [Google Scholar] [CrossRef]
- Park, Y.-K.; Wang, L.; Giampietro, A.; Lai, B.; Lee, J.E.; Ge, K. Distinct Roles of Transcription Factors KLF4, Krox20, and Peroxisome Proliferator-Activated Receptor γ in Adipogenesis. Mol. Cell Biol. 2017, 37, e00554-16. [Google Scholar] [CrossRef]
- Zhang, C.; He, Y.; Okutsu, M.; Ong, L.C.; Jin, Y.; Zheng, L.; Chow, P.; Yu, S.; Zhang, M.; Yan, Z. Autophagy is involved in adipogenic differentiation by repressesing proteasome-dependent PPARγ2 degradation. Am. J. Physiol. Metab. 2013, 305, E530–E539. [Google Scholar] [CrossRef] [PubMed]
- Dimova, E.Y.; Kietzmann, T. The MAPK pathway and HIF-1 are involved in the induction of the human PAI-1 gene expression by insulin in the human hepatoma cell line HepG2. Ann. N. Y. Acad. Sci. 2006, 1090, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.-S.; Chan, E.C.; Higuchi, M.; Dusting, G.J.; Jiang, F. Redox mechanisms in regulation of adipocyte differentiation: Beyond a general stress response. Cells 2012, 1, 976–993. [Google Scholar] [CrossRef] [PubMed]
- Sikkeland, J. The Role of Mitogen Activated Protein Kinase (MAPK) Phosphatases (MKPs) in Adipogenesis: A Study of Vaccinia H1-Related (VHR) Phosphatase. Master’s Thesis, University of Oslo, Oslo, Norway, 2008. [Google Scholar]
- Dembinska-Kiec, A.; Mykkänen, O.; Kiec-Wilk, B.; Mykkänen, H. Antioxidant phytochemicals against type 2 diabetes. Br. J. Nutr. 2008, 99, ES109–ES117. [Google Scholar] [CrossRef] [PubMed]
- Rosen, E.D.; MacDougald, O.A. Adipocyte differentiation from the inside out. Nat. Rev. Mol. Cell Biol. 2006, 7, 885–896. [Google Scholar] [CrossRef] [PubMed]
- Oishi, Y.; Manabe, I.; Tobe, K.; Tsushima, K.; Shindo, T.; Fujiu, K.; Nishimura, G.; Maemura, K.; Yamauchi, T.; Kubota, N. Krüppel-like transcription factor KLF5 is a key regulator of adipocyte differentiation. Cell Metab. 2005, 1, 27–39. [Google Scholar] [CrossRef]
- Sue, N.; Jack, B.H.A.; Eaton, S.A.; Pearson, R.C.M.; Funnell, A.P.W.; Turner, J.; Czolij, R.; Denyer, G.; Bao, S.; Molero-Navajas, J.C. Targeted disruption of the basic Krüppel-like factor gene (Klf3) reveals a role in adipogenesis. Mol. Cell. Biol. 2008, 28, 3967–3978. [Google Scholar] [CrossRef]
- Rosen, E.D. The transcriptional basis of adipocyte development. Prostaglandins Leukot. Essent. Fat. Acids 2005, 73, 31–34. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.P. Role of PPAR $γ$, transcriptional cofactors, and adiponectin in the regulation of nutrient metabolism, adipogenesis and insulin action: View from the chair. Int. J. Obes. 2005, 29, S3–S4. [Google Scholar] [CrossRef] [PubMed]
- Exley, M.A.; Hand, L.; O’Shea, D.; Lynch, L. Interplay between the immune system and adipose tissue in obesity. J. Endocrinol. 2014, 223, R41–R48. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, D.; Zhu, Q.; Gao, X.; Yang, S.; Xu, A.; Wu, D. Inhibitory effects of A-769662, a novel activator of AMP-activated protein kinase, on 3T3-L1 adipogenesis. Biol. Pharm. Bull. 2009, 32, 993–998. [Google Scholar] [CrossRef] [PubMed]
- Dubois, S.G.; Heilbronn, L.K.; Smith, S.R.; Albu, J.B.; Kelley, D.E.; Ravussin, E.; Group, L.A.A.R. Decreased expression of adipogenic genes in obese subjects with type 2 diabetes. Obesity 2006, 14, 1543–1552. [Google Scholar] [CrossRef] [PubMed]
- Di Rosa, C.; Lattanzi, G.; Taylor, S.F.; Manfrini, S.; Khazrai, Y.M. Very low calorie ketogenic diets in overweight and obesity treatment: Effects on anthropometric parameters, body composition, satiety, lipid profile and microbiota. Obes. Res. Clin. Pract. 2020, 14, 491–503. [Google Scholar] [CrossRef]
- Gurzov, E.N.; Stanley, W.J.; Pappas, E.G.; Thomas, H.E.; Gough, D.J. The JAK/STAT pathway in obesity and diabetes. FEBS J. 2016, 283, 3002–3015. [Google Scholar] [CrossRef]
- Grohmann, M.; Wiede, F.; Dodd, G.T.; Gurzov, E.N.; Ooi, G.J.; Butt, T.; Rasmiena, A.A.; Kaur, S.; Gulati, T.; Goh, P.K. Obesity drives STAT-1-dependent NASH and STAT-3-dependent HCC. Cell 2018, 175, 1289–1306. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Muenzberg, H.; Gavrilova, O.; Reed, J.A.; Berryman, D.; Villanueva, E.C.; Louis, G.W.; Leinninger, G.M.; Bertuzzi, S.; Seeley, R.J. Loss of cytokine-STAT5 signaling in the CNS and pituitary gland alters energy balance and leads to obesity. PLoS ONE 2008, 3, e1639. [Google Scholar] [CrossRef]
- Barclay, J.L.; Nelson, C.N.; Ishikawa, M.; Murray, L.A.; Kerr, L.M.; McPhee, T.R.; Powell, E.E.; Waters, M.J. GH-dependent STAT5 signaling plays an important role in hepatic lipid metabolism. Endocrinology 2011, 152, 181–192. [Google Scholar] [CrossRef]
- Ghilardi, N.; Ziegler, S.; Wiestner, A.; Stoffel, R.; Heim, M.H.; Skoda, R.C. Defective STAT signaling by the leptin receptor in diabetic mice. Proc. Natl. Acad. Sci. USA 1996, 93, 6231–6235. [Google Scholar] [CrossRef]
- Das, U.N. Essential fatty acids: Biochemistry, physiology and pathology. Biotechnol. J. Healthc. Nutr. Technol. 2006, 1, 420–439. [Google Scholar] [CrossRef]
- Deng, X.; Zhang, W.; O-Sullivan, I.S.; Williams, J.B.; Dong, Q.; Park, E.A.; Raghow, R.; Unterman, T.G.; Elam, M.B. FoxO1 inhibits sterol regulatory element-binding protein-1c (SREBP-1c) gene expression via transcription factors Sp1 and SREBP-1c. J. Biol. Chem. 2012, 287, 20132–20143. [Google Scholar] [CrossRef] [PubMed]
- Polak, P.; Cybulski, N.; Feige, J.N.; Auwerx, J.; Rüegg, M.A.; Hall, M.N. Adipose-specific knockout of raptor results in lean mice with enhanced mitochondrial respiration. Cell Metab. 2008, 8, 399–410. [Google Scholar] [CrossRef]
- Fajas, L.; Landsberg, R.L.; Huss-Garcia, Y.; Sardet, C.; Lees, J.A.; Auwerx, J. E2Fs regulate adipocyte differentiation. Dev. Cell 2002, 3, 39–49. [Google Scholar] [CrossRef]
- Fajas, L.; Schoonjans, K.; Gelman, L.; Kim, J.B.; Najib, J.; Martin, G.; Fruchart, J.-C.; Briggs, M.; Spiegelman, B.M.; Auwerx, J. Regulation of peroxisome proliferator-activated receptor $γ$ expression by adipocyte differentiation and determination factor 1/sterol regulatory element binding protein 1: Implications for adipocyte differentiation and metabolism. Mol. Cell. Biol. 1999, 19, 5495–5503. [Google Scholar] [CrossRef] [PubMed]
- Naaz, A.; Holsberger, D.R.; Iwamoto, G.A.; Nelson, A.; Kiyokawa, H.; Cooke, P.S. Loss of cyclin-dependent kinase inhibitors produces adipocyte hyperplasia and obesity. FASEB J. 2004, 18, 1925–1927. [Google Scholar] [CrossRef] [PubMed]
- Boqué, N.; Campión, J.; Milagro, F.I.; Moreno-Aliaga, M.-J.; Martinez, J.A. Some cyclin-dependent kinase inhibitors-related genes are regulated by vitamin C in a model of diet-induced obesity. Biol. Pharm. Bull. 2009, 32, 1462–1468. [Google Scholar]
- Iqbal, N.J.; Lu, Z.; Liu, S.M.; Schwartz, G.J.; Chua, S., Jr.; Zhu, L. Cyclin-dependent kinase 4 is a preclinical target for diet-induced obesity. JCI Insight 2018, 3, e123000. [Google Scholar] [CrossRef] [PubMed]
- Eto, I. Expression of p27 (Kip1), a Cyclin-Dependent Kinase Inhibitor, in Human Peripheral Blood Mononuclear Cells is Inversely Associated with Potential Carcinogenic Risk in Obese Type 2 Diabetic Individuals Relative to Lean Normal Controls. Am. J. Mol. Biol. 2014, 4, 47868. [Google Scholar] [CrossRef][Green Version]
- Tobler, K.; Freudenthaler, A.; Baumgartner-Parzer, S.M.; Wolzt, M.; Ludvik, B.; Nansalmaa, E.; Nowotny, P.J.; Seidinger, D.; Steiner, S.; Luger, A. Reduction of both number and proliferative activity of human endothelial progenitor cells in obesity. Int. J. Obes. 2010, 34, 687–700. [Google Scholar] [CrossRef]
- Lu, Z.; Marcelin, G.; Bauzon, F.; Wang, H.; Fu, H.; Le Dun, S.; Zhao, H.; Li, X.; Jo, Y.; Wardlaw, S. pRb is an obesity suppressor in hypothalamus and high-fat diet inhibits pRb in this location. EMBO J. 2013, 32, 844–857. [Google Scholar] [CrossRef] [PubMed]
- Auld, C.A.; Morrison, R.F. Evidence for cytosolic p27 (Kip1) ubiquitylation and degradation during adipocyte hyperplasia. Obesity 2006, 14, 2136–2144. [Google Scholar] [CrossRef]
- Xie, Q.; Peng, S.; Tao, L.; Ruan, H.; Yang, Y.; Li, T.M.; Adams, U.; Meng, S.; Bi, X.; Dong, M.Q.; et al. E2F transcription factor 1 regulates cellular and organismal senescence by inhibiting forkhead box O transcription factors. J. Biol. Chem. 2014, 289, 34205–34213. [Google Scholar] [CrossRef]
- Isakson, P.; Hammarstedt, A.; Gustafson, B.; Smith, U. Impaired preadipocyte differentiation in human abdominal obesity: Role of Wnt, tumor necrosis factor-$α$, and inflammation. Diabetes 2009, 58, 1550–1557. [Google Scholar] [CrossRef]
- Liu, Z.; Brooks, R.S.; Ciappio, E.D.; Kim, S.J.; Crott, J.W.; Bennett, G.; Greenberg, A.S.; Mason, J.B. Diet-induced obesity elevates colonic TNF-$α$ in mice and is accompanied by an activation of Wnt signaling: A mechanism for obesity-associated colorectal cancer. J. Nutr. Biochem. 2012, 23, 1207–1213. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-R.; Lazarenko, O.P.; Wu, X.; Tong, Y.; Blackburn, M.L.; Shankar, K.; Badger, T.M.; Ronis, M.J.J. Obesity reduces bone density associated with activation of PPARγ and suppression of Wnt/β-catenin in rapidly growing male rats. PLoS ONE 2010, 5, e13704. [Google Scholar] [CrossRef] [PubMed]
- Davis, D.B.; Lavine, J.A.; Suhonen, J.I.; Krautkramer, K.A.; Rabaglia, M.E.; Sperger, J.M.; Fernandez, L.A.; Yandell, B.S.; Keller, M.P.; Wang, I.-M. FoxM1 is up-regulated by obesity and stimulates β-cell proliferation. Mol. Endocrinol. 2010, 24, 1822–1834. [Google Scholar] [CrossRef]
- Benzler, J.; Andrews, Z.B.; Pracht, C.; Stöhr, S.; Shepherd, P.R.; Grattan, D.R.; Tups, A. Hypothalamic WNT signalling is impaired during obesity and reinstated by leptin treatment in male mice. Endocrinology 2013, 154, 4737–4745. [Google Scholar] [CrossRef]
- Tong, J.F.; Yan, X.; Zhu, M.J.; Ford, S.P.; Nathanielsz, P.W.; Du, M. Maternal obesity downregulates myogenesis and $β$-catenin signaling in fetal skeletal muscle. Am. J. Physiol. Metab. 2009, 296, E917–E924. [Google Scholar] [CrossRef]
- Liu, B.; Chen, Y.; Cheng, H.; Wang, Y.; Rui, H.; Yang, M.; Dong, H.; Han, D.; Dong, J. The protective effects of curcumin on obesity-related glomerulopathy are associated with inhibition of Wnt/$β$-catenin signaling activation in podocytes. Evid. Based Complement. Altern. Med. 2015, 2015, 827472. [Google Scholar] [CrossRef] [PubMed]
- Vanlint, S. Vitamin D and obesity. Nutrients 2013, 5, 949–956. [Google Scholar] [CrossRef]
- Pourshahidi, L.K. Vitamin D and obesity: Current perspectives and future directions. Proc. Nutr. Soc. 2015, 74, 115–124. [Google Scholar] [CrossRef]
- Kubota, N.; Terauchi, Y.; Miki, H.; Tamemoto, H.; Yamauchi, T.; Komeda, K.; Satoh, S.; Nakano, R.; Ishii, C.; Sugiyama, T. PPAR$γ$ mediates high-fat diet–induced adipocyte hypertrophy and insulin resistance. Mol. Cell 1999, 4, 597–609. [Google Scholar] [CrossRef]
- Ando, H.; Takamura, T.; Matsuzawa-Nagata, N.; Shima, K.R.; Eto, T.; Misu, H.; Shiramoto, M.; Tsuru, T.; Irie, S.; Fujimura, A. Clock gene expression in peripheral leucocytes of patients with type 2 diabetes. Diabetologia 2009, 52, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Tong, Q.; Dalgin, G.; Xu, H.; Ting, C.-N.; Leiden, J.M.; Hotamisligil, G.S. Function of GATA transcription factors in preadipocyte-adipocyte transition. Science 2000, 290, 134–138. [Google Scholar] [CrossRef]
- Wang, P.; Xu, S.; Li, W.; Wang, F.; Yang, Z.; Jiang, L.; Wang, Q.; Huang, M.; Zhou, P. Salvianolic acid B inhibited PPAR$γ$ expression and attenuated weight gain in mice with high-fat diet-induced obesity. Cell. Physiol. Biochem. 2014, 34, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Al-Mansoori, L.; Al-Jaber, H.; Madani, A.Y.; Mazloum, N.A.; Agouni, A.; Ramanjaneya, M.; Abou-Samra, A.-B.; Elrayess, M.A. Suppression of GATA-3 increases adipogenesis, reduces inflammation and improves insulin sensitivity in 3T3L-1 preadipocytes. Cell. Signal. 2020, 75, 109735. [Google Scholar] [CrossRef]
- Montani, J.P.; Antic, V.; Yang, Z.; Dulloo, A. Pathways from obesity to hypertension: From the perspective of a vicious triangle. Int. J. Obes. 2002, 26, S28–S38. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Hu, C.; Hsu, C.-K.; Zhang, Q.; Bi, C.; Asnicar, M.; Hsiung, H.M.; Fox, N.; Slieker, L.J.; Yang, D.D. Targeted disruption of the melanin-concentrating hormone receptor-1 results in hyperphagia and resistance to diet-induced obesity. Endocrinology 2002, 143, 2469–2477. [Google Scholar] [CrossRef]
- Farmer, S.R. The forkhead transcription factor Foxo1: A possible link between obesity and insulin resistance. Mol. Cell 2003, 11, 6–8. [Google Scholar] [CrossRef]
- Plum, L.; Lin, H.V.; Dutia, R.; Tanaka, J.; Aizawa, K.S.; Matsumoto, M.; Kim, A.J.; Cawley, N.X.; Paik, J.-H.; Loh, Y.P. The obesity susceptibility gene Cpe links FoxO1 signaling in hypothalamic pro-opiomelanocortin neurons with regulation of food intake. Nat. Med. 2009, 15, 1195–1201. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, L.; Wong, C.C.L.; Li, G.; Xu, T.; Pajvani, U.; Park, S.K.R.; Wronska, A.; Chen, B.-X.; Marks, A.R.; Fukamizu, A. Calcium signaling through CaMKII regulates hepatic glucose production in fasting and obesity. Cell Metab. 2012, 15, 739–751. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Behl, T.; Kaur, I.; Sehgal, A.; Singh, S.; Zengin, G.; Negrut, N.; Nistor-Cseppento, D.C.; Pavel, F.M.; Corb Aron, R.A.; Bungau, S. Exploring the Genetic Conception of Obesity via the Dual Role of FoxO. Int. J. Mol. Sci. 2021, 22, 3179. https://doi.org/10.3390/ijms22063179
Behl T, Kaur I, Sehgal A, Singh S, Zengin G, Negrut N, Nistor-Cseppento DC, Pavel FM, Corb Aron RA, Bungau S. Exploring the Genetic Conception of Obesity via the Dual Role of FoxO. International Journal of Molecular Sciences. 2021; 22(6):3179. https://doi.org/10.3390/ijms22063179
Chicago/Turabian StyleBehl, Tapan, Ishnoor Kaur, Aayush Sehgal, Sukhbir Singh, Gokhan Zengin, Nicoleta Negrut, Delia Carmen Nistor-Cseppento, Flavia Maria Pavel, Raluca Anca Corb Aron, and Simona Bungau. 2021. "Exploring the Genetic Conception of Obesity via the Dual Role of FoxO" International Journal of Molecular Sciences 22, no. 6: 3179. https://doi.org/10.3390/ijms22063179
APA StyleBehl, T., Kaur, I., Sehgal, A., Singh, S., Zengin, G., Negrut, N., Nistor-Cseppento, D. C., Pavel, F. M., Corb Aron, R. A., & Bungau, S. (2021). Exploring the Genetic Conception of Obesity via the Dual Role of FoxO. International Journal of Molecular Sciences, 22(6), 3179. https://doi.org/10.3390/ijms22063179