
Cushing’s Syndrome Effects on the Thyroid


 ,
, 
Abstract
1. Introduction
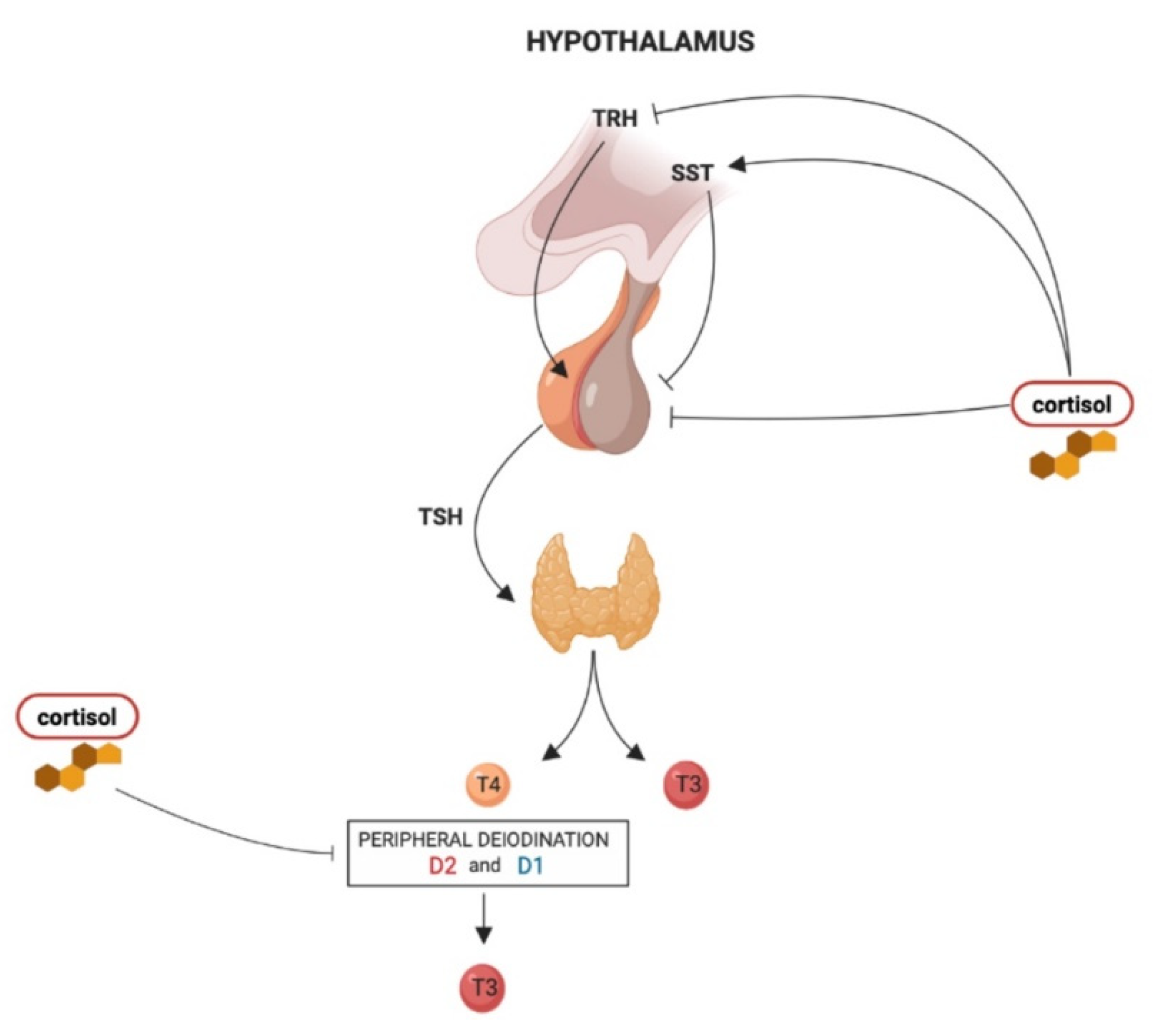
2. Effects of Glucocorticoids on Hypothalamus–Pituitary–Thyroid Axis
3. Thyroid Function Dynamic Changes in Cushing’s Syndrome
4. Exacerbation of Thyroid Disease after Surgery for Cushing’s Syndrome
5. Drugs Used for Cushing’s Syndrome and Their Effects on Thyroid Function
5.1. Effects of ACTH Directed Drugs: Pasireotide and Cabergoline on HPT Axis Activity
5.2. Effects of Steroidogenesis Inhibitors on HPT Axis Activity
5.3. Effects of Glucocorticoid Receptors Antagonist Mifepristone on HPT Axis Activity
6. Conclusions
Author Contributions
Funding
Informed Consent Statement
Conflicts of Interest
References
- Pivonello, R.; De Martino, M.C.; De Leo, M.; Lombardi, G.; Colao, A. Cushing’s syndrome. Endocrinol. Metab. Clin. N. Am. 2008, 37, 135–149. [Google Scholar] [CrossRef] [PubMed]
- Pivonello, R.; Isidori, A.M.; De Martino, M.C.; Newell-Price, J.; Biller, B.M.K.; Colao, A. Complications of cushing’s syndrome: State of the art. Lancet Diabetes Endocrinol. 2016, 4, 611–629. [Google Scholar] [CrossRef]
- Resmini, E.; Santos, A.; Webb, S.M. Cortisol excess and the brain. Front. Horm. Res. 2016, 46, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Lado-Abeal, J.; Rodriguez-Arnao, J.; Newell-Price, J.D.; Perry, L.A.; Grossman, A.B.; Besser, G.M.; Trainer, P.J. Menstrual abnormalities in women with cushing’s disease are correlated with hypercortisolemia rather than raised circulating androgen levels. J. Clin. Endocrinol. Metab. 1998, 83, 3083–3088. [Google Scholar] [CrossRef]
- Takahashi, H.; Bando, H.; Zhang, C.; Yamasaki, R.; Saito, S. Mechanism of impaired growth hormone secretion in patients with cushing’s disease. Acta Endocrinol. 1992, 127, 13–17. [Google Scholar] [CrossRef]
- Giustina, A.; Bossoni, S.; Bussi, A.R.; Pozzi, A.; Wehrenberg, W.B. Effect of galanin on the growth hormone (gh) response to gh-releasing hormone in patients with cushing’s disease. Endocr. Res. 1993, 19, 47–56. [Google Scholar] [CrossRef]
- Hollenberg, A.N. The role of the thyrotropin-releasing hormone (trh) neuron as a metabolic sensor. Thyroid 2008, 18, 131–139. [Google Scholar] [CrossRef]
- Paragliola, R.M.; Corsello, A.; Concolino, P.; Ianni, F.; Papi, G.; Pontecorvi, A.; Corsello, S.M. Iodothyronine deiodinases and reduced sensitivity to thyroid hormones. Front. Biosci. 2020, 25, 201–228. [Google Scholar]
- Sugrue, M.L.; Vella, K.R.; Morales, C.; Lopez, M.E.; Hollenberg, A.N. The thyrotropin-releasing hormone gene is regulated by thyroid hormone at the level of transcription in vivo. Endocrinology 2010, 151, 793–801. [Google Scholar] [CrossRef]
- Joseph-Bravo, P.; Jaimes-Hoy, L.; Charli, J.L. Regulation of trh neurons and energy homeostasis-related signals under stress. J. Endocrinol. 2015, 224, R139–R159. [Google Scholar] [CrossRef]
- Cintra, A.; Fuxe, K.; Wikstro¨m, A.C.; Visser, T.; Gustafsson, J.A. Evidence for thyrotropin-releasing hormone and glucocorticoid receptor-immunoreactive neurons in various preoptic and hypothalamic nuclei of the male rat. Brain Res. 1990, 506, 139–144. [Google Scholar] [CrossRef]
- Kovacs, K.; Rotondo, F.; Stefaneanu, L.; Fereidooni, F.; Horvath, E.; Lloyd, R.V.; Scheithauer, B.W. Glucocorticoid receptor expression in nontumorous human pituitaries and pituitary adenomas. Endocr. Pathol. 2000, 11, 267–275. [Google Scholar] [CrossRef]
- Kakucska, I.; Qi, Y.; Lechan, R.M. Changes in adrenal status affect hypothalamic thyrotropin-releasing hormone gene expression in parallel with corticotropin-releasing hormone. Endocrinology 1995, 136, 2795–2802. [Google Scholar] [CrossRef]
- Alkemade, A.; Unmehopa, U.A.; Wiersinga, W.M.; Swaab, D.F.; Fliers, E. Glucocorticoids decrease thyrotropin-releasing hormone messenger ribonucleic acid expression in the paraventricular nucleus of the human hypothalamus. J. Clin. Endocrinol. Metab. 2005, 90, 323–327. [Google Scholar] [CrossRef]
- Alkemade, A.; Unmehopa, U.A.; Brouwer, J.P.; Hoogendijk, W.J.; Wiersinga, W.M.; Swaab, D.F.; Fliers, E. Decreased thyrotropin-releasing hormone gene expression in the hypothalamic paraventricular nucleus of patients with major depression. Mol. Psychiatry 2003, 8, 838–839. [Google Scholar] [CrossRef][Green Version]
- Ahlquist, J.; Franklyn, J.; Ramsden, D.; Sheppard, M. The influence of dexamethasone on serum thyrotrophin and thyrotrophin synthesis in the rat. Mol. Cell. Endocrinol. 1989, 64, 55–61. [Google Scholar] [CrossRef]
- Taylor, A.D.; Philip, J.G.; John, C.D.; Cover, P.O.; Morris, J.F.; Flower, R.J.; Buckingham, J.C. Annexin 1 (lipocortin 1) mediates the glucocorticoid inhibition of cyclic adenosine 3′, 5′-monophosphate-stimulated prolactin secretion. Endocrinology 2000, 141, 2209–2219. [Google Scholar] [CrossRef] [PubMed]
- Traverso, V.; Christian, H.C.; Morris, J.F.; Buckingham, J.C. Lipocortin 1 (annexin 1): A candidate paracrine agent localized in pituitary folliculo-stellate cells. Endocrinology 1999, 140, 4311–4319. [Google Scholar] [CrossRef] [PubMed]
- Eigler, T.; Ben-Shlomo, A. Somatostatin system: Molecular mechanisms regulating anterior pituitary hormones. J. Mol. Endocrinol. 2014, 53, R1–R19. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, K.; Ishizuka, T.; Shimizu, C.; Ito, Y.; Wakabayashi, I. Increased hypothalamic somatostatin mrna following dexamethasone administration in rats. Acta Endocrinol. 1992, 127, 416–419. [Google Scholar] [CrossRef]
- Adriaanse, R.; Brabant, G.; Endert, E.; Wiersinga, W.M. Pulsatile thyrotropin secretion in patients with cushing’s syndrome. Metabolism 1994, 43, 782–786. [Google Scholar] [CrossRef]
- Bartalena, L.; Martino, E.; Petrini, L.; Velluzzi, F.; Loviselli, A.; Grasso, L.; Mammoli, C.; Pinchera, A. The nocturnal serum thyrotropin surge is abolished in patients with adrenocorticotropin (acth)-dependent or acth-independent cushing’s syndrome. J. Clin. Endocrinol. Metab. 1991, 72, 1195–1199. [Google Scholar] [CrossRef] [PubMed]
- Tamada, D.; Kitamura, T.; Takahara, M.; Tanaka, T.; Takeda, M.; Otsuki, M.; Shimomura, I. Tsh ratio as a novel diagnostic method for cushing’s syndrome. Endocr. J. 2018, 65, 841–848. [Google Scholar] [CrossRef] [PubMed]
- Duick, D.S.; Wahner, H.W. Thyroid axis in patients with cushing’s syndrome. Arch. Intern. Med. 1979, 139, 767–772. [Google Scholar] [CrossRef] [PubMed]
- Mathioudakis, N.; Thapa, S.; Wand, G.S.; Salvatori, R. Acth-secreting pituitary microadenomas are associated with a higher prevalence of central hypothyroidism compared to other microadenoma types. Clin. Endocrinol. 2012, 77, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Heyma, P.; Larkins, R.G. Glucocorticoids decrease in conversion of thyroxine into 3, 5, 3′-tri-iodothyronine by isolated rat renal tubules. Clin. Sci. 1982, 62, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Shekhar, S.; McGlotten, R.; Auh, S.; Rother, K.I.; Nieman, L.K. The hypothalamic-pituitary-thyroid axis in cushing syndrome before and after curative surgery. J. Clin. Endocrinol. Metab. 2020. [Google Scholar] [CrossRef]
- Roelfsema, F.; Pereira, A.M.; Biermasz, N.R.; Frolich, M.; Keenan, D.M.; Veldhuis, J.D.; Romijn, J.A. Diminished and irregular tsh secretion with delayed acrophase in patients with cushing’s syndrome. Eur. J. Endocrinol. 2009, 161, 695–703. [Google Scholar] [CrossRef]
- Dogansen, S.C.; Yalin, G.Y.; Canbaz, B.; Tanrikulu, S.; Yarman, S. Dynamic changes of central thyroid functions in the management of cushing’s syndrome. Arch. Endocrinol. Metab. 2018, 62, 164–171. [Google Scholar] [CrossRef]
- Colao, A.; Pivonello, R.; Faggiano, A.; Filippella, M.; Ferone, D.; Di Somma, C.; Cerbone, G.; Marzullo, P.; Fenzi, G.; Lombardi, G. Increased prevalence of thyroid autoimmunity in patients successfully treated for cushing’s disease. Clin. Endocrinol. 2000, 53, 13–19. [Google Scholar] [CrossRef]
- Xiang, B.; Tao, R.; Liu, X.; Zhu, X.; He, M.; Ma, Z.; Yang, Y.; Zhang, Z.; Li, Y.; Yao, Z.; et al. A study of thyroid functions in patients with cushing’s syndrome: A single-center experience. Endocr. Connect 2019, 8, 1176–1185. [Google Scholar] [CrossRef]
- Krassas, G.E.; Pontikides, N.; Kaltsas, T.; Papadopoulou, P.; Paunkovic, J.; Paunkovic, N.; Duntas, L.H. Disturbances of menstruation in hypothyroidism. Clin. Endocrinol. 1999, 50, 655–659. [Google Scholar] [CrossRef] [PubMed]
- Cai, R.; Zhou, W.; Jiang, L.; Jiang, Y.; Su, T.; Zhang, C.; Zhou, W.; Ning, G.; Wang, W. Association between thyroid function and serum cortisol in cortisol-producing adenoma patients. Endocrine 2020, 69, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Niepomniszcze, H.; Pitoia, F.; Katz, S.B.; Chervin, R.; Bruno, O.D. Primary thyroid disorders in endogenous cushing’s syndrome. Eur. J. Endocrinol. 2002, 147, 305–311. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pivonello, R.; De Martino, M.C.; De Leo, M.; Tauchmanova, L.; Faggiano, A.; Lombardi, G.; Colao, A. Cushing’s syndrome: Aftermath of the cure. Arq. Bras. Endocrinol. Metabol. 2007, 51, 1381–1391. [Google Scholar] [CrossRef]
- Takasu, N.; Komiya, I.; Nagasawa, Y.; Asawa, T.; Yamada, T. Exacerbation of autoimmune thyroid dysfunction after unilateral adrenalectomy in patients with cushing’s syndrome due to an adrenocortical adenoma. N. Engl. J. Med. 1990, 322, 1708–1712. [Google Scholar] [CrossRef]
- Takasu, N.; Ohara, N.; Yamada, T.; Komiya, I. Development of autoimmune thyroid dysfunction after bilateral adrenalectomy in a patient with carney’s complex and after removal of acth-producing pituitary adenoma in a patient with cushing’s disease. J. Endocrinol. Investig. 1993, 16, 697–702. [Google Scholar] [CrossRef]
- McGregor, A.M. Immunoendocrine interactions and autoimmunity. N. Engl. J. Med. 1990, 322, 1739–1741. [Google Scholar] [CrossRef]
- Tatsi, C.; Keil, M.; Lyssikatos, C.; Belyavskaya, E.; Stratakis, C.A.; Lodish, M.B. Incidence of autoimmune and related disorders after resolution of endogenous cushing syndrome in children. Horm. Metab. Res. 2018, 50, 290–295. [Google Scholar] [CrossRef]
- Candrina, R.; Di Stefano, O. Exacerbation of celiac disease after cure of cushing’s disease. Am. J. Med. 1993, 95. [Google Scholar] [CrossRef]
- Yakushiji, F.; Kita, M.; Hiroi, N.; Ueshiba, H.; Monma, I.; Miyachi, Y. Exacerbation of rheumatoid arthritis after removal of adrenal adenoma in cushing’s syndrome. Endocr. J. 1995, 42, 219–223. [Google Scholar] [CrossRef][Green Version]
- Russo, L.; Vitti, P.; Pinchera, A.; Marino, M. Exacerbation of autoimmune thyroiditis following bilateral adrenalectomy for cushing’s syndrome. Thyroid 2010, 20, 669–670. [Google Scholar] [CrossRef] [PubMed]
- Hiromatsu, Y.; Eguchi, H.; Nakamura, Y.; Mukohara, K. A case of graves’ disease after adrenalectomy for cushing’s syndrome. Intern. Med. 2020. [Google Scholar] [CrossRef]
- Sigmarsdottir, A.A.; Olafsson, I.H.; Kjartansson, O.; Bjarnason, R. Thyrotoxicosis in a 13-year-old girl following pituitary adenectomy for cushing’s disease. Clin. Case Rep. 2017, 5, 1341–1343. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Arikan, E.; Guldiken, S.; Altun, B.U.; Kara, M.; Tugrul, A. Exacerbations of graves’ disease after unilateral adrenalectomy for cushing’s syndrome. J. Endocrinol. Investig. 2004, 27, 574–576. [Google Scholar] [CrossRef] [PubMed]
- Morita, H.; Isaji, M.; Mune, T.; Daido, H.; Isomura, Y.; Sarui, H.; Tanahashi, T.; Takeda, N.; Ishizuka, T.; Yasuda, K. Transient graves disease developing after surgery for cushing disease. Am. J. Med. Sci. 2002, 323, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Mussig, K.; Gallwitz, B.; Haring, H.U.; Seif, F.J. Manifestation of thyroid autoimmunity in patients successfully treated for hypercortisolism. Clin. Endocrinol. 2004, 61, 284. [Google Scholar] [CrossRef]
- Stojic-Vukanic, Z.; Rauski, A.; Kosec, D.; Radojevic, K.; Pilipovic, I.; Leposavic, G. Dysregulation of t-cell development in adrenal glucocorticoid-deprived rats. Exp. Biol. Med. 2009, 234, 1067–1074. [Google Scholar] [CrossRef] [PubMed]
- Vita, R.; Lapa, D.; Trimarchi, F.; Benvenga, S. Stress triggers the onset and the recurrences of hyperthyroidism in patients with graves’ disease. Endocrine 2015, 48, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Desailloud, R.; Hober, D. Viruses and thyroiditis: An update. Virol. J. 2009, 6, 5. [Google Scholar] [CrossRef]
- Efstathiadou, Z.A.; Sykja, A.; Anagnostis, P.; Panagiotou, A.; Kita, M. Occurrence of de quervain’s thyroiditis after resolution of hypercortisolism following pasireotide treatment for cushing’s disease and surgery for an adrenocortical adenoma: Report of two cases. Eur. Thyroid. J. 2014, 3, 69–72. [Google Scholar] [CrossRef]
- Elenkov, I.J. Glucocorticoids and the th1/th2 balance. Ann. N. Y. Acad. Sci. 2004, 1024, 138–146. [Google Scholar] [CrossRef]
- Tamada, D.; Onodera, T.; Kitamura, T.; Yamamoto, Y.; Hayashi, Y.; Murata, Y.; Otsuki, M.; Shimomura, I. Hyperthyroidism due to thyroid-stimulating hormone secretion after surgery for cushing’s syndrome: A novel cause of the syndrome of inappropriate secretion of thyroid-stimulating hormone. J. Clin. Endocrinol. Metab. 2013, 98, 2656–2662. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.J.; Fiering, S.N.; Pallud, S.E.; Parlow, A.F.; St Germain, D.L.; Galton, V.A. Targeted disruption of the type 2 selenodeiodinase gene (dio2) results in a phenotype of pituitary resistance to t4. Mol. Endocrinol. 2001, 15, 2137–2148. [Google Scholar] [CrossRef] [PubMed]
- Nieman, L.K.; Biller, B.M.; Findling, J.W.; Murad, M.H.; Newell-Price, J.; Savage, M.O.; Tabarin, A.; Endocrine, S. Treatment of cushing’s syndrome: An endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 2015, 100, 2807–2831. [Google Scholar] [CrossRef]
- Gatto, F.; Arvigo, M.; Ferone, D. Somatostatin receptor expression and patients’ response to targeted medical treatment in pituitary tumors: Evidences and controversies. J. Endocrinol. Investig. 2020, 43, 1543–1553. [Google Scholar] [CrossRef] [PubMed]
- Kienitz, T.; Quinkler, M.; Strasburger, C.J.; Ventz, M. Long-term management in five cases of tsh-secreting pituitary adenomas: A single center study and review of the literature. Eur. J. Endocrinol. 2007, 157, 39–46. [Google Scholar] [CrossRef]
- Bruns, C.; Lewis, I.; Briner, U.; Meno-Tetang, G.; Weckbecker, G. Som230: A novel somatostatin peptidomimetic with broad somatotropin release inhibiting factor (srif) receptor binding and a unique antisecretory profile. Eur. J. Endocrinol. 2002, 146, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Gatto, F.; Arvigo, M.; Amaru, J.; Campana, C.; Cocchiara, F.; Graziani, G.; Bruzzone, E.; Giusti, M.; Boschetti, M.; Ferone, D. Cell specific interaction of pasireotide: Review of preclinical studies in somatotroph and corticotroph pituitary cells. Pituitary 2019, 22, 89–99. [Google Scholar] [CrossRef]
- Pivonello, R.; Ferrigno, R.; De Martino, M.C.; Simeoli, C.; Di Paola, N.; Pivonello, C.; Barba, L.; Negri, M.; De Angelis, C.; Colao, A. Medical treatment of cushing’s disease: An overview of the current and recent clinical trials. Front. Endocrinol. 2020, 11, 648. [Google Scholar] [CrossRef]
- Murray, R.D.; Kim, K.; Ren, S.G.; Lewis, I.; Weckbecker, G.; Bruns, C.; Melmed, S. The novel somatostatin ligand (som230) regulates human and rat anterior pituitary hormone secretion. J. Clin. Endocrinol. Metab. 2004, 89, 3027–3032. [Google Scholar] [CrossRef][Green Version]
- Giusti, M.; Lomeo, A.; Torre, R.; Sghedoni, D.; Mazzocchi, G.; Durante, R.; Giordano, G. Effect of subacute cabergoline treatment on prolactin, thyroid stimulating hormone and growth hormone response to simultaneous administration of thyrotrophin-releasing hormone and growth hormone-releasing hormone in hyperprolactinaemic women. Clin. Endocrinol. 1989, 30, 315–321. [Google Scholar] [CrossRef]
- Daniel, E.; Newell-Price, J.D. Therapy of endocrine disease: Steroidogenesis enzyme inhibitors in cushing’s syndrome. Eur. J. Endocrinol. 2015, 172, R263–R280. [Google Scholar] [CrossRef] [PubMed]
- Samuels, M.H. Effects of metyrapone administration on thyrotropin secretion in healthy subjects--a clinical research center study. J. Clin. Endocrinol. Metab. 2000, 85, 3049–3052. [Google Scholar] [CrossRef]
- Comby, F.; Lagorce, J.F.; Buxeraud, J.; Raby, C. Antithyroid action of ketoconazole: In-vitro studies and rat in-vivo studies. J. Pharm. Pharmacol. 1994, 46, 50–53. [Google Scholar] [CrossRef] [PubMed]
- Caksen, H.; Tutus, A.; Kurtoglu, S.; Ozturk, F.; Okumus, Y.; Coksevim, B. Low dose ketoconazole therapy and thyroid functions in rats. Acta Medica 2002, 45, 177–179. [Google Scholar]
- Paragliola, R.M.; Corsello, A.; Locantore, P.; Papi, G.; Pontecorvi, A.; Corsello, S.M. Medical approaches in adrenocortical carcinoma. Biomedicines 2020, 8, 551. [Google Scholar] [CrossRef] [PubMed]
- Zatelli, M.C.; Gentilin, E.; Daffara, F.; Tagliati, F.; Reimondo, G.; Carandina, G.; Ambrosio, M.R.; Terzolo, M.; Degli Uberti, E.C. Therapeutic concentrations of mitotane (o,p’-ddd) inhibit thyrotroph cell viability and tsh expression and secretion in a mouse cell line model. Endocrinology 2010, 151, 2453–2461. [Google Scholar] [CrossRef] [PubMed]
- Vikner, M.E.; Krogh, J.; Daugaard, G.; Andreassen, M. Metabolic and hormonal side effects of mitotane treatment for adrenocortical carcinoma: A retrospective study in 50 danish patients. Clin. Endocrinol. 2021, 94, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Daffara, F.; De Francia, S.; Reimondo, G.; Zaggia, B.; Aroasio, E.; Porpiglia, F.; Volante, M.; Termine, A.; Di Carlo, F.; Dogliotti, L.; et al. Prospective evaluation of mitotane toxicity in adrenocortical cancer patients treated adjuvantly. Endocr. Relat. Cancer 2008, 15, 1043–1053. [Google Scholar] [CrossRef]
- Takiyama, Y.; Tanaka, H.; Takiyama, Y.; Makino, I. The effects of hydrocortisone and ru486 (mifepristone) on iodide uptake in porcine thyroid cells in primary culture. Endocrinology 1994, 135, 1972–1979. [Google Scholar] [CrossRef]
- Heikinheimo, O.; Ranta, S.; Grunberg, S.; Lähteenmäki, P.; Spitz, I.M. Alterations in the pituitary-thyroid and pituitary-adrenal axes—consequences of long-term mifepristone treatment. Metabolism 1997, 46, 292–296. [Google Scholar] [CrossRef]
- Fleseriu, M.; Biller, B.M.; Findling, J.W.; Molitch, M.E.; Schteingart, D.E.; Gross, C.; Investigators, S.S. Mifepristone, a glucocorticoid receptor antagonist, produces clinical and metabolic benefits in patients with cushing’s syndrome. J. Clin. Endocrinol. Metab. 2012, 97, 2039–2049. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.R.; East, H.E.; Eilerman, B.S.; Gordon, M.B.; King, E.E.; Knecht, L.A.; Salke, B.; Samson, S.L.; Yuen, K.C.J.; Yau, H. Clinical management of patients with cushing syndrome treated with mifepristone: Consensus recommendations. Clin. Diabetes Endocrinol. 2020, 6, 18. [Google Scholar] [CrossRef] [PubMed]
- Guarda, F.J.; Findling, J.; Yuen, K.C.J.; Fleseriu, M.; Nachtigall, L.B. Mifepristone increases thyroid hormone requirements in patients with central hypothyroidism: A multicenter study. J. Endocr. Soc. 2019, 3, 1707–1714. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Drug | Test Results | Condition Mimicked | ||
|---|---|---|---|---|
| TSH | FT4 | FT3 | ||
| Pasireotide | NR | NR | NR | - |
| Cabergoline | NR | NR | NR | - |
| Metyrapone | 35% increase compared to baseline * | Normal | Normal | - |
| Ketoconazole | NR | NR | NR | - |
| Mitotane | Normal | Low | Normal | Central hypothyroidism |
| Mifepristone | High | Normal | NR | Subclinical hypothyroidism |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paragliola, R.M.; Corsello, A.; Papi, G.; Pontecorvi, A.; Corsello, S.M. Cushing’s Syndrome Effects on the Thyroid. Int. J. Mol. Sci. 2021, 22, 3131. https://doi.org/10.3390/ijms22063131
Paragliola RM, Corsello A, Papi G, Pontecorvi A, Corsello SM. Cushing’s Syndrome Effects on the Thyroid. International Journal of Molecular Sciences. 2021; 22(6):3131. https://doi.org/10.3390/ijms22063131
Chicago/Turabian StyleParagliola, Rosa Maria, Andrea Corsello, Giampaolo Papi, Alfredo Pontecorvi, and Salvatore Maria Corsello. 2021. "Cushing’s Syndrome Effects on the Thyroid" International Journal of Molecular Sciences 22, no. 6: 3131. https://doi.org/10.3390/ijms22063131
APA StyleParagliola, R. M., Corsello, A., Papi, G., Pontecorvi, A., & Corsello, S. M. (2021). Cushing’s Syndrome Effects on the Thyroid. International Journal of Molecular Sciences, 22(6), 3131. https://doi.org/10.3390/ijms22063131