
Ring-Selective Fragmentation in the Tirapazamine Molecule upon Low-Energy Electron Attachment


Abstract
:1. Introduction
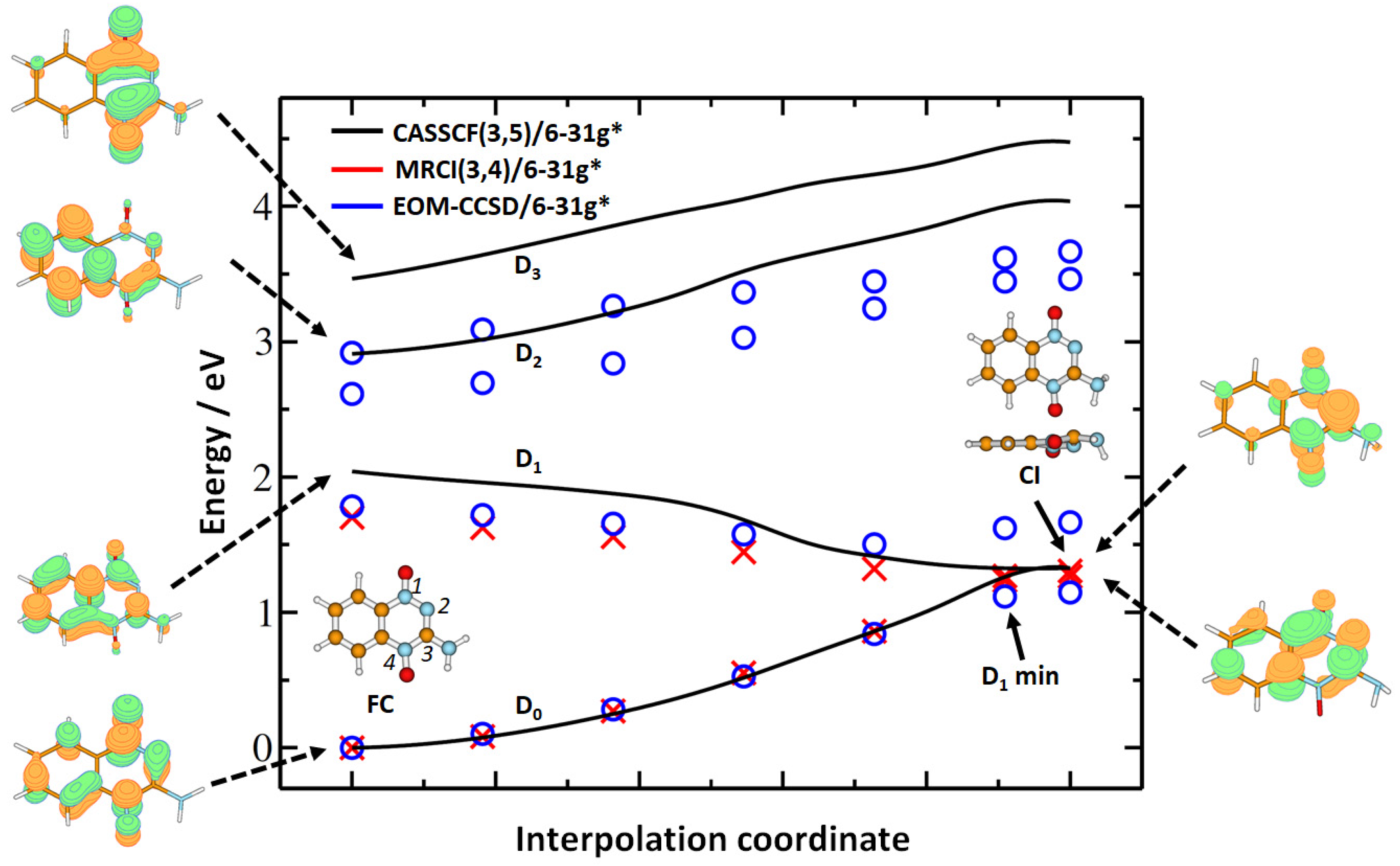
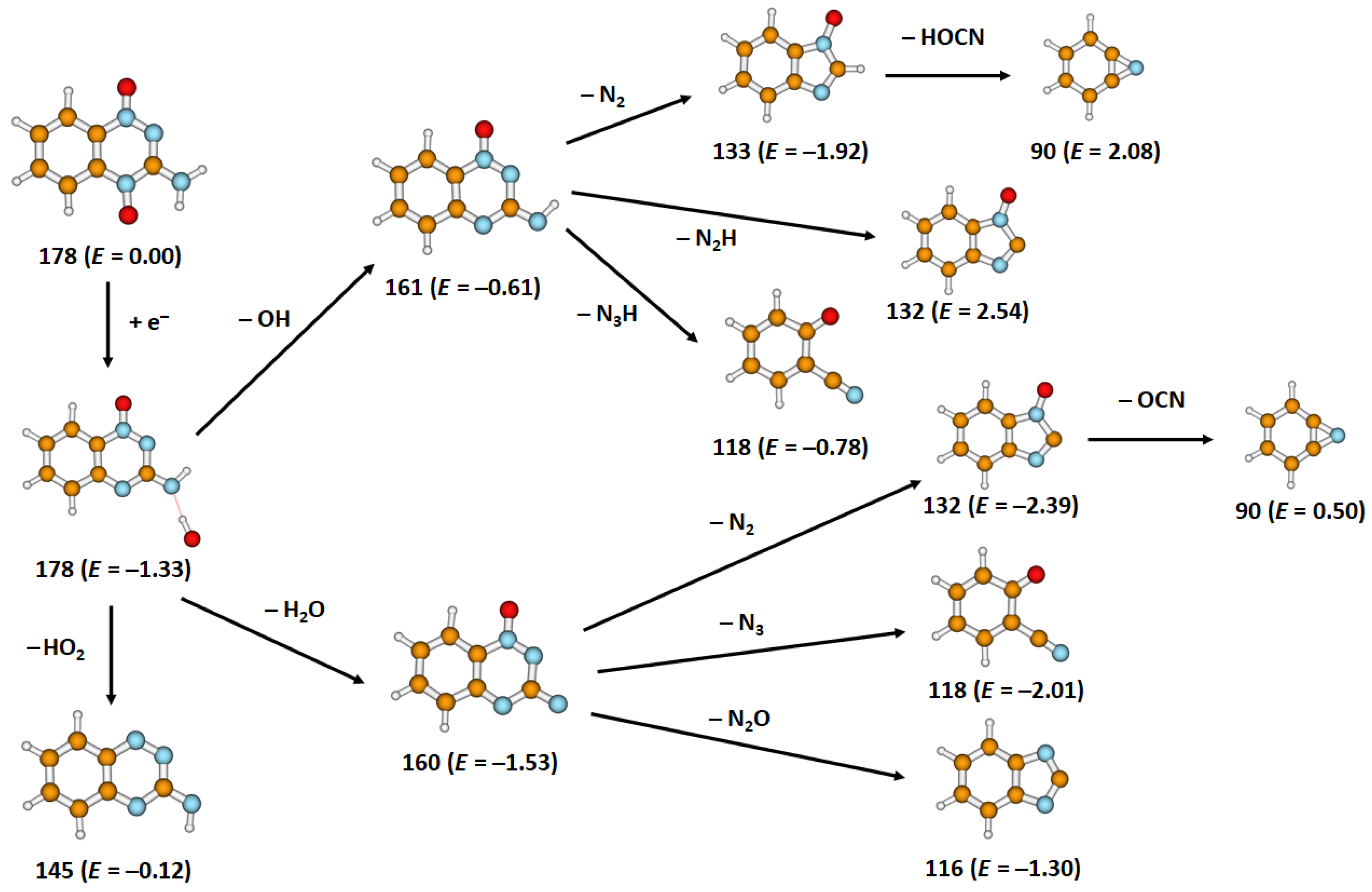
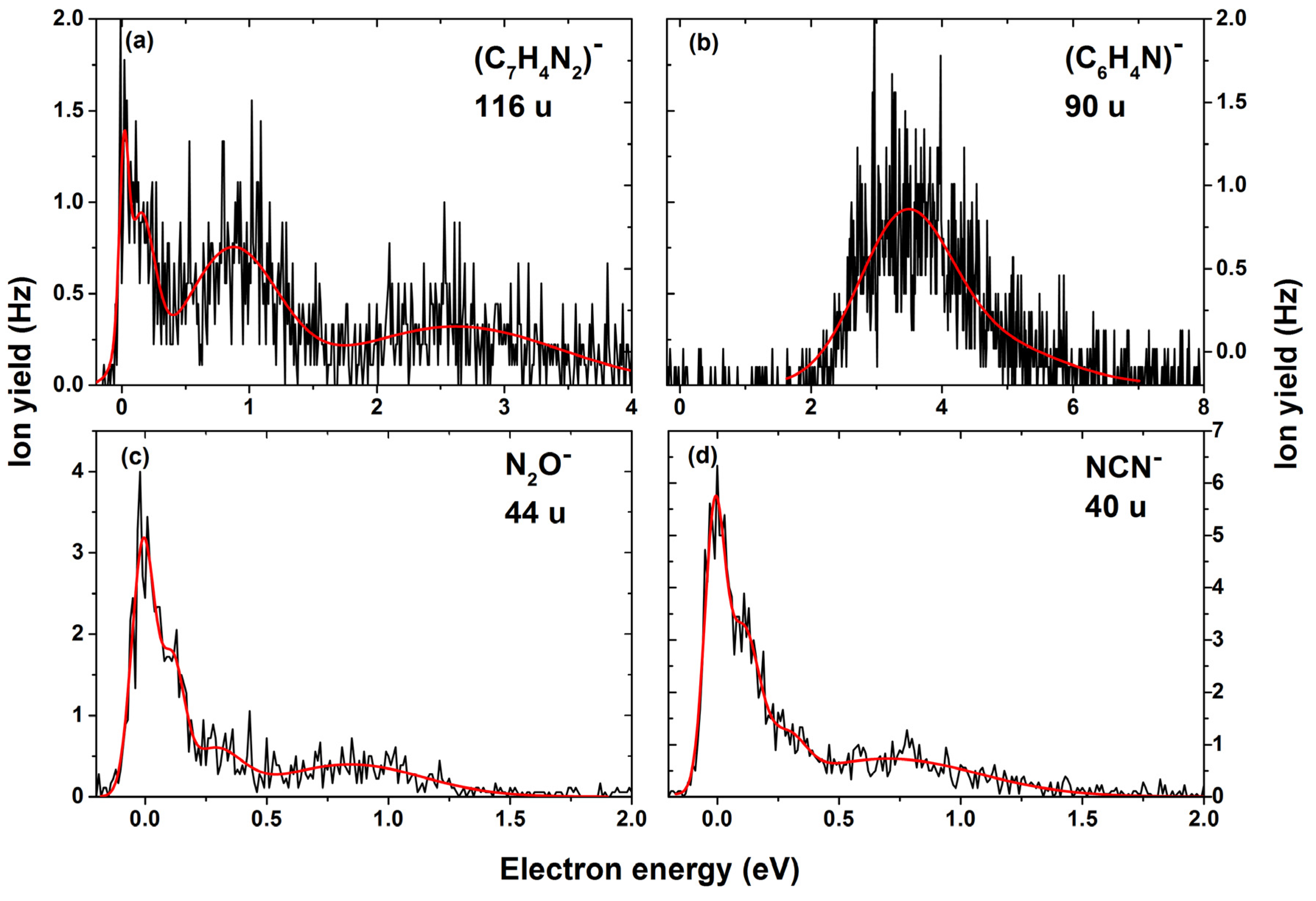
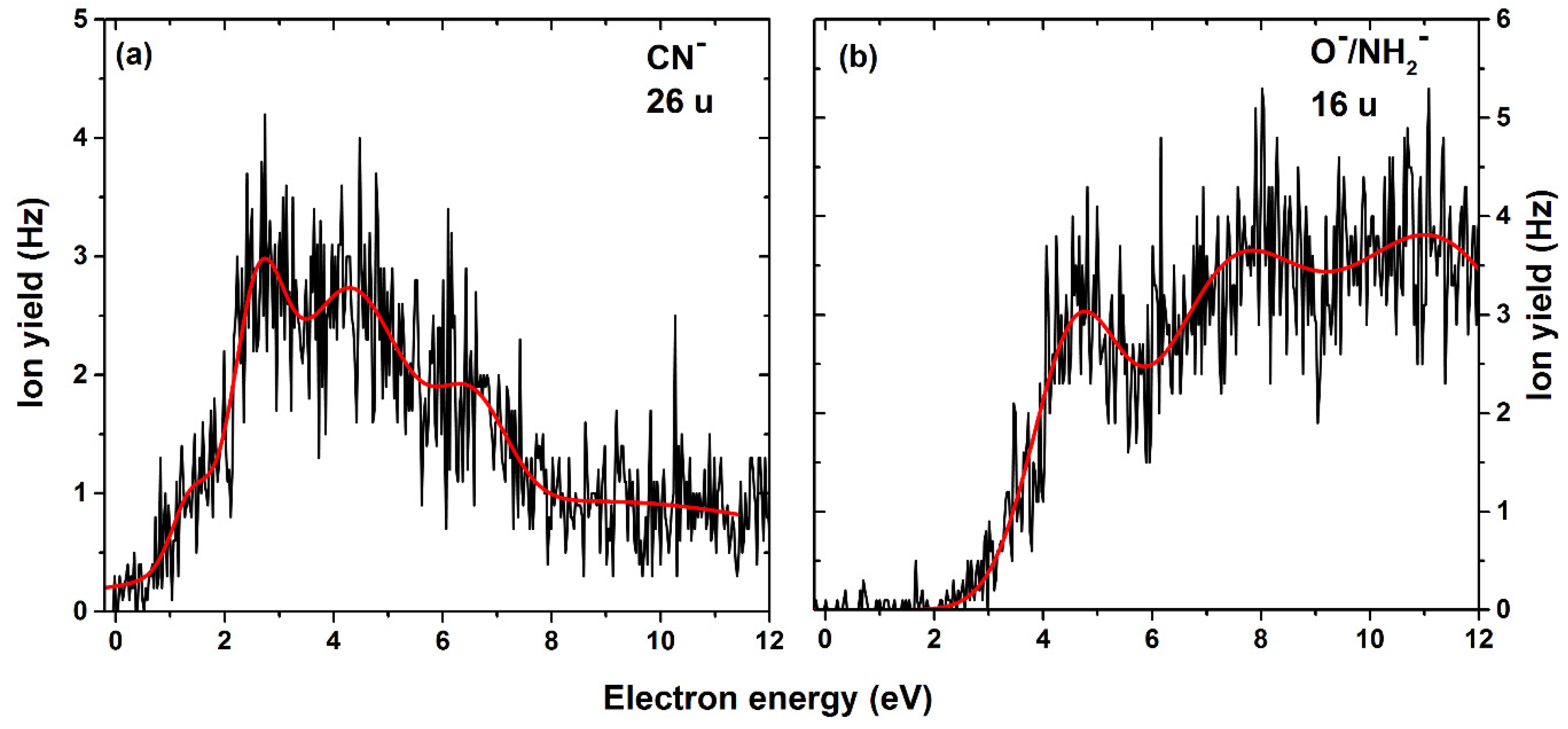
2. Results
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Oronsky, B.T.; Knox, S.J.; Scicinski, J. Six Degrees of Separation: The Oxygen Effect in the Development of Radiosensitizers. Transl. Oncol. 2011, 4, 189–198. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.M.; Wilson, W.R. Exploiting tumour hypoxia in cancer treatment. Nat. Rev. Cancer 2004, 4, 437–447. [Google Scholar] [CrossRef]
- Vaupel, P.; Mayer, A. Hypoxia in cancer: Significance and impact on clinical outcome. Cancer Metastasis Rev. 2007, 26, 225–239. [Google Scholar] [CrossRef]
- Raviraj, J.; Bokkasam, V.K.; Kumar, V.S.; Reddy, U.S.; Suman, V. Radiosensitizers, radioprotectors, and radiation mitigators. Indian J. Dent. Res. 2014, 25, 83. [Google Scholar] [CrossRef]
- Wang, H.; Mu, X.; He, H.; Zhang, X.D. Cancer Radiosensitizers. Trends Pharmacol. Sci. 2018, 39, 24–48. [Google Scholar] [CrossRef]
- Seiwert, T.Y.; Salama, J.K.; Vokes, E.E. The concurrent chemoradiation paradigm—General principles. Nat. Clin. Pract. Oncol. 2007, 4, 86–100. [Google Scholar] [CrossRef]
- Wardman, P. Chemical radiosensitizers for use in radiotherapy. Clin. Oncol. 2007, 19, 397–417. [Google Scholar] [CrossRef] [PubMed]
- Meißner, R.; Kočišek, J.; Feketeová, L.; Fedor, J.; Fárník, M.; Limão-Vieira, P.; Illenberger, E.; Denifl, S. Low-energy electrons transform the nimorazole molecule into a radiosensitiser. Nat. Commun. 2019, 10, 2388. [Google Scholar] [CrossRef] [PubMed]
- Overgaard, J.; Hansen, H.S.; Overgaard, M.; Bastholt, L.; Berthelsen, A.; Specht, L.; Lindeløv, B.; Jørgensen, K. A randomized double-blind phase III study of nimorazole as a hypoxic radiosensitizer of primary radiotherapy in supraglottic larynx and pharynx carcinoma. Results of the Danish Head and Neck Cancer Study (DAHANCA) Protocol 5-85. Radiother. Oncol. 1998, 46, 135–146. [Google Scholar] [CrossRef]
- Hay, M.P.; Pruijn, F.B.; Gamage, S.A.; Liyanage, H.D.S.; Kovacs, M.S.; Patterson, A.V.; Wilson, W.R.; Brown, J.M.; Denny, W.A. DNA-Targeted 1,2,4-Benzotriazine 1,4-Dioxides: Potent Analogues of the Hypoxia-Selective Cytotoxin Tirapazamine. J. Med. Chem. 2004, 47, 475–488. [Google Scholar] [CrossRef]
- Brown, J.M. The Hypoxic Cell: A Target for Selective Cancer Therapy. Cancer Res. 1999, 59, 5863–5870. [Google Scholar]
- Junnotula, V.; Sarkar, U.; Sinha, S.; Gates, K.S. Initiation of DNA strand cleavage by 1, 2, 4-benzotriazine 1, 4-dioxide antitumor agents: Mechanistic insight from studies of 3-methyl-1, 2, 4-benzotriazine 1, 4-dioxide. J. Am. Chem. Soc. 2009, 131, 1015–1024. [Google Scholar] [CrossRef] [Green Version]
- Li, L.C.; Zha, D.; Zhu, Y.Q.; Xu, M.H.; Wong, N.B. Theoretical study of the mechanism of hydroxyl radical release from tirapazamine’s undergoing enzymatic catalysis. Chem. Phys. Lett. 2005, 408, 329–334. [Google Scholar] [CrossRef]
- Zagorevskii, D.; Song, M.; Breneman, C.; Yuan, Y.; Fuchs, T.; Gates, K.S.; Greenlief, C.M. A mass spectrometry study of tirapazamine and its metabolites: Insights into the mechanism of metabolic transformations and the characterization of reaction intermediates. J. Am. Soc. Mass Spectrom. 2003, 14, 881–892. [Google Scholar] [CrossRef] [Green Version]
- Shinde, S.S.; Maroz, A.; Hay, M.P.; Patterson, A.V.; Denny, W.A.; Anderson, R.F. Characterization of radicals formed following enzymatic reduction of 3-substituted analogues of the hypoxia-selective cytotoxin 3-amino-1, 2, 4-benzotriazine 1, 4-dioxide (tirapazamine). J. Am. Chem. Soc. 2010, 132, 2591–2599. [Google Scholar] [CrossRef]
- Anderson, R.F.; Shinde, S.S.; Hay, M.P.; Gamage, S.A.; Denny, W.A. Activation of 3-amino-1, 2, 4-benzotriazine 1, 4-dioxide antitumor agents to oxidizing species following their one-electron reduction. J. Am. Chem. Soc. 2003, 125, 748–756. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.F.; Shinde, S.S.; Hay, M.P.; Denny, W.A. Potentiation of the cytotoxicity of the anticancer agent tirapazamine by benzotriazine N-oxides: The role of redox equilibria. J. Am. Chem. Soc. 2006, 128, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.D.D.; Weinfeld, M. Dual action of tirapazamine in the induction of DNA strand breaks. Cancer Res. 1996, 56, 1584–1590. [Google Scholar] [PubMed]
- Daniels, J.S.; Gates, K.S. DNA cleavage by the antitumor agent 3-amino-1,2,4-benzotriazine 1,4-dioxide (SR4233): Evidence for involvement of hydroxyl radical. J. Am. Chem. Soc. 1996, 118, 3380–3385. [Google Scholar] [CrossRef]
- Adams, G.E.; Dewey, D.L. Hydrated electrons and radiobiological sensitization. Biochem. Biophys. Res. Commun. 1963, 12. [Google Scholar] [CrossRef]
- Pimblott, S.M.; LaVerne, J.A. Production of low-energy electrons by ionizing radiation. Radiat. Phys. Chem. 2007, 76, 1244–1247. [Google Scholar] [CrossRef]
- Kumar, A.; Becker, D.; Adhikary, A.; Sevilla, M.D. Reaction of electrons with DNA: Radiation damage to radiosensitization. Int. J. Mol. Sci. 2019, 20, 3998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alizadeh, E.; Sanche, L. Precursors of solvated electrons in radiobiological physics and chemistry. Chem. Rev. 2012, 112, 5578–5602. [Google Scholar] [CrossRef] [PubMed]
- Boudaïffa, B.; Cloutier, P.; Hunting, D.; Huels, M.A.; Sanche, L. Resonant formation of DNA strand breaks by low-energy (3 to 20 eV) electrons. Science 2000, 287, 1658–1660. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Wang, F.; Denisov, S.A.; Adhikary, A.; Mostafavi, M. Reactivity of prehydrated electrons toward nucleobases and nucleotides in aqueous solution. Sci. Adv. 2017, 3, e1701669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arthur-Baidoo, E.; Ameixa, J.; Ziegler, P.; Ferreira da Silva, F.; Ončák, M.; Denifl, S. Reactions in Tirapazamine Induced by the Attachment of Low-Energy Electrons: Dissociation Versus Roaming of OH. Angew. Chem.-Int. Ed. 2020, 59, 17177–17181. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Glaser, R.; Gates, K.S. On the reaction mechanism of tirapazamine reduction chemistry: Unimolecular N-OH homolysis, stepwise dehydration, or triazene ring-opening. Chem. Res. Toxicol. 2012, 25, 634–645. [Google Scholar] [CrossRef]
- Yin, J.; Glaser, R.; Gates, K.S. Electron and spin-density analysis of tirapazamine reduction chemistry. Chem. Res. Toxicol. 2012, 25, 620–633. [Google Scholar] [CrossRef]
- Chomicz, L.; Zdrowowicz, M.; Kasprzykowski, F.; Rak, J.; Buonaugurio, A.; Wang, Y.; Bowen, K.H. How to find out whether a 5-substituted uracil could be a potential DNA radiosensitizer. J. Phys. Chem. Lett. 2013, 4, 2853–2857. [Google Scholar] [CrossRef]
- Ameixa, J.; Arthur-Baidoo, E.; Meißner, R.; Makurat, S.; Kozak, W.; Butowska, K.; Ferreira Da Silva, F.; Rak, J.; Denifl, S. Low-energy electron-induced decomposition of 5-trifluoromethanesulfonyl-uracil: A potential radiosensitizer. J. Chem. Phys. 2018, 149, 164307. [Google Scholar] [CrossRef] [Green Version]
- Spisz, P.; Zdrowowicz, M.; Kozak, W.; Chomicz-Mańka, L.; Falkiewicz, K.; Makurat, S.; Sikorski, A.; Wyrzykowski, D.; Rak, J.; Arthur-Baidoo, E.; et al. Uracil-5-yl O-Sulfamate: An Illusive Radiosensitizer. Pitfalls in Modeling the Radiosensitizing Derivatives of Nucleobases. J. Phys. Chem. B 2020, 124, 5600–5613. [Google Scholar] [CrossRef] [PubMed]
- Ptasińska, S.; Denifl, S.; Scheier, P.; Märk, T.D. Inelastic electron interaction (attachment/ionization) with deoxyribose. J. Chem. Phys. 2004, 120, 8505–8511. [Google Scholar] [CrossRef] [PubMed]
- Bald, I.; Langer, J.; Tegeder, P.; Ingólfsson, O. From isolated molecules through clusters and condensates to the building blocks of life. Int. J. Mass Spectrom. 2008, 277, 4–25. [Google Scholar] [CrossRef]
- Gorfinkiel, J.D.; Ptasinska, S. Electron scattering from molecules and molecular aggregates of biological relevance. J. Phys. B At. Mol. Opt. Phys. 2017, 50, 182001. [Google Scholar] [CrossRef] [Green Version]
- Ribar, A.; Fink, K.; Probst, M.; Huber, S.E.; Feketeová, L.; Denifl, S. Isomer Selectivity in Low-Energy Electron Attachment to Nitroimidazoles. Chem.-Eur. J. 2017, 23, 12892–12899. [Google Scholar] [CrossRef]
- Ptasinska, S.; Denifl, S.; Scheier, P.; Illenberger, E.; Märk, T.D. Bond- and site-selective loss of H atoms from nucleobases by very-low-energy electrons (<3 eV). Angew. Chem.-Int. Ed. 2005, 44, 6941–6943. [Google Scholar] [CrossRef]
- Gallup, G.A.; Aflatooni, K.; Burrow, P.D. Dissociative electron attachment near threshold, thermal attachment rates, and vertical attachment energies of chloroalkanes. J. Chem. Phys. 2003. [Google Scholar] [CrossRef] [Green Version]
- Meißner, R.; Feketeová, L.; Bayer, A.; Postler, J.; Limão-Vieira, P.; Denifl, S. Positive and negative ions of the amino acid histidine formed in low-energy electron collisions. J. Mass Spectrom. 2019, 54, 802–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, H.; Jørgensen, P. Coupled cluster response functions. J. Chem. Phys. 1990, 93, 3333–3344. [Google Scholar] [CrossRef]
- Stanton, J.F.; Bartlett, R.J. The equation of motion coupled-cluster method. A systematic biorthogonal approach to molecular excitation energies, transition probabilities, and excited state properties. J. Chem. Phys. 1993, 98, 7029–7039. [Google Scholar] [CrossRef]
- Krylov, A.I. Equation-of-motion coupled-cluster methods for open-shell and electronically excited species: The Hitchhiker’s guide to fock space. Annu. Rev. Phys. Chem. 2008, 59, 433–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werner, H.J.; Knowles, P.J.; Knizia, G.; Manby, F.R.; Schütz, M.; Celani, P.; Korona, T.; Lindh, R.; Mitrushenkov, A.; Rauhut, G.; et al. MOLPRO, version 2012.1; A package of ab initio programs; University of Cardiff Chemistry Consultants (UC3): Cardiff, UK, 2012.
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mass (u) | Anion | Neutral Counterparts | Resonance Positions (eV) | Thresholds (eV) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | Exp | Theory | |||
| 178 | C7H6N4O2− | 0 | 0.1 | 0.3 | - | - | - | - | ~0 | −1.28 | |
| 177 a | C7H5N4O2− | H | 0 | 0.1 | 0.3 | 0.9 | - | - | - | ~0 | −0.92 |
| 162 a | C7H4N3O2− | NH2 | 0.3 | 0.7 | - | - | - | - | - | ~0 | −1.71 |
| 161 a | C7H5N4O− | OH | 0 | 0.1 | 0.3 | 0.9 | - | - | - | ~0 | −0.61 |
| 145 | C7H5N4− | HO2 | 0.4 | 1.0 | 1.7 | 4.5 | 5.3 | - | - | ~0 | −0.12 |
| 133 | C7H5N2O− | OH + N2 | 0.0 | 0.1 | 0.3 | 0.8 | 2.4 | - | - | ~0 | −1.92 |
| 132 | C7H4N2O− | H2O + N2/ OH + N2H | 0.0 | 0.1 | 0.3 | 0.8 | 2.4 | 2.8 | 3.2 | ~0 | −2.39/ 2.54 |
| 118 | C7H4NO− | OH + N3H/ H2O + N3 | 0.0 | 0.1 | 2.5 | 3.2 | 3.9 | - | - | ~0 | −0.78/ −2.01 |
| 116 | C7H4N2− | H2O + N2O | 0.0 | 0.1 | 0.9 | 2.6 | - | - | - | ~0 | −1.30 |
| 90 | C6H4N− | OH + N2 + HOCN/H2O + N2 + NCO | 3.6 | 4.7 | - | - | - | - | - | 2.1 | 2.08/ 0.50 |
| 44 | N2O− | C7H6N2O | 0.0 | 0.1 | 0.3 | 0.9 | - | - | - | ~0 | −0.12 |
| 40 | NCN− | C6H6N2O2 | 0.0 | 0.1 | 0.3 | 0.7 | - | - | - | ~0 | −0.79 |
| 26 | CN− | C6H6N3O2/ C6H6NO2 + N2 | 1.4 | 2.6 | 4.2 | 6.5 | - | - | - | 0.7 | 0.58/ −2.64 |
| 16 | O− NH2− | C7H6N4O C7H4N3O2 | 4.6 | 7.3 | 11.1 | - | - | - | - | 2.7 | 0.81 1.19 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arthur-Baidoo, E.; Ameixa, J.; Ončák, M.; Denifl, S. Ring-Selective Fragmentation in the Tirapazamine Molecule upon Low-Energy Electron Attachment. Int. J. Mol. Sci. 2021, 22, 3159. https://doi.org/10.3390/ijms22063159
Arthur-Baidoo E, Ameixa J, Ončák M, Denifl S. Ring-Selective Fragmentation in the Tirapazamine Molecule upon Low-Energy Electron Attachment. International Journal of Molecular Sciences. 2021; 22(6):3159. https://doi.org/10.3390/ijms22063159
Chicago/Turabian StyleArthur-Baidoo, Eugene, Joao Ameixa, Milan Ončák, and Stephan Denifl. 2021. "Ring-Selective Fragmentation in the Tirapazamine Molecule upon Low-Energy Electron Attachment" International Journal of Molecular Sciences 22, no. 6: 3159. https://doi.org/10.3390/ijms22063159
APA StyleArthur-Baidoo, E., Ameixa, J., Ončák, M., & Denifl, S. (2021). Ring-Selective Fragmentation in the Tirapazamine Molecule upon Low-Energy Electron Attachment. International Journal of Molecular Sciences, 22(6), 3159. https://doi.org/10.3390/ijms22063159