
Pseudomonas aeruginosa: An Audacious Pathogen with an Adaptable Arsenal of Virulence Factors


Abstract

1. Introduction
2. Pseudomonas aeruginosa Virulence Factors: A Wealth of Weaponry
2.1. The Outer Membrane: Lipopolysaccharide and Proteins
2.1.1. Lipopolysaccharide
2.1.2. Outer Membrane Proteins
Porins: OprF, OprH, and OprD Superfamily
Lipoproteins
2.2. Biofilm Formation
Alginate
2.3. Flagellum
2.4. Type IV Pili
2.5. Protein Secretion Systems
2.5.1. Type III Secretion System
Effector-Dependent Pathogenicity
Effector-Independent Pathogenicity
2.6. Other Released Products
2.6.1. Exotoxin A
2.6.2. Proteolytic Enzymes
2.6.3. Lipolytic Enzymes
2.6.4. Pyocyanin
2.7. Other Bacterial Products
2.7.1. Rhamnolipids
2.7.2. Antioxidant Enzymes
2.8. Iron Acquisition Systems
Siderophores: Pyoverdine and Pyochelin
2.9. Quorum Sensing
2.9.1. Acyl-Homoserine Lactone QS Systems: Las and Rhl
2.9.2. The Quinolone QS System: Pqs
2.9.3. The Novel QS System: Iqs
3. CF Lung Environment
4. Bacterial Adaptation within the Lung
4.1. Emergence of Hypermutators
4.2. Phenotypic Diversity and Morphology Variants
4.3. Mucoid Phenotype Switch and Sessile-Biofilm Lifestyle
4.4. Loss of O-Antigen and Structural Modifications of Lipid A
4.5. Lack of Motility and Non-Flagellated, Non-Piliated Phenotype
4.6. Selection against T3SS and Loss of Cytotoxicity
4.7. Reduced Communication Systems
4.8. Specialised Metabolism
4.9. Change of Iron Uptake Strategy
4.10. Acquisition of Antibiotic Resistance
5. Genomic and Phenotypic Approaches to the Study of P. aeruginosa Adaptation within the CF Lung
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Riquelme, S.A.; Liimatta, K.; Wong Fok Lung, T.; Fields, B.; Ahn, D.; Chen, D.; Lozano, C.; Sáenz, Y.; Uhlemann, A.C.; Kahl, B.C.; et al. Pseudomonas aeruginosa Utilizes Host-Derived Itaconate to Redirect Its Metabolism to Promote Biofilm Formation. Cell Metab. 2020, 31, 1091–1106. [Google Scholar] [CrossRef]
- Fernández-Barat, L.; Ferrer, M.; De Rosa, F.; Gabarrús, A.; Esperatti, M.; Terraneo, S.; Rinaudo, M.; Li Bassi, G.; Torres, A. Intensive care unit-acquired pneumonia due to Pseudomonas aeruginosa with and without multidrug resistance. J. Infect. 2017, 74, 142–152. [Google Scholar] [CrossRef]
- Wunderink, R.G.; Waterer, G. Advances in the causes and management of community acquired pneumonia in adults. BMJ 2017, 358, j2471. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Nuñez, M.; Marti, S.; Puig, C.; Perez-Brocal, V.; Millares, L.; Santos, S.; Ardanuy, C.; Moya, A.; Liñares, J.; Monsó, E. Bronchial microbiome, PA biofilm-forming capacity and exacerbation in severe COPD patients colonized by P. aeruginosa. Future Microbiol. 2017, 12, 379–392. [Google Scholar] [CrossRef] [PubMed]
- Winstanley, C.; O’Brien, S.; Brockhurst, M.A. Pseudomonas aeruginosa Evolutionary Adaptation and Diversification in Cystic Fibrosis Chronic Lung Infections. Trends Microbiol. 2016, 24, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Kubes, J.N.; Fridkin, S.K. Factors affecting the geographic variability of antibiotic-resistant healthcare-associated infections in the United States using the CDC Antibiotic Resistance Patient Safety Atlas. Infect. Control. Hosp. Epidemiol. 2019, 40, 597–599. [Google Scholar] [CrossRef] [PubMed]
- Wiehlmann, L.; Wagner, G.; Cramer, N.; Siebert, B.; Gudowius, P.; Morales, G.; Köhler, T.; van Delden, C.; Weinel, C.; Slickers, P.; et al. Population structure of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2007, 104, 8101–8106. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Guidelines for the Prevention and Control of Carbapenem-Resistant Acinetobacter baumannii and Pseudomonas aeruginosa in Health Care Facilities; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef]
- Botelho, J.; Grosso, F.; Peixe, L. Antibiotic resistance in Pseudomonas aeruginosa—Mechanisms, epidemiology and evolution. Drug Resist. Updat. 2019, 44, 100640. [Google Scholar] [CrossRef]
- Moradali, M.F.; Ghods, S.; Rehm, B.H. Lifestyle: A Paradigm for Adaptation, Survival, and Persistence. Front. Cell Infect. Microbiol. 2017, 7, 39. [Google Scholar] [CrossRef]
- Maurice, N.M.; Bedi, B.; Sadikot, R.T. Pseudomonas aeruginosa Biofilms: Host Response and Clinical Implications in Lung Infections. Am. J. Respir. Cell Mol. Biol. 2018, 58, 428–439. [Google Scholar] [CrossRef]
- Francis, V.I.; Stevenson, E.C.; Porter, S.L. Two-component systems required for virulence in Pseudomonas aeruginosa. FEMS Microbiol. Lett. 2017, 364. [Google Scholar] [CrossRef]
- Sainz-Mejías, M.; Jurado-Martín, I.; McClean, S. Understanding Pseudomonas aeruginosa-Host Interactions: The Ongoing Quest for an Efficacious Vaccine. Cells 2020, 9, 2617. [Google Scholar] [CrossRef]
- Riquelme, S.A.; Ahn, D.; Prince, A. Pseudomonas aeruginosa and Klebsiella pneumoniae Adaptation to Innate Immune Clearance Mechanisms in the Lung. J. Innate Immun. 2018, 10, 442–454. [Google Scholar] [CrossRef]
- Elborn, J.S. Cystic fibrosis. Lancet 2016, 388, 2519–2531. [Google Scholar] [CrossRef]
- Riquelme, S.A.; Hopkins, B.D.; Wolfe, A.L.; DiMango, E.; Kitur, K.; Parsons, R.; Prince, A. Cystic Fibrosis Transmembrane Conductance Regulator Attaches Tumor Suppressor PTEN to the Membrane and Promotes Anti Pseudomonas aeruginosa Immunity. Immunity 2017, 47, 1169–1181. [Google Scholar] [CrossRef] [PubMed]
- Reece, E.; Segurado, R.; Jackson, A.; McClean, S.; Renwick, J.; Greally, P. Co-colonisation with Aspergillus fumigatus and Pseudomonas aeruginosa is associated with poorer health in cystic fibrosis patients: An Irish registry analysis. BMC Pulm. Med. 2017, 17, 70. [Google Scholar] [CrossRef]
- Cigana, C.; Lorè, N.I.; Riva, C.; De Fino, I.; Spagnuolo, L.; Sipione, B.; Rossi, G.; Nonis, A.; Cabrini, G.; Bragonzi, A. Tracking the immunopathological response to Pseudomonas aeruginosa during respiratory infections. Sci. Rep. 2016, 6, 21465. [Google Scholar] [CrossRef]
- Smith, E.E.; Buckley, D.G.; Wu, Z.; Saenphimmachak, C.; Hoffman, L.R.; D’Argenio, D.A.; Miller, S.I.; Ramsey, B.W.; Speert, D.P.; Moskowitz, S.M.; et al. Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proc. Natl. Acad. Sci. USA 2006, 103, 8487–8492. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, P.N.; Koch, G.; Thompson, J.A.; Xavier, K.B.; Cool, R.H.; Quax, W.J. The multiple signaling systems regulating virulence in Pseudomonas aeruginosa. Microbiol. Mol. Biol. Rev. 2012, 76, 46–65. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, D.; Schneper, L.; Kumari, H.; Mathee, K. A dynamic and intricate regulatory network determines Pseudomonas aeruginosa virulence. Nucleic Acids Res. 2013, 41, 1–20. [Google Scholar] [CrossRef]
- Montor, W.R.; Huang, J.; Hu, Y.; Hainsworth, E.; Lynch, S.; Kronish, J.W.; Ordonez, C.L.; Logvinenko, T.; Lory, S.; LaBaer, J. Genome-wide study of Pseudomonas aeruginosa outer membrane protein immunogenicity using self-assembling protein microarrays. Infect. Immun. 2009, 77, 4877–4886. [Google Scholar] [CrossRef] [PubMed]
- Remans, K.; Vercammen, K.; Bodilis, J.; Cornelis, P. Genome-wide analysis and literature-based survey of lipoproteins in Pseudomonas aeruginosa. Microbiology 2010, 156, 2597–2607. [Google Scholar] [CrossRef] [PubMed]
- Bianconi, I.; Alcalá-Franco, B.; Scarselli, M.; Dalsass, M.; Buccato, S.; Colaprico, A.; Marchi, S.; Masignani, V.; Bragonzi, A. Genome-Based Approach Delivers Vaccine Candidates Against Pseudomonas aeruginosa. Front. Immunol. 2018, 9, 3021. [Google Scholar] [CrossRef]
- Pang, Z.; Raudonis, R.; Glick, B.R.; Lin, T.J.; Cheng, Z. Antibiotic resistance in Pseudomonas aeruginosa: Mechanisms and alternative therapeutic strategies. Biotechnol. Adv. 2019, 37, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, R.F.; Sá-Correia, I.; Valvano, M.A. Lipopolysaccharide modification in Gram-negative bacteria during chronic infection. FEMS Microbiol. Rev. 2016, 40, 480–493. [Google Scholar] [CrossRef]
- Huszczynski, S.M.; Lam, J.S.; Khursigara, C.M. The Role of Pseudomonas aeruginosa Lypopolisaccharide in Bacterial Pathogenesis and Phisiology. Pathogens 2019, 9, 6. [Google Scholar] [CrossRef] [PubMed]
- King, J.D.; Kocíncová, D.; Westman, E.L.; Lam, J.S. Lipopolysaccharide biosynthesis in Pseudomonas aeruginosa. Innate Immun. 2009, 15, 261–312. [Google Scholar] [CrossRef]
- Yan, F.; Li, W.; Jono, H.; Li, Q.; Zhang, S.; Li, J.D.; Shen, H. Reactive oxygen species regulate Pseudomonas aeruginosa lipopolysaccharide-induced MUC5AC mucin expression via PKC-NADPH oxidase-ROS-TGF-alpha signaling pathways in human airway epithelial cells. Biochem. Biophys. Res. Commun. 2008, 366, 513–519. [Google Scholar] [CrossRef]
- Li, W.; Yan, F.; Zhou, H.; Lin, X.; Wu, Y.; Chen, C.; Zhou, N.; Chen, Z.; Li, J.D.; Shen, H.P. aeruginosa lipopolysaccharide-induced MUC5AC and CLCA3 expression is partly through Duox1 in vitro and in vivo. PLoS ONE 2013, 8, e63945. [Google Scholar] [CrossRef]
- Eutamene, H.; Theodorou, V.; Schmidlin, F.; Tondereau, V.; Garcia-Villar, R.; Salvador-Cartier, C.; Chovet, M.; Bertrand, C.; Bueno, L. LPS-induced lung inflammation is linked to increased epithelial permeability: Role of MLCK. Eur. Respir. J. 2005, 25, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Wieland, C.W.; Siegmund, B.; Senaldi, G.; Vasil, M.L.; Dinarello, C.A.; Fantuzzi, G. Pulmonary inflammation induced by Pseudomonas aeruginosa lipopolysaccharide, phospholipase C, and exotoxin A: Role of interferon regulatory factor 1. Infect. Immun. 2002, 70, 1352–1358. [Google Scholar] [CrossRef]
- Lam, J.S.; Taylor, V.L.; Islam, S.T.; Hao, Y.; Kocíncová, D. Genetic and Functional Diversity of Pseudomonas aeruginosa Lipopolysaccharide. Front. Microbiol. 2011, 2, 118. [Google Scholar] [CrossRef] [PubMed]
- Park, B.S.; Lee, J.O. Recognition of lipopolysaccharide pattern by TLR4 complexes. Exp. Mol. Med. 2013, 45, e66. [Google Scholar] [CrossRef] [PubMed]
- Florez, C.; Raab, J.E.; Cooke, A.C.; Schertzer, J.W. Membrane Distribution of the Pseudomonas Quinolone Signal Modulates Outer Membrane Vesicle Production in Pseudomonas aeruginosa. mBio 2017, 8. [Google Scholar] [CrossRef]
- Alshalchi, S.A.; Anderson, G.G. Expression of the lipopolysaccharide biosynthesis gene lpxD affects biofilm formation of Pseudomonas aeruginosa. Arch. Microbiol. 2015, 197, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Pieterse, E.; Rother, N.; Yanginlar, C.; Hilbrands, L.B.; van der Vlag, J. Neutrophils Discriminate between Lipopolysaccharides of Different Bacterial Sources and Selectively Release Neutrophil Extracellular Traps. Front. Immunol. 2016, 7, 484. [Google Scholar] [CrossRef]
- Murphy, K.; Park, A.J.; Hao, Y.; Brewer, D.; Lam, J.S.; Khursigara, C.M. Influence of O polysaccharides on biofilm development and outer membrane vesicle biogenesis in Pseudomonas aeruginosa PAO1. J. Bacteriol. 2014, 196, 1306–1317. [Google Scholar] [CrossRef]
- Lindhout, T.; Lau, P.C.Y.; Brewer, D.; Lam, J.S. Truncation in the core oligosaccharide of lipopolysaccharide affects flagella-mediated motility in Pseudomonas aeruginosa PAO1 via modulation of cell surface attachment. Microbiology 2009, 155, 3449–3460. [Google Scholar] [CrossRef] [PubMed]
- Jamasbi, R.J.; Taylor, N.M. Correlation Between the Lipopolysaccharide Expression and Adhesiveness of Clinical Isolates of Pseudomonas aeruginosa. Lab. Med. 2010, 41, 24–30. [Google Scholar] [CrossRef]
- Chevalier, S.; Bouffartigues, E.; Bodilis, J.; Maillot, O.; Lesouhaitier, O.; Feuilloley, M.G.J.; Orange, N.; Dufour, A.; Cornelis, P. Structure, function and regulation of Pseudomonas aeruginosa porins. FEMS Microbiol. Rev. 2017, 41, 698–722. [Google Scholar] [CrossRef] [PubMed]
- Cassin, E.K.; Tseng, B.S. Pushing beyond the Envelope: The Potential Roles of OprF in Pseudomonas aeruginosa Biofilm Formation and Pathogenicity. J. Bacteriol. 2019, 201, e00050-19. [Google Scholar] [CrossRef] [PubMed]
- Navare, A.T.; Chavez, J.D.; Zheng, C.; Weisbrod, C.R.; Eng, J.K.; Siehnel, R.; Singh, P.K.; Manoil, C.; Bruce, J.E. Probing the protein interaction network of Pseudomonas aeruginosa cells by chemical cross-linking mass spectrometry. Structure 2015, 23, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Fito-Boncompte, L.; Chapalain, A.; Bouffartigues, E.; Chaker, H.; Lesouhaitier, O.; Gicquel, G.; Bazire, A.; Madi, A.; Connil, N.; Véron, W.; et al. Full virulence of Pseudomonas aeruginosa requires OprF. Infect. Immun. 2011, 79, 1176–1186. [Google Scholar] [CrossRef] [PubMed]
- Bukhari, S.I.; Aleanizy, F.S. Association of OprF mutant and disturbance of biofilm and pyocyanin virulence in Pseudomonas aeruginosa. Saudi Pharm. J. 2020, 28, 196–200. [Google Scholar] [CrossRef] [PubMed]
- McClean, S. Eight stranded B-barrel and related outer membrane proteins: Role in bacterial pathogenesis. Protein Pept. Lett. 2012, 19, 1013–1025. [Google Scholar] [CrossRef]
- Song, F.; Wang, H.; Sauer, K.; Ren, D. Cyclic-di-GMP and OprF Are Involved in the Response of Pseudomonas aeruginosa to Substrate Material Stiffness during Attachment on Polydimethylsiloxane (PDMS). Front. Microbiol. 2018, 9, 110. [Google Scholar] [CrossRef]
- Bouffartigues, E.; Moscoso, J.A.; Duchesne, R.; Rosay, T.; Fito-Boncompte, L.; Gicquel, G.; Maillot, O.; Bénard, M.; Bazire, A.; Brenner-Weiss, G.; et al. The absence of the Pseudomonas aeruginosa OprF protein leads to increased biofilm formation through variation in c-di-GMP level. Front. Microbiol. 2015, 6, 630. [Google Scholar] [CrossRef]
- Garai, P.; Berry, L.; Moussouni, M.; Bleves, S.; Blanc-Potard, A.B. Killing from the inside: Intracellular role of T3SS in the fate of Pseudomonas aeruginosa within macrophages revealed by mgtC and OprF mutants. PLoS Pathog. 2019, 15, e1007812. [Google Scholar] [CrossRef]
- Mishra, M.; Ressler, A.; Schlesinger, L.S.; Wozniak, D.J. Identification of OprF as a complement component C3 binding acceptor molecule on the surface of Pseudomonas aeruginosa. Infect. Immun. 2015, 83, 3006–3014. [Google Scholar] [CrossRef]
- Moussouni, M.; Berry, L.; Sipka, T.; Nguyen-Chi, M.; Blanc-Potard, A.B. Pseudomonas aeruginosa OprF plays a role in resistance to macrophage clearance during acute infection. Sci. Rep. 2021, 11, 359. [Google Scholar] [CrossRef] [PubMed]
- Qadi, M.; Lopez-Causapé, C.; Izquierdo-Rabassa, S.; Mateu Borrás, M.; Goldberg, J.B.; Oliver, A.; Albertí, S. Surfactant Protein A Recognizes Outer Membrane Protein OprH on Pseudomonas aeruginosa Isolates from Individuals With Chronic Infection. J. Infect. Dis. 2016, 214, 1449–1455. [Google Scholar] [CrossRef] [PubMed]
- Paulsson, M.; Su, Y.C.; Ringwood, T.; Uddén, F.; Riesbeck, K. Pseudomonas aeruginosa uses multiple receptors for adherence to laminin during infection of the respiratory tract and skin wounds. Sci. Rep. 2019, 9, 18168. [Google Scholar] [CrossRef]
- Arhin, A.; Boucher, C. The outer membrane protein OprQ and adherence of Pseudomonas aeruginosa to human fibronectin. Microbiology 2010, 156, 1415–1423. [Google Scholar] [CrossRef] [PubMed]
- Panmanee, W.; Gomez, F.; Witte, D.; Pancholi, V.; Britigan, B.E.; Hassett, D.J. The peptidoglycan-associated lipoprotein OprL helps protect a Pseudomonas aeruginosa mutant devoid of the transactivator OxyR from hydrogen peroxide-mediated killing during planktonic and biofilm culture. J. Bacteriol. 2008, 190, 3658–3669. [Google Scholar] [CrossRef] [PubMed]
- Auda, I.G.; Ali Salman, I.M.; Auda, J.G. Efflux pumps of Gram-negative bacteria in brief. Gene Rep. 2020, 20. [Google Scholar] [CrossRef]
- Kucharska, I.; Liang, B.; Ursini, N.; Tamm, L.K. Molecular Interactions of Lipopolysaccharide with an Outer Membrane Protein from Pseudomonas aeruginosa Probed by Solution NMR. Biochemistry 2016, 55, 5061–5072. [Google Scholar] [CrossRef]
- Lee, J.; Patel, D.S.; Kucharska, I.; Tamm, L.K.; Im, W. Refinement of OprH-LPS Interactions by Molecular Simulations. Biophys. J. 2017, 112, 346–355. [Google Scholar] [CrossRef]
- Lee, K.; Yoon, S.S. Pseudomonas aeruginosa Biofilm, a Programmed Bacterial Life for Fitness. J. Microbiol. Biotechnol. 2017, 27, 1053–1064. [Google Scholar] [CrossRef]
- Yan, S.; Wu, G. Can Biofilm Be Reversed Through Quorum Sensing in Pseudomonas aeruginosa? Front. Microbiol. 2019, 10, 1582. [Google Scholar] [CrossRef] [PubMed]
- Mann, E.E.; Wozniak, D.J. Pseudomonas biofilm matrix composition and niche biology. FEMS Microbiol. Rev. 2012, 36, 893–916. [Google Scholar] [CrossRef]
- Wei, Q.; Ma, L.Z. Biofilm matrix and its regulation in Pseudomonas aeruginosa. Int. J. Mol. Sci. 2013, 14, 20983–21005. [Google Scholar] [CrossRef] [PubMed]
- Rosenau, F.; Isenhardt, S.; Gdynia, A.; Tielker, D.; Schmidt, E.; Tielen, P.; Schobert, M.; Jahn, D.; Wilhelm, S.; Jaeger, K.E. Lipase LipC affects motility, biofilm formation and rhamnolipid production in Pseudomonas aeruginosa. FEMS Microbiol. Lett. 2010, 309, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Tielen, P.; Rosenau, F.; Wilhelm, S.; Jaeger, K.E.; Flemming, H.C.; Wingender, J. Extracellular enzymes affect biofilm formation of mucoid Pseudomonas aeruginosa. Microbiology 2010, 156, 2239–2252. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.; Gdynia, A.; Tielen, P.; Rosenau, F.; Jaeger, K.E. The autotransporter esterase EstA of Pseudomonas aeruginosa is required for rhamnolipid production, cell motility, and biofilm formation. J. Bacteriol. 2007, 189, 6695–6703. [Google Scholar] [CrossRef]
- Tielen, P.; Kuhn, H.; Rosenau, F.; Jaeger, K.E.; Flemming, H.C.; Wingender, J. Interaction between extracellular lipase LipA and the polysaccharide alginate of Pseudomonas aeruginosa. BMC Microbiol. 2013, 13, 159. [Google Scholar] [CrossRef] [PubMed]
- Cooke, A.C.; Florez, C.; Dunshee, E.B.; Lieber, A.D.; Terry, M.L.; Light, C.J.; Schertzer, J.W. Quinolone Signal-Induced Outer Membrane Vesicles Enhance Biofilm Dispersion in Pseudomonas aeruginosa. mSphere 2020, 5, e01109-20. [Google Scholar] [CrossRef]
- Miller, C.L.; Romero, M.; Karna, S.L.; Chen, T.; Heeb, S.; Leung, K.P. RsmW, Pseudomonas aeruginosa small non-coding RsmA-binding RNA upregulated in biofilm versus planktonic growth conditions. BMC Microbiol. 2016, 16, 155. [Google Scholar] [CrossRef]
- Thöming, J.G.; Tomasch, J.; Preusse, M.; Koska, M.; Grahl, N.; Pohl, S.; Willger, S.D.; Kaever, V.; Müsken, M.; Häussler, S. Parallel evolutionary paths to produce more than one Pseudomonas aeruginosa biofilm phenotype. NPJ Biofilms Microbiomes 2020, 6, 2. [Google Scholar] [CrossRef]
- Cross, A.R.; Raghuram, V.; Wang, Z.; Dey, D.; Goldberg, J.B. Overproduction of the AlgT Sigma Factor Is Lethal to Mucoid Pseudomonas aeruginosa. J. Bacteriol. 2020, 202, e00445-20. [Google Scholar] [CrossRef]
- Ryder, C.; Byrd, M.; Wozniak, D.J. Role of polysaccharides in Pseudomonas aeruginosa biofilm development. Curr. Opin. Microbiol. 2007, 10, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Ghafoor, A.; Hay, I.D.; Rehm, B.H. Role of exopolysaccharides in Pseudomonas aeruginosa biofilm formation and architecture. Appl. Environ. Microbiol. 2011, 77, 5238–5246. [Google Scholar] [CrossRef]
- Orgad, O.; Oren, Y.; Walker, S.L.; Herzberg, M. The role of alginate in Pseudomonas aeruginosa EPS adherence, viscoelastic properties and cell attachment. Biofouling 2011, 27, 787–798. [Google Scholar] [CrossRef]
- Strateva, T.; Mitov, I. Contribution of an arsenal of virulence factors to pathogenesis of Pseudomonas aeruginosa infections Ann. Microbiol. 2011, 61, 717–732. [Google Scholar] [CrossRef]
- Leid, J.G.; Willson, C.J.; Shirtliff, M.E.; Hassett, D.J.; Parsek, M.R.; Jeffers, A.K. The exopolysaccharide alginate protects Pseudomonas aeruginosa biofilm bacteria from IFN-gamma-mediated macrophage killing. J. Immunol. 2005, 175, 7512–7518. [Google Scholar] [CrossRef]
- Rybtke, M.; Jensen, P.; Nielsen, C.H.; Tolker-Nielsen, T. The Extracellular Polysaccharide Matrix of Pseudomonas aeruginosa Biofilms Is a Determinant of Polymorphonuclear Leukocyte Responses. Infect. Immun. 2020, 89, e00631-20. [Google Scholar] [CrossRef]
- Goltermann, L.; Tolker-Nielsen, T. Importance of the Exopolysaccharide Matrix in Antimicrobial Tolerance of Pseudomonas aeruginosa Aggregates. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef]
- Haiko, J.; Westerlund-Wikström, B. The role of the bacterial flagellum in adhesion and virulence. Biology 2013, 2, 1242–1267. [Google Scholar] [CrossRef] [PubMed]
- Sampedro, I.; Parales, R.E.; Krell, T.; Hill, J.E. Pseudomonas chemotaxis. FEMS Microbiol. Rev. 2015, 39, 17–46. [Google Scholar] [CrossRef]
- Song, W.S.; Yoon, S.I. Crystal structure of FliC flagellin from Pseudomonas aeruginosa and its implication in TLR5 binding and formation of the flagellar filament. Biochem. Biophys. Res. Commun. 2014, 444, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Schwarzer, C.; Fischer, H.; Machen, T.E. Chemotaxis and Binding of Pseudomonas aeruginosa to Scratch-Wounded Human Cystic Fibrosis Airway Epithelial Cells. PLoS ONE 2016, 11, e0150109. [Google Scholar] [CrossRef]
- Duan, Q.; Zhou, M.; Zhu, L.; Zhu, G. Flagella and bacterial pathogenicity. J. Basic Microbiol. 2013, 53, 1–8. [Google Scholar] [CrossRef]
- Bucior, I.; Pielage, J.F.; Engel, J.N. Pseudomonas aeruginosa pili and flagella mediate distinct binding and signaling events at the apical and basolateral surface of airway epithelium. PLoS Pathog. 2012, 8, e1002616. [Google Scholar] [CrossRef]
- Ketko, A.K.; Lin, C.; Moore, B.B.; LeVine, A.M. Surfactant protein A binds flagellin enhancing phagocytosis and IL-1β production. PLoS ONE 2013, 8, e82680. [Google Scholar] [CrossRef]
- Zhang, S.; McCormack, F.X.; Levesque, R.C.; O’Toole, G.A.; Lau, G.W. The flagellum of Pseudomonas aeruginosa is required for resistance to clearance by surfactant protein A. PLoS ONE 2007, 2, e564. [Google Scholar] [CrossRef] [PubMed]
- Guttenplan, S.B.; Kearns, D.B. Regulation of flagellar motility during biofilm formation. FEMS Microbiol. Rev. 2013, 37, 849–871. [Google Scholar] [CrossRef]
- Jacobsen, T.; Bardiaux, B.; Francetic, O.; Izadi-Pruneyre, N.; Nilges, M. Structure and function of minor pilins of type IV pili. Med. Microbiol. Immunol. 2020, 209, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Horna, G.; Quezada, K.; Ramos, S.; Mosqueda, N.; Rubio, M.; Guerra, H.; Ruiz, J. Specific type IV pili groups in clinical isolates of Pseudomonas aeruginosa. Int. Microbiol. 2019, 22, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Burrows, L.L. Pseudomonas aeruginosa twitching motility: Type IV pili in action. Annu. Rev. Microbiol. 2012, 66, 493–520. [Google Scholar] [CrossRef] [PubMed]
- Talà, L.; Fineberg, A.; Kukura, P.; Persat, A. Pseudomonas aeruginosa orchestrates twitching motility by sequential control of type IV pili movements. Nat. Microbiol. 2019, 4, 774–780. [Google Scholar] [CrossRef] [PubMed]
- Heiniger, R.W.; Winther-Larsen, H.C.; Pickles, R.J.; Koomey, M.; Wolfgang, M.C. Infection of human mucosal tissue by Pseudomonas aeruginosa requires sequential and mutually dependent virulence factors and a novel pilus-associated adhesin. Cell Microbiol. 2010, 12, 1158–1173. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.D.; Garrett, C.K.; Bond, J.E.; Coggan, K.A.; Wolfgang, M.C.; Redinbo, M.R. Pseudomonas aeruginosa PilY1 binds integrin in an RGD- and calcium-dependent manner. PLoS ONE 2011, 6, e29629. [Google Scholar] [CrossRef] [PubMed]
- Siryaporn, A.; Kuchma, S.L.; O’Toole, G.A.; Gitai, Z. Surface attachment induces Pseudomonas aeruginosa virulence. Proc. Natl. Acad. Sci. USA 2014, 111, 16860–16865. [Google Scholar] [CrossRef] [PubMed]
- Marko, V.A.; Kilmury, S.L.N.; MacNeil, L.T.; Burrows, L.L. Pseudomonas aeruginosa type IV minor pilins and PilY1 regulate virulence by modulating FimS-AlgR activity. PLoS Pathog. 2018, 14, e1007074. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.K.; Sheth, H.B.; Wong, W.Y.; Sherburne, R.; Paranchych, W.; Hodges, R.S.; Lingwood, C.A.; Krivan, H.; Irvin, R.T. The binding of Pseudomonas aeruginosa pili to glycosphingolipids is a tip-associated event involving the C-terminal region of the structural pilin subunit. Mol. Microbiol. 1994, 11, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Craig, L.; Pique, M.E.; Tainer, J.A. Type IV pilus structure and bacterial pathogenicity. Nat. Rev. Microbiol. 2004, 2, 363–378. [Google Scholar] [CrossRef]
- Schroeder, T.H.; Zaidi, T.; Pier, G.B. Lack of adherence of clinical isolates of Pseudomonas aeruginosa to asialo-GM(1) on epithelial cells. Infect. Immun. 2001, 69, 719–729. [Google Scholar] [CrossRef]
- Tolker-Nielsen, T. Pseudomonas aeruginosa biofilm infections: From molecular biofilm biology to new treatment possibilities. APMIS 2014, 138, 1–51. [Google Scholar] [CrossRef]
- Van Schaik, E.J.; Giltner, C.L.; Audette, G.F.; Keizer, D.W.; Bautista, D.L.; Slupsky, C.M.; Sykes, B.D.; Irvin, R.T. DNA binding: A novel function of Pseudomonas aeruginosa type IV pili. J. Bacteriol. 2005, 187, 1455–1464. [Google Scholar] [CrossRef]
- Tan, R.M.; Kuang, Z.; Hao, Y.; Lau, G.W. Type IV pilus of Pseudomonas aeruginosa confers resistance to antimicrobial activities of the pulmonary surfactant protein-A. J. Innate Immun. 2014, 6, 227–239. [Google Scholar] [CrossRef]
- Arlehamn, C.S.; Evans, T.J. Pseudomonas aeruginosa pilin activates the inflammasome. Cell Microbiol. 2011, 13, 388–401. [Google Scholar] [CrossRef]
- Bleves, S.; Viarre, V.; Salacha, R.; Michel, G.P.; Filloux, A.; Voulhoux, R. Protein secretion systems in Pseudomonas aeruginosa: A wealth of pathogenic weapons. Int. J. Med. Microbiol. 2010, 300, 534–543. [Google Scholar] [CrossRef]
- Pena, R.T.; Blasco, L.; Ambroa, A.; González-Pedrajo, B.; Fernández-García, L.; López, M.; Bleriot, I.; Bou, G.; García-Contreras, R.; Wood, T.K.; et al. Relationship Between Quorum Sensing and Secretion Systems. Front. Microbiol. 2019, 10, 1100. [Google Scholar] [CrossRef]
- Zhao, K.; Li, W.; Li, J.; Ma, T.; Wang, K.; Yuan, Y.; Li, J.S.; Xie, R.; Huang, T.; Zhang, Y.; et al. TesG is a type I secretion effector of Pseudomonas aeruginosa that suppresses the host immune response during chronic infection. Nat. Microbiol. 2019, 4, 459–469. [Google Scholar] [CrossRef]
- Anantharajah, A.; Mingeot-Leclercq, M.P.; Van Bambeke, F. Targeting the Type Three Secretion System in Pseudomonas aeruginosa. Trends Pharmacol. Sci. 2016, 37, 734–749. [Google Scholar] [CrossRef]
- Sana, T.G.; Berni, B.; Bleves, S. The T6SSs of Pseudomonas aeruginosa Strain PAO1 and Their Effectors: Beyond Bacterial-Cell Targeting. Front. Cell Infect. Microbiol. 2016, 6, 61. [Google Scholar] [CrossRef]
- Goure, J.; Pastor, A.; Faudry, E.; Chabert, J.; Dessen, A.; Attree, I. The V antigen of Pseudomonas aeruginosa is required for assembly of the functional PopB/PopD translocation pore in host cell membranes. Infect. Immun. 2004, 72, 4741–4750. [Google Scholar] [CrossRef]
- Williams McMackin, E.A.; Djapgne, L.; Corley, J.M.; Yahr, T.L. Fitting Pieces into the Puzzle of Pseudomonas aeruginosa Type III Secretion System Gene Expression. J. Bacteriol. 2019, 201. [Google Scholar] [CrossRef] [PubMed]
- Burstein, D.; Satanower, S.; Simovitch, M.; Belnik, Y.; Zehavi, M.; Yerushalmi, G.; Ben-Aroya, S.; Pupko, T.; Banin, E. Novel type III effectors in Pseudomonas aeruginosa. mBio 2015, 6, e00161. [Google Scholar] [CrossRef] [PubMed]
- Ince, D.; Sutterwala, F.S.; Yahr, T.L. Secretion of Flagellar Proteins by the Pseudomonas aeruginosa Type III Secretion-Injectisome System. J. Bacteriol. 2015, 197, 2003–2011. [Google Scholar] [CrossRef] [PubMed]
- Neeld, D.; Jin, Y.; Bichsel, C.; Jia, J.; Guo, J.; Bai, F.; Wu, W.; Ha, U.H.; Terada, N.; Jin, S. Pseudomonas aeruginosa injects NDK into host cells through a type III secretion system. Microbiology 2014, 160, 1417–1426. [Google Scholar] [CrossRef] [PubMed]
- Javanmardi, F.; Emami, A.; Pirbonyeh, N.; Keshavarzi, A.; Rajaee, M. A systematic review and meta-analysis on Exo-toxins prevalence in hospital acquired Pseudomonas aeruginosa isolates. Infect. Genet. Evol. 2019, 75, 104037. [Google Scholar] [CrossRef]
- Shaver, C.M.; Hauser, A.R. Relative contributions of Pseudomonas aeruginosa ExoU, ExoS, and ExoT to virulence in the lung. Infect. Immun. 2004, 72, 6969–6977. [Google Scholar] [CrossRef] [PubMed]
- Howell, H.A.; Logan, L.K.; Hauser, A.R. Type III secretion of ExoU is critical during early Pseudomonas aeruginosa pneumonia. mBio 2013, 4, e00032-13. [Google Scholar] [CrossRef] [PubMed]
- Peña, C.; Cabot, G.; Gómez-Zorrilla, S.; Zamorano, L.; Ocampo-Sosa, A.; Murillas, J.; Almirante, B.; Pomar, V.; Aguilar, M.; Granados, A.; et al. Influence of virulence genotype and resistance profile in the mortality of Pseudomonas aeruginosa bloodstream infections. Clin. Infect. Dis. 2015, 60, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Hauser, A.R. The type III secretion system of Pseudomonas aeruginosa: Infection by injection. Nat. Rev. Microbiol. 2009, 7, 654–665. [Google Scholar] [CrossRef]
- De Lima, C.D.; Calegari-Silva, T.C.; Pereira, R.M.; Santos, S.A.; Lopes, U.G.; Plotkowski, M.C.; Saliba, A.M. ExoU activates NF-κB and increases IL-8/KC secretion during Pseudomonas aeruginosa infection. PLoS ONE 2012, 7, e41772. [Google Scholar] [CrossRef]
- Pazos, M.A.; Lanter, B.B.; Yonker, L.M.; Eaton, A.D.; Pirzai, W.; Gronert, K.; Bonventre, J.V.; Hurley, B.P. Pseudomonas aeruginosa ExoU augments neutrophil transepithelial migration. PLoS Pathog. 2017, 13, e1006548. [Google Scholar] [CrossRef]
- Wagener, B.M.; Anjum, N.; Christiaans, S.C.; Banks, M.E.; Parker, J.C.; Threet, A.T.; Walker, R.R.; Isbell, K.D.; Moser, S.A.; Stevens, T.; et al. Exoenzyme Y Contributes to End-Organ Dysfunction Caused by Pseudomonas aeruginosa Pneumonia in Critically Ill Patients: An Exploratory Study. Toxins 2020, 12, 369. [Google Scholar] [CrossRef]
- Beckert, U.; Wolter, S.; Hartwig, C.; Bähre, H.; Kaever, V.; Ladant, D.; Frank, D.W.; Seifert, R. ExoY from Pseudomonas aeruginosa is a nucleotidyl cyclase with preference for cGMP and cUMP formation. Biochem. Biophys. Res. Commun. 2014, 450, 870–874. [Google Scholar] [CrossRef]
- Stevens, T.C.; Ochoa, C.D.; Morrow, K.A.; Robson, M.J.; Prasain, N.; Zhou, C.; Alvarez, D.F.; Frank, D.W.; Balczon, R.; Stevens, T. The Pseudomonas aeruginosa exoenzyme Y impairs endothelial cell proliferation and vascular repair following lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 306, L915–L924. [Google Scholar] [CrossRef]
- Mancl, J.M.; Suarez, C.; Liang, W.G.; Kovar, D.R.; Tang, W.J. Pseudomonas aeruginosa exoenzyme Y directly bundles actin filaments. J. Biol. Chem. 2020, 295, 3506–3517. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Zhou, Y.; Liu, F.; Liu, H.; Tan, H.; Jin, S.; Wu, W.; Ge, B. Bacterial Nucleotidyl Cyclase Inhibits the Host Innate Immune Response by Suppressing TAK1 Activation. Infect. Immun. 2017, 85. [Google Scholar] [CrossRef] [PubMed]
- Armentrout, E.I.; Kundracik, E.C.; Rietsch, A. Cell-type-specific hypertranslocation of effectors by the Pseudomonas aeruginosa type III secretion system. Mol. Microbiol. 2020. [Google Scholar] [CrossRef]
- Sun, J.; Barbieri, J.T. Pseudomonas aeruginosa ExoT ADP-ribosylates CT10 regulator of kinase (Crk) proteins. J. Biol. Chem. 2003, 278, 32794–32800. [Google Scholar] [CrossRef]
- Vourc’h, M.; Roquilly, A.; Broquet, A.; David, G.; Hulin, P.; Jacqueline, C.; Caillon, J.; Retiere, C.; Asehnoune, K. Exoenzyme T Plays a Pivotal Role in the IFN-γ Production after Pseudomonas Challenge in IL-12 Primed Natural Killer Cells. Front. Immunol. 2017, 8, 1283. [Google Scholar] [CrossRef]
- Sarges, E.D.S.N.; Rodrigues, Y.C.; Furlaneto, I.P.; de Melo, M.V.H.; Brabo, G.L.D.C.; Lopes, K.C.M.; Quaresma, A.J.P.G.; Lima, L.N.G.C.; Lima, K.V.B. Type III Secretion System Virulotypes and Their Association with Clinical Features of Cystic Fibrosis Patients. Infect. Drug Resist. 2020, 13, 3771–3781. [Google Scholar] [CrossRef] [PubMed]
- Rangel, S.M.; Diaz, M.H.; Knoten, C.A.; Zhang, A.; Hauser, A.R. Correction: The Role of ExoS in Dissemination of Pseudomonas aeruginosa during Pneumonia. PLoS Pathog. 2015, 11, e1005163. [Google Scholar] [CrossRef] [PubMed]
- Rangel, S.M.; Logan, L.K.; Hauser, A.R. The ADP-ribosyltransferase domain of the effector protein ExoS inhibits phagocytosis of Pseudomonas aeruginosa during pneumonia. mBio 2014, 5, e01080-14. [Google Scholar] [CrossRef]
- Vareechon, C.; Zmina, S.E.; Karmakar, M.; Pearlman, E.; Rietsch, A. Pseudomonas aeruginosa Effector ExoS Inhibits ROS Production in Human Neutrophils. Cell Host Microbe 2017, 21, 611–618. [Google Scholar] [CrossRef]
- Epelman, S.; Stack, D.; Bell, C.; Wong, E.; Neely, G.G.; Krutzik, S.; Miyake, K.; Kubes, P.; Zbytnuik, L.D.; Ma, L.L.; et al. Different domains of Pseudomonas aeruginosa exoenzyme S activate distinct TLRs. J. Immunol. 2004, 173, 2031–2040. [Google Scholar] [CrossRef] [PubMed]
- Galle, M.; Jin, S.; Bogaert, P.; Haegman, M.; Vandenabeele, P.; Beyaert, R. The Pseudomonas aeruginosa type III secretion system has an exotoxin S/T/Y independent pathogenic role during acute lung infection. PLoS ONE 2012, 7, e41547. [Google Scholar] [CrossRef] [PubMed]
- Faure, E.; Mear, J.B.; Faure, K.; Normand, S.; Couturier-Maillard, A.; Grandjean, T.; Balloy, V.; Ryffel, B.; Dessein, R.; Chignard, M.; et al. Pseudomonas aeruginosa type-3 secretion system dampens host defense by exploiting the NLRC4-coupled inflammasome. Am. J. Respir. Crit. Care Med. 2014, 189, 799–811. [Google Scholar] [CrossRef]
- Michalska, M.; Wolf, P. Pseudomonas Exotoxin A: Optimized by evolution for effective killing. Front. Microbiol. 2015, 6, 963. [Google Scholar] [CrossRef]
- Wolf, P.; Elsässer-Beile, U. Pseudomonas exotoxin A: From virulence factor to anti-cancer agent. Int. J. Med. Microbiol. 2009, 299, 161–176. [Google Scholar] [CrossRef]
- Du, X.; Youle, R.J.; FitzGerald, D.J.; Pastan, I. Pseudomonas exotoxin A-mediated apoptosis is Bak dependent and preceded by the degradation of Mcl-1. Mol. Cell Biol. 2010, 30, 3444–3452. [Google Scholar] [CrossRef]
- Schultz, M.J.; Rijneveld, A.W.; Florquin, S.; Speelman, P.; VAN Deventer, S.J.H.; VAN DER Poll, T. Impairment of host defence by exotoxin A in Pseudomonas aeruginosa pneumonia in mice. J. Med. Microbiol. 2001, 50, 822–827. [Google Scholar] [CrossRef][Green Version]
- Schultz, M.J.; Speelman, P.; Zaat, S.A.; Hack, C.E.; van Deventer, S.J.; van der Poll, T. The effect of Pseudomonas exotoxin A on cytokine production in whole blood exposed to Pseudomonas aeruginosa. FEMS Immunol. Med. Microbiol. 2000, 29, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Galdino, A.C.M.; Branquinha, M.H.; Santos, A.L.S.; Viganor, L. Pseudomonas aeruginosa and its arsenal of proteases: Weapons to battle the host. In Pathophysiological Aspects of Proteases; Springer Nature: Singapore, 2017; pp. 381–397. [Google Scholar]
- Li, X.H.; Lee, J.H. Quorum sensing-dependent post-secretional activation of extracellular proteases in Pseudomonas aeruginosa. J. Biol. Chem. 2019, 294, 19635–19644. [Google Scholar] [CrossRef]
- Gellatly, S.L.; Hancock, R.E. Pseudomonas aeruginosa: New insights into pathogenesis and host defenses. Pathog. Dis. 2013, 67, 159–173. [Google Scholar] [CrossRef] [PubMed]
- Nomura, K.; Obata, K.; Keira, T.; Miyata, R.; Hirakawa, S.; Takano, K.; Kohno, T.; Sawada, N.; Himi, T.; Kojima, T. Pseudomonas aeruginosa elastase causes transient disruption of tight junctions and downregulation of PAR-2 in human nasal epithelial cells. Respir. Res. 2014, 15, 21. [Google Scholar] [CrossRef]
- Mariencheck, W.I.; Alcorn, J.F.; Palmer, S.M.; Wright, J.R. Pseudomonas aeruginosa elastase degrades surfactant proteins A and D. Am. J. Respir. Cell Mol. Biol. 2003, 28, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Kuang, Z.; Hao, Y.; Walling, B.E.; Jeffries, J.L.; Ohman, D.E.; Lau, G.W. Pseudomonas aeruginosa elastase provides an escape from phagocytosis by degrading the pulmonary surfactant protein-A. PLoS ONE 2011, 6, e27091. [Google Scholar] [CrossRef]
- Saint-Criq, V.; Villeret, B.; Bastaert, F.; Kheir, S.; Hatton, A.; Cazes, A.; Xing, Z.; Sermet-Gaudelus, I.; Garcia-Verdugo, I.; Edelman, A.; et al. LasB protease impairs innate immunity in mice and humans by targeting a lung epithelial cystic fibrosis transmembrane regulator-IL-6-antimicrobial-repair pathway. Thorax 2018, 73, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lee, K.M.; Park, S.; Cho, Y.; Lee, E.; Park, J.H.; Shin, O.S.; Son, J.; Yoon, S.S.; Yu, J.W. Bacterial secretant from Pseudomonas aeruginosa Dampens inflammasome activation in a Quorum sensing-Dependent Manner. Front. Immunol. 2017, 8, 333. [Google Scholar] [CrossRef] [PubMed]
- Casilag, F.; Lorenz, A.; Krueger, J.; Klawonn, F.; Weiss, S.; Häussler, S. The LasB Elastase of Pseudomonas aeruginosa Acts in Concert with Alkaline Protease AprA To Prevent Flagellin-Mediated Immune Recognition. Infect. Immun. 2016, 84, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Bastaert, F.; Kheir, S.; Saint-Criq, V.; Villeret, B.; Dang, P.M.; El-Benna, J.; Sirard, J.C.; Voulhoux, R.; Sallenave, J.M. LasB Subverts Alveolar Macrophage Activity by Interfering with Bacterial Killing Through Downregulation of Innate Immune Defense, Reactive Oxygen Species Generation, and Complement Activation. Front. Immunol. 2018, 9, 1675. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; He, X.; Xie, W.; Xiong, J.; Sheng, H.; Guo, S.; Huang, C.; Zhang, D.; Zhang, K. Elastase LasB of Pseudomonas aeruginosa promotes biofilm formation partly through rhamnolipid-mediated regulation. Can. J. Microbiol. 2014, 60, 227–235. [Google Scholar] [CrossRef]
- Krishnan, G.; Sethumadhavan, A.; Muthusamy, S.; Mani, M. Antibiotic resistant clinical isolates of Pseudomonas aeruginosa harbor LasA gene. Internet J. Microbiol. 2019, 16. [Google Scholar] [CrossRef]
- Laarman, A.J.; Bardoel, B.W.; Ruyken, M.; Fernie, J.; Milder, F.J.; van Strijp, J.A.; Rooijakkers, S.H. Pseudomonas aeruginosa alkaline protease blocks complement activation via the classical and lectin pathways. J. Immunol. 2012, 188, 386–393. [Google Scholar] [CrossRef] [PubMed]
- Bardoel, B.W.; van der Ent, S.; Pel, M.J.; Tommassen, J.; Pieterse, C.M.; van Kessel, K.P.; van Strijp, J.A. Pseudomonas evades immune recognition of flagellin in both mammals and plants. PLoS Pathog. 2011, 7, e1002206. [Google Scholar] [CrossRef]
- Butterworth, M.B.; Zhang, L.; Heidrich, E.M.; Myerburg, M.M.; Thibodeau, P.H. Activation of the epithelial sodium channel (ENaC) by the alkaline protease from Pseudomonas aeruginosa. J. Biol. Chem. 2012, 287, 32556–32565. [Google Scholar] [CrossRef] [PubMed]
- Iiyama, K.; Takahashi, E.; Lee, J.M.; Mon, H.; Morishita, M.; Kusakabe, T.; Yasunaga-Aoki, C. Alkaline protease contributes to pyocyanin production in Pseudomonas aeruginosa. FEMS Microbiol. Lett. 2017, 364. [Google Scholar] [CrossRef] [PubMed]
- Conibear, T.C.R.; Willcox, M.D.P.; Flanagan, J.L.; Zhu, H. Characterization of protease IV expression in Pseudomonas aeruginosa clinical isolates. J. Med. Microbiol. 2012, 61, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Malloy, J.L.; Veldhuizen, R.A.; Thibodeaux, B.A.; O’Callaghan, R.J.; Wright, J.R. Pseudomonas aeruginosa protease IV degrades surfactant proteins and inhibits surfactant host defense and biophysical functions. Am. J. Physiol. Lung Cell Mol. Physiol. 2005, 288, L409–L418. [Google Scholar] [CrossRef]
- Smith, L.; Rose, B.; Tingpej, P.; Zhu, H.; Conibear, T.; Manos, J.; Bye, P.; Elkins, M.; Willcox, M.; Bell, S.; et al. Protease IV production in Pseudomonas aeruginosa from the lungs of adults with cystic fibrosis. J. Med. Microbiol. 2006, 55, 1641–1644. [Google Scholar] [CrossRef]
- Guillon, A.; Brea, D.; Morello, E.; Tang, A.; Jouan, Y.; Ramphal, R.; Korkmaz, B.; Perez-Cruz, M.; Trottein, F.; O’Callaghan, R.J.; et al. Pseudomonas aeruginosa proteolytically alters the interleukin 22-dependent lung mucosal defense. Virulence 2017, 8, 810–820. [Google Scholar] [CrossRef]
- Bradshaw, J.L.; Caballero, A.R.; Bierdeman, M.A.; Adams, K.V.; Pipkins, H.R.; Tang, A.; O’Callaghan, R.J.; McDaniel, L.S. Pseudomonas aeruginosa Protease IV Exacerbates Pneumococcal Pneumonia and Systemic Disease. mSphere 2018, 3. [Google Scholar] [CrossRef]
- Park, S.J.; Kim, S.K.; So, Y.I.; Park, H.Y.; Li, X.H.; Yeom, D.H.; Lee, M.N.; Lee, B.L.; Lee, J.H. Protease IV, a quorum sensing-dependent protease of Pseudomonas aeruginosa modulates insect innate immunity. Mol. Microbiol. 2014, 94, 1298–1314. [Google Scholar] [CrossRef] [PubMed]
- Verma, N.; Dollinger, P.; Kovacic, F.; Jaeger, K.E.; Gohlke, H. The Membrane-Integrated Steric Chaperone Lif Facilitates Active Site Opening of Pseudomonas aeruginosa Lipase A. J. Comput. Chem. 2020, 41, 500–512. [Google Scholar] [CrossRef]
- Bofill, C.; Prim, N.; Mormeneo, M.; Manresa, A.; Pastor, F.I.; Diaz, P. Differential behaviour of Pseudomonas sp. 42A2 LipC, a lipase showing greater versatility than its counterpart LipA. Biochimie 2010, 92, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Berdiev, N.S.; Ziyavitdinov, J.F.; Asrorov, A.M.; Olimjonov, S.S.; Salikhov, S.I. Characterization of a novel lipase from Pseudomonas aeruginosa. Nova Biotechnol. Chim 2019, 18, 44–51. [Google Scholar] [CrossRef][Green Version]
- Kipnis, E.; Sawa, T.; Wiener-Kronish, J. Targeting mechanisms of Pseudomonas aeruginosa pathogenesis. Med. Mal. Infect. 2006, 36, 78–91. [Google Scholar] [CrossRef]
- Wargo, M.J.; Gross, M.J.; Rajamani, S.; Allard, J.L.; Lundblad, L.K.; Allen, G.B.; Vasil, M.L.; Leclair, L.W.; Hogan, D.A. Hemolytic phospholipase C inhibition protects lung function during Pseudomonas aeruginosa infection. Am. J. Respir. Crit. Care Med. 2011, 184, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Bezzerri, V.; d’Adamo, P.; Rimessi, A.; Lanzara, C.; Crovella, S.; Nicolis, E.; Tamanini, A.; Athanasakis, E.; Tebon, M.; Bisoffi, G.; et al. Phospholipase C-β3 is a key modulator of IL-8 expression in cystic fibrosis bronchial epithelial cells. J. Immunol. 2011, 186, 4946–4958. [Google Scholar] [CrossRef]
- Hall, S.; McDermott, C.; Anoopkumar-Dukie, S.; McFarland, A.J.; Forbes, A.; Perkins, A.V.; Davey, A.K.; Chess-Williams, R.; Kiefel, M.J.; Arora, D.; et al. Cellular Effects of Pyocyanin, a Secreted Virulence Factor of Pseudomonas aeruginosa. Toxins 2016, 8, 236. [Google Scholar] [CrossRef] [PubMed]
- Zeng, B.; Wang, C.; Zhang, P.; Guo, Z.; Chen, L.; Duan, K. Heat shock protein DnaJ in Pseudomonas aeruginosa affects biofilm formation via pyocyanin production. Microorganisms 2020, 8, 395. [Google Scholar] [CrossRef] [PubMed]
- Managò, A.; Becker, K.A.; Carpinteiro, A.; Wilker, B.; Soddemann, M.; Seitz, A.P.; Edwards, M.J.; Grassmé, H.; Szabò, I.; Gulbins, E. Pseudomonas aeruginosa pyocyanin induces neutrophil death via mitochondrial reactive oxygen species and mitochondrial acid sphingomyelinase. Antioxid. Redox Signal. 2015, 22, 1097–1110. [Google Scholar] [CrossRef] [PubMed]
- Alfiniyah, C.; Bees, M.A.; Wood, A.J. Quorum machinery: Effect of the las system in rhl regulation of P. aeruginosa. AIP Conf. Proc. 2019, 2192, 060001. [Google Scholar] [CrossRef]
- Halldorsson, S.; Gudjonsson, T.; Gottfredsson, M.; Singh, P.K.; Gudmundsson, G.H.; Baldursson, O. Azithromycin maintains airway epithelial integrity during Pseudomonas aeruginosa infection. Am. J. Respir. Cell Mol. Biol. 2010, 42, 62–68. [Google Scholar] [CrossRef]
- Zulianello, L.; Canard, C.; Köhler, T.; Caille, D.; Lacroix, J.S.; Meda, P. Rhamnolipids are virulence factors that promote early infiltration of primary human airway epithelia by Pseudomonas aeruginosa. Infect. Immun. 2006, 74, 3134–3137. [Google Scholar] [CrossRef]
- Köhler, T.; Guanella, R.; Carlet, J.; van Delden, C. Quorum sensing-dependent virulence during Pseudomonas aeruginosa colonisation and pneumonia in mechanically ventilated patients. Thorax 2010, 65, 703–710. [Google Scholar] [CrossRef][Green Version]
- Raya, A.; Sodagari, M.; Pinzon, N.M.; He, X.; Zhang Newby, B.M.; Ju, L.K. Effects of rhamnolipids and shear on initial attachment of Pseudomonas aeruginosa PAO1 in glass flow chambers. Environ. Sci. Pollut Res. Int. 2010, 17, 1529–1538. [Google Scholar] [CrossRef] [PubMed]
- Nickzad, A.; Déziel, E. The involvement of rhamnolipids in microbial cell adhesion and biofilm development—An approach for control? Lett. Appl. Microbiol. 2014, 58, 447–453. [Google Scholar] [CrossRef]
- Davey, M.E.; Caiazza, N.C.; O’Toole, G.A. Rhamnolipid surfactant production affects biofilm architecture in Pseudomonas aeruginosa PAO1. J. Bacteriol. 2003, 185, 1027–1036. [Google Scholar] [CrossRef]
- Murray, T.S.; Kazmierczak, B.I. Pseudomonas aeruginosa exhibits sliding motility in the absence of type IV pili and flagella. J. Bacteriol. 2008, 190, 2700–2708. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, J.; Richardson, A.P.; Lépine, F.; Déziel, E. Self-produced extracellular stimuli modulate the Pseudomonas aeruginosa swarming motility behaviour. Environ. Microbiol. 2007, 9, 2622–2630. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Yu, S.; Zhang, Z.; Wei, Q.; Yan, L.; Ai, G.; Liu, H.; Ma, L.Z. Coordination of swarming motility, biosurfactant synthesis, and biofilm matrix exopolysaccharide production in Pseudomonas aeruginosa. Appl. Environ. Microbiol. 2014, 80, 6724–6732. [Google Scholar] [CrossRef] [PubMed]
- Glick, R.; Gilmour, C.; Tremblay, J.; Satanower, S.; Avidan, O.; Déziel, E.; Greenberg, E.P.; Poole, K.; Banin, E. Increase in rhamnolipid synthesis under iron-limiting conditions influences surface motility and biofilm formation in Pseudomonas aeruginosa. J. Bacteriol. 2010, 192, 2973–2980. [Google Scholar] [CrossRef]
- Jensen, P.Ø.; Bjarnsholt, T.; Phipps, R.; Rasmussen, T.B.; Calum, H.; Christoffersen, L.; Moser, C.; Williams, P.; Pressler, T.; Givskov, M.; et al. Rapid necrotic killing of polymorphonuclear leukocytes is caused by quorum-sensing-controlled production of rhamnolipid by Pseudomonas aeruginosa. Microbiology 2007, 153, 1329–1338. [Google Scholar] [CrossRef]
- Alhede, M.; Bjarnsholt, T.; Jensen, P.; Phipps, R.K.; Moser, C.; Christophersen, L.; Christensen, L.D.; van Gennip, M.; Parsek, M.; Høiby, N.; et al. Pseudomonas aeruginosa recognizes and responds aggressively to the presence of polymorphonuclear leukocytes. Microbiology 2009, 155, 3500–3508. [Google Scholar] [CrossRef]
- Dössel, J.; Meyer-Hoffert, U.; Schröder, J.M.; Gerstel, U. Pseudomonas aeruginosa-derived rhamnolipids subvert the host innate immune response through manipulation of the human beta-defensin-2 expression. Cell Microbiol. 2012, 14, 1364–1375. [Google Scholar] [CrossRef]
- Malhotra, S.; Hayes, D.; Wozniak, D.J. Cystic Fibrosis and Pseudomonas aeruginosa: The Host-Microbe Interface. Clin. Microbiol. Rev. 2019, 32, e00138-18. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Lee, B.Y.; Lau, G.W.; Cho, Y.H. IscR modulates catalase A (KatA) activity, peroxide resistance and full virulence of Pseudomonas aeruginosa PA14. J. Microbiol. Biotechnol. 2009, 19, 1520–1526. [Google Scholar] [CrossRef] [PubMed]
- Heo, Y.J.; Chung, I.Y.; Cho, W.J.; Lee, B.Y.; Kim, J.H.; Choi, K.H.; Lee, J.W.; Hassett, D.J.; Cho, Y.H. The major catalase gene (katA) of Pseudomonas aeruginosa PA14 is under both positive and negative control of the global transactivator OxyR in response to hydrogen peroxide. J. Bacteriol. 2010, 192, 381–390. [Google Scholar] [CrossRef]
- Khakimova, M.; Ahlgren, H.G.; Harrison, J.J.; English, A.M.; Nguyen, D. The stringent response controls catalases in Pseudomonas aeruginosa and is required for hydrogen peroxide and antibiotic tolerance. J. Bacteriol. 2013, 195, 2011–2020. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.H.; Choi, Y.S.; Cho, Y.H. Unusual properties of catalase A (KatA) of Pseudomonas aeruginosa PA14 are associated with its biofilm peroxide resistance. J. Bacteriol. 2008, 190, 2663–2670. [Google Scholar] [CrossRef]
- Lee, J.S.; Heo, Y.J.; Lee, J.K.; Cho, Y.H. KatA, the major catalase, is critical for osmoprotection and virulence in Pseudomonas aeruginosa PA14. Infect. Immun. 2005, 73, 4399–4403. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Panmanee, W.; Wilson, J.J.; Mahtani, H.K.; Li, Q.; Vanderwielen, B.D.; Makris, T.M.; Rogers, M.; McDaniel, C.; Lipscomb, J.D.; et al. Catalase (KatA) plays a role in protection against anaerobic nitric oxide in Pseudomonas aeruginosa. PLoS ONE 2014, 9, e91813. [Google Scholar] [CrossRef] [PubMed]
- Dauner, M.; Skerra, A. Scavenging Bacterial Siderophores with Engineered Lipocalin Proteins as an Alternative Antimicrobial Strategy. ChemBioChem 2020, 21, 601–606. [Google Scholar] [CrossRef]
- Cornelis, P.; Dingemans, J. Pseudomonas aeruginosa adapts its iron uptake strategies in function of the type of infections. Front. Cell Infect. Microbiol. 2013, 3, 75. [Google Scholar] [CrossRef]
- Bonneau, A.; Roche, B.; Schalk, I.J. Iron acquisition in Pseudomonas aeruginosa by the siderophore pyoverdine: An intricate interacting network including periplasmic and membrane proteins. Sci. Rep. 2020, 10, 120. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.; Kirienko, N.V. Interdependence between iron acquisition and biofilm formation in Pseudomonas aeruginosa. J. Microbiol. 2018, 56, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Cornelis, P.; Matthijs, S.; Van Oeffelen, L. Iron uptake regulation in Pseudomonas aeruginosa. Biometals 2009, 22, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Zhang, L. The hierarchy quorum sensing network in Pseudomonas aeruginosa. Protein Cell 2015, 6, 26–41. [Google Scholar] [CrossRef]
- Wade, D.S.; Calfee, M.W.; Rocha, E.R.; Ling, E.A.; Engstrom, E.; Coleman, J.P.; Pesci, E.C. Regulation of Pseudomonas quinolone signal synthesis in Pseudomonas aeruginosa. J. Bacteriol. 2005, 187, 4372–4380. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Wu, J.; Deng, Y.; Wang, J.; Wang, C.; Chang, C.; Dong, Y.; Williams, P.; Zhang, L.H. A cell-cell communication signal integrates quorum sensing and stress response. Nat. Chem. Biol. 2013, 9, 339–343. [Google Scholar] [CrossRef]
- Kariminik, A.; Baseri-Salehi, M.; Kheirkhah, B. Pseudomonas aeruginosa quorum sensing modulates immune responses: An updated review article. Immunol. Lett. 2017, 190, 1–6. [Google Scholar] [CrossRef]
- Ueda, A.; Wood, T.K. Connecting quorum sensing, c-di-GMP, pel polysaccharide, and biofilm formation in Pseudomonas aeruginosa through tyrosine phosphatase TpbA (PA3885). PLoS Pathog. 2009, 5, e1000483. [Google Scholar] [CrossRef]
- Sana, T.G.; Hachani, A.; Bucior, I.; Soscia, C.; Garvis, S.; Termine, E.; Engel, J.; Filloux, A.; Bleves, S. The second type VI secretion system of Pseudomonas aeruginosa strain PAO1 is regulated by quorum sensing and Fur and modulates internalization in epithelial cells. J. Biol. Chem. 2012, 287, 27095–27105. [Google Scholar] [CrossRef]
- Maura, D.; Hazan, R.; Kitao, T.; Ballok, A.E.; Rahme, L.G. Evidence for Direct Control of Virulence and Defense Gene Circuits by the Pseudomonas aeruginosa Quorum Sensing Regulator, MvfR. Sci. Rep. 2016, 6, 34083. [Google Scholar] [CrossRef]
- Schwarzer, C.; Ravishankar, B.; Patanwala, M.; Shuai, S.; Fu, Z.; Illek, B.; Fischer, H.; Machen, T.E. Thapsigargin blocks Pseudomonas aeruginosa homoserine lactone-induced apoptosis in airway epithelia. Am. J. Physiol. Cell Physiol. 2014, 306, C844–C855. [Google Scholar] [CrossRef]
- Schwarzer, C.; Fu, Z.; Patanwala, M.; Hum, L.; Lopez-Guzman, M.; Illek, B.; Kong, W.; Lynch, S.V.; Machen, T.E. Pseudomonas aeruginosa biofilm-associated homoserine lactone C12 rapidly activates apoptosis in airway epithelia. Cell Microbiol. 2012, 14, 698–709. [Google Scholar] [CrossRef]
- Song, D.; Meng, J.; Cheng, J.; Fan, Z.; Chen, P.; Ruan, H.; Tu, Z.; Kang, N.; Li, N.; Xu, Y.; et al. Pseudomonas aeruginosa quorum-sensing metabolite induces host immune cell death through cell surface lipid domain dissolution. Nat. Microbiol. 2019, 4, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Möker, N.; Dean, C.R.; Tao, J. Pseudomonas aeruginosa increases formation of multidrug-tolerant persister cells in response to quorum-sensing signaling molecules. J. Bacteriol. 2010, 192, 1946–1955. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Cheng, J. Quorum Sensing in Pseudomonas aeruginosa and Its Relationship to Biofilm Development; American Chemical Society (USA): Washington, DC, USA, 2019; Volume 1323, pp. 1–16. [Google Scholar]
- Bleves, S.; Soscia, C.; Nogueira-Orlandi, P.; Lazdunski, A.; Filloux, A. Quorum sensing negatively controls type III secretion regulon expression in Pseudomonas aeruginosa PAO1. J. Bacteriol. 2005, 187, 3898–3902. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Cheng, J.; Wang, Y.; Shen, X. The Pseudomonas Quinolone Signal (PQS): Not Just for Quorum Sensing Anymore. Front. Cell Infect. Microbiol. 2018, 8, 230. [Google Scholar] [CrossRef]
- García-Reyes, S.; Soberón-Chávez, G.; Cocotl-Yanez, M. The third quorum-sensing system of Pseudomonas aeruginosa: Pseudomonas quinolone signal and the enigmatic PqsE protein. J. Med. Microbiol. 2020, 69, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Cornelis, P. Putting an end to the Pseudomonas aeruginosa IQS controversy. Microbiol. Open 2020, 9, e962. [Google Scholar] [CrossRef]
- Wang, J.; Wang, C.; Yu, H.B.; Dela Ahator, S.; Wu, X.; Lv, S.; Zhang, L.H. Bacterial quorum-sensing signal IQS induces host cell apoptosis by targeting POT1-p53 signalling pathway. Cell Microbiol. 2019, 21, e13076. [Google Scholar] [CrossRef]
- Schick, A.; Kassen, R. Rapid diversification of Pseudomonas aeruginosa in cystic fibrosis lung-like conditions. Proc. Natl. Acad. Sci. USA 2018, 115, 10714–10719. [Google Scholar] [CrossRef] [PubMed]
- Berical, A.; Lee, R.E.; Randell, S.H.; Hawkins, F. Challenges Facing Airway Epithelial Cell-Based Therapy for Cystic Fibrosis. Front. Pharmacol. 2019, 10, 74. [Google Scholar] [CrossRef]
- Rossi, E.; La Rosa, R.; Bartell, J.A.; Marvig, R.L.; Haagensen, J.A.J.; Sommer, L.M.; Molin, S.; Johansen, H.K. Pseudomonas aeruginosa adaptation and evolution in patients with cystic fibrosis. Nat. Rev. Microbiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Bhagirath, A.Y.; Li, Y.; Somayajula, D.; Dadashi, M.; Badr, S.; Duan, K. Cystic fibrosis lung environment and Pseudomonas aeruginosa infection. BMC Pulm. Med. 2016, 16, 174. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.A.; Morelli, P.; Galietta, L.J.; Colin, A.A. Airway microenvironment alterations and pathogen growth in cystic fibrosis. Pediatric Pulmonol. 2019, 54, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.J.; LiPuma, J.J. The Microbiome in Cystic Fibrosis. Clin. Chest Med. 2016, 37, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Beswick, E.; Amich, J.; Gago, S. Factoring in the Complexity of the Cystic Fibrosis Lung to Understand Aspergillus fumigatus and Pseudomonas aeruginosa Interactions. Pathogens 2020, 9, 639. [Google Scholar] [CrossRef]
- Williams, H.D.; Behrends, V.; Bundy, J.G.; Ryall, B.; Zlosnik, J.E. Hypertonic Saline Therapy in Cystic Fibrosis: Do Population Shifts Caused by the Osmotic Sensitivity of Infecting Bacteria Explain the Effectiveness of this Treatment? Front. Microbiol. 2010, 1, 120. [Google Scholar] [CrossRef]
- Venkatakrishnan, V.; Thaysen-Andersen, M.; Chen, S.C.; Nevalainen, H.; Packer, N.H. Cystic fibrosis and bacterial colonization define the sputum N-glycosylation phenotype. Glycobiology 2015, 25, 88–100. [Google Scholar] [CrossRef]
- Vankeerberghen, A.; Cuppens, H.; Cassiman, J.J. The cystic fibrosis transmembrane conductance regulator: An intriguing protein with pleiotropic functions. J. Cyst. Fibros. 2002, 1, 13–29. [Google Scholar] [CrossRef]
- Saint-Criq, V.; Gray, M.A. Role of CFTR in epithelial physiology. Cell Mol. Life Sci. 2017, 74, 93–115. [Google Scholar] [CrossRef] [PubMed]
- Pezzulo, A.A.; Tang, X.X.; Hoegger, M.J.; Abou Alaiwa, M.H.; Ramachandran, S.; Moninger, T.O.; Karp, P.H.; Wohlford-Lenane, C.L.; Haagsman, H.P.; van Eijk, M.; et al. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature 2012, 487, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Erra Díaz, F.; Dantas, E.; Geffner, J. Unravelling the Interplay between Extracellular Acidosis and Immune Cells. Mediat. Inflamm. 2018, 2018, 1218297. [Google Scholar] [CrossRef]
- Møller, S.A.; Jensen, P.; Høiby, N.; Ciofu, O.; Kragh, K.N.; Bjarnsholt, T.; Kolpen, M. Hyperbaric oxygen treatment increases killing of aggregating Pseudomonas aeruginosa isolates from cystic fibrosis patients. J. Cyst. Fibros. 2019, 18, 657–664. [Google Scholar] [CrossRef]
- Riquelme, S.A.; Lozano, C.; Moustafa, A.M.; Liimatta, K.; Tomlinson, K.L.; Britto, C.; Khanal, S.; Gill, S.K.; Narechania, A.; Azcona-Gutiérrez, J.M.; et al. CFTR-PTEN-dependent mitochondrial metabolic dysfunction promotes Pseudomonas aeruginosa airway infection. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Chmiel, J.F.; Aksamit, T.R.; Chotirmall, S.H.; Dasenbrook, E.C.; Elborn, J.S.; LiPuma, J.J.; Ranganathan, S.C.; Waters, V.J.; Ratjen, F.A. Antibiotic management of lung infections in cystic fibrosis. I. The microbiome, methicillin-resistant Staphylococcus aureus, gram-negative bacteria, and multiple infections. Ann. Am. Thorac. Soc. 2014, 11, 1120–1129. [Google Scholar]
- Quinn, R.A.; Adem, S.; Mills, R.H.; Comstock, W.; DeRight Goldasich, L.; Humphrey, G.; Aksenov, A.A.; Melnik, A.V.; da Silva, R.; Ackermann, G.; et al. Neutrophilic proteolysis in the cystic fibrosis lung correlates with a pathogenic microbiome. Microbiome 2019, 7, 23. [Google Scholar] [CrossRef]
- Guillot, L.; Beucher, J.; Tabary, O.; Le Rouzic, P.; Clement, A.; Corvol, H. Lung disease modifier genes in cystic fibrosis. Int. J. Biochem. Cell Biol. 2014, 52, 83–93. [Google Scholar] [CrossRef]
- Di Paola, M.; Park, A.J.; Ahmadi, S.; Roach, E.J.; Wu, Y.S.; Struder-Kypke, M.; Lam, J.S.; Bear, C.E.; Khursigara, C.M. SLC6A14 is a genetic modifier of cystic fibrosis that regulates Pseudomonas aeruginosa attachment to human bronchial epithelial cells. mBio 2017, 8. [Google Scholar] [CrossRef]
- Park, J.E.; Yung, R.; Stefanowicz, D.; Shumansky, K.; Akhabir, L.; Durie, P.R.; Corey, M.; Zielenski, J.; Dorfman, R.; Daley, D.; et al. Cystic fibrosis modifier genes related to Pseudomonas aeruginosa infection. Genes Immun. 2011, 12, 370–377. [Google Scholar] [CrossRef]
- Zeitlin, P.L. Cystic fibrosis and estrogens: A perfect storm. J. Clin. Investig. 2008, 118, 3841–3844. [Google Scholar] [CrossRef]
- Sweezey, N.B.; Ratjen, F. The cystic fibrosis gender gap: Potential roles of estrogen. Pediatric Pulmonol. 2014, 49, 309–317. [Google Scholar] [CrossRef]
- Saint-Criq, V.; Harvey, B.J. Estrogen and the cystic fibrosis gender gap. Steroids 2014, 81, 4–8. [Google Scholar] [CrossRef] [PubMed]
- Chotirmall, S.H.; Smith, S.G.; Gunaratnam, C.; Cosgrove, S.; Dimitrov, B.D.; O’Neill, S.J.; Harvey, B.J.; Greene, C.M.; McElvaney, N.G. Effect of estrogen on Pseudomonas mucoidy and exacerbations in cystic fibrosis. N. Engl. J. Med. 2012, 366, 1978–1986. [Google Scholar] [CrossRef] [PubMed]
- Tyrrell, J.; Harvey, B.J. Sexual dimorphism in the microbiology of the CF ‘Gender Gap’: Estrogen modulation of Pseudomonas aeruginosa virulence. Steroids 2020, 156, 108575. [Google Scholar] [CrossRef]
- Folkesson, A.; Jelsbak, L.; Yang, L.; Johansen, H.K.; Ciofu, O.; Høiby, N.; Molin, S. Adaptation of Pseudomonas aeruginosa to the cystic fibrosis airway: An evolutionary perspective. Nat. Rev. Microbiol. 2012, 10, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Bianconi, I.; Jeukens, J.; Freschi, L.; Alcalá-Franco, B.; Facchini, M.; Boyle, B.; Molinaro, A.; Kukavica-Ibrulj, I.; Tümmler, B.; Levesque, R.C.; et al. Comparative genomics and biological characterization of sequential Pseudomonas aeruginosa isolates from persistent airways infection. BMC Genom. 2015, 16, 1105. [Google Scholar] [CrossRef] [PubMed]
- Cattoir, V.; Narasimhan, G.; Skurnik, D.; Aschard, H.; Roux, D.; Ramphal, R.; Jyot, J.; Lory, S. Transcriptional response of mucoid Pseudomonas aeruginosa to human respiratory mucus. mBio 2013, 3, e00410-12. [Google Scholar] [CrossRef] [PubMed]
- Oliver, A.; Mena, A. Bacterial hypermutation in cystic fibrosis, not only for antibiotic resistance. Clin. Microbiol. Infect. 2010, 16, 798–808. [Google Scholar] [CrossRef]
- Colque, C.A.; Albarracín Orio, A.G.; Feliziani, S.; Marvig, R.L.; Tobares, A.R.; Johansen, H.K.; Molin, S.; Smania, A.M. Hypermutator Pseudomonas aeruginosa Exploits Multiple Genetic Pathways To Develop Multidrug Resistance during Long-Term Infections in the Airways of Cystic Fibrosis Patients. Antimicrob. Agents Chemother. 2020, 64, e02142-19. [Google Scholar] [CrossRef]
- Feliziani, S.; Marvig, R.L.; Luján, A.M.; Moyano, A.J.; Di Rienzo, J.A.; Krogh Johansen, H.; Molin, S.; Smania, A.M. Coexistence and within-host evolution of diversified lineages of hypermutable Pseudomonas aeruginosa in long-term cystic fibrosis infections. PLoS Genet. 2014, 10, e1004651. [Google Scholar] [CrossRef]
- López-Causapé, C.; Rojo-Molinero, E.; Macià, M.D.; Oliver, A. The problems of antibiotic resistance in cystic fibrosis and solutions. Expert Rev. Respir. Med. 2015, 9, 73–88. [Google Scholar] [CrossRef]
- Fothergill, J.L.; Walshaw, M.J.; Winstanley, C. Transmissible strains of Pseudomonas aeruginosa in cystic fibrosis lung infections. Eur. Respir. J. 2012, 40, 227–238. [Google Scholar] [CrossRef]
- Workentine, M.L.; Sibley, C.D.; Glezerson, B.; Purighalla, S.; Norgaard-Gron, J.C.; Parkins, M.D.; Rabin, H.R.; Surette, M.G. Phenotypic heterogeneity of Pseudomonas aeruginosa populations in a cystic fibrosis patient. PLoS ONE 2013, 8, e60225. [Google Scholar] [CrossRef]
- Markussen, T.; Marvig, R.L.; Gómez-Lozano, M.; Aanæs, K.; Burleigh, A.E.; Høiby, N.; Johansen, H.K.; Molin, S.; Jelsbak, L. Environmental heterogeneity drives within-host diversification and evolution of Pseudomonas aeruginosa. mBio 2014, 5, e01592-14. [Google Scholar] [CrossRef]
- Jorth, P.; Staudinger, B.J.; Wu, X.; Hisert, K.B.; Hayden, H.; Garudathri, J.; Harding, C.L.; Radey, M.C.; Rezayat, A.; Bautista, G.; et al. Regional Isolation Drives Bacterial Diversification within Cystic Fibrosis Lungs. Cell Host Microbe 2015, 18, 307–319. [Google Scholar] [CrossRef]
- Malone, J.G. Role of small colony variants in persistence of Pseudomonas aeruginosa infections in cystic fibrosis lungs. Infect. Drug Resist. 2015, 8, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Harrison, J.J.; Almblad, H.; Irie, Y.; Wolter, D.J.; Eggleston, H.C.; Randall, T.E.; Kitzman, J.O.; Stackhouse, B.; Emerson, J.C.; Mcnamara, S.; et al. Elevated exopolysaccharide levels in Pseudomonas aeruginosa flagellar mutants have implications for biofilm growth and chronic infections. PLoS Genet. 2020, 16, e1008848. [Google Scholar] [CrossRef] [PubMed]
- Pestrak, M.J.; Chaney, S.B.; Eggleston, H.C.; Dellos-Nolan, S.; Dixit, S.; Mathew-Steiner, S.S.; Roy, S.; Parsek, M.R.; Sen, C.K.; Wozniak, D.J. Pseudomonas aeruginosa rugose small-colony variants evade host clearance, are hyper-inflammatory, and persist in multiple host environments. PLoS Pathog. 2018, 14, e1006842. [Google Scholar] [CrossRef] [PubMed]
- Bartell, J.A.; Cameron, D.R.; Mojsoska, B.; Haagensen, J.A.J.; Pressler, T.; Sommer, L.M.; Lewis, K.; Molin, S.; Johansen, H.K. Bacterial persisters in long-term infection: Emergence and fitness in a complex host environment. PLoS Pathog. 2020, 16, e1009112. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, S.; Hayes, D.; Wozniak, D.J. Mucoid Pseudomonas aeruginosa and regional inflammation in the cystic fibrosis lung. J. Cyst. Fibros. 2019, 18, 796–803. [Google Scholar] [CrossRef] [PubMed]
- Candido Caçador, N.; Paulino da Costa Capizzani, C.; Gomes Monteiro Marin Torres, L.A.; Galetti, R.; Ciofu, O.; da Costa Darini, A.L.; Høiby, N. Adaptation of Pseudomonas aeruginosa to the chronic phenotype by mutations in the algTmucABD operon in isolates from Brazilian cystic fibrosis patients. PLoS ONE 2018, 13, e0208013. [Google Scholar] [CrossRef]
- Freschi, L.; Bertelli, C.; Jeukens, J.; Moore, M.P.; Kukavica-Ibrulj, I.; Emond-Rheault, J.G.; Hamel, J.; Fothergill, J.L.; Tucker, N.P.; McClean, S.; et al. Genomic characterisation of an international Pseudomonas aeruginosa reference panel indicates that the two major groups draw upon distinct mobile gene pools. FEMS Microbiol. Lett. 2018, 365. [Google Scholar] [CrossRef]
- Smith, D.J.; Lamont, I.L.; Anderson, G.J.; Reid, D.W. Targeting iron uptake to control Pseudomonas aeruginosa infections in cystic fibrosis. Eur. Respir. J. 2013, 42, 1723–1736. [Google Scholar] [CrossRef]
- Jones, C.J.; Wozniak, D.J. Psl Produced by Mucoid Pseudomonas aeruginosa Contributes to the Establishment of Biofilms and Immune Evasion. mBio 2017, 8, e00864-17. [Google Scholar] [CrossRef]
- Rao, J.; Damron, F.H.; Basler, M.; Digiandomenico, A.; Sherman, N.E.; Fox, J.W.; Mekalanos, J.J.; Goldberg, J.B. Comparisons of Two Proteomic Analyses of Non-Mucoid and Mucoid Pseudomonas aeruginosa Clinical Isolates from a Cystic Fibrosis Patient. Front. Microbiol. 2011, 2, 162. [Google Scholar] [CrossRef]
- Chen, L.; Zou, Y.; Kronfl, A.A.; Wu, Y. Type VI secretion system of Pseudomonas aeruginosa is associated with biofilm formation but not environmental adaptation. Microbiologyopen 2020, 9, e991. [Google Scholar] [CrossRef]
- Malhotra, S.; Limoli, D.H.; English, A.E.; Parsek, M.R.; Wozniak, D.J. Mixed Communities of Mucoid and Nonmucoid Pseudomonas aeruginosa Exhibit Enhanced Resistance to Host Antimicrobials. mBio 2018, 9, e00275-18. [Google Scholar] [CrossRef]
- Cullen, L.; Weiser, R.; Olszak, T.; Maldonado, R.F.; Moreira, A.S.; Slachmuylders, L.; Brackman, G.; Paunova-Krasteva, T.S.; Zarnowiec, P.; Czerwonka, G.; et al. Phenotypic characterization of an international Pseudomonas aeruginosa reference panel: Strains of cystic fibrosis (CF) origin show less in vivo virulence than non-CF strains. Microbiology 2015, 161, 1961–1977. [Google Scholar] [CrossRef]
- Cross, A.R.; Goldberg, J.B. Remodeling of O Antigen in Mucoid Pseudomonas aeruginosa via Transcriptional Repression of wzz2. mBio 2019, 10, e02914-18. [Google Scholar] [CrossRef]
- Di Lorenzo, F.; Silipo, A.; Bianconi, I.; Lore, N.I.; Scamporrino, A.; Sturiale, L.; Garozzo, D.; Lanzetta, R.; Parrilli, M.; Bragonzi, A.; et al. Persistent cystic fibrosis isolate Pseudomonas aeruginosa strain RP73 exhibits an under-acylated LPS structure responsible of its low inflammatory activity. Mol. Immunol. 2015, 63, 166–175. [Google Scholar] [CrossRef]
- SenGupta, S.; Hittle, L.E.; Ernst, R.K.; Uriarte, S.M.; Mitchell, T.C. A Pseudomonas aeruginosa hepta-acylated lipid A variant associated with cystic fibrosis selectively activates human neutrophils. J. Leukoc. Biol. 2016, 100, 1047–1059. [Google Scholar] [CrossRef]
- Tart, A.H.; Blanks, M.J.; Wozniak, D.J. The AlgT-dependent transcriptional regulator AmrZ (AlgZ) inhibits flagellum biosynthesis in mucoid, nonmotile Pseudomonas aeruginosa cystic fibrosis isolates. J. Bacteriol. 2006, 188, 6483–6489. [Google Scholar] [CrossRef] [PubMed]
- Wolfgang, M.C.; Jyot, J.; Goodman, A.L.; Ramphal, R.; Lory, S. Pseudomonas aeruginosa regulates flagellin expression as part of a global response to airway fluid from cystic fibrosis patients. Proc. Natl. Acad. Sci. USA 2004, 101, 6664–6668. [Google Scholar] [CrossRef] [PubMed]
- Cigana, C.; Lorè, N.I.; Bernardini, M.L.; Bragonzi, A. Dampening Host Sensing and Avoiding Recognition in Pseudomonas aeruginosa Pneumonia. J. Biomed. Biotechnol. 2011, 2011, 852513. [Google Scholar] [CrossRef]
- Huus, K.E.; Joseph, J.; Zhang, L.; Wong, A.; Aaron, S.D.; Mah, T.F.; Sad, S. Clinical Isolates of Pseudomonas aeruginosa from Chronically Infected Cystic Fibrosis Patients Fail to Activate the Inflammasome during Both Stable Infection and Pulmonary Exacerbation. J. Immunol. 2016, 196, 3097–3108. [Google Scholar] [CrossRef] [PubMed]
- Jyot, J.; Sonawane, A.; Wu, W.; Ramphal, R. Genetic mechanisms involved in the repression of flagellar assembly by Pseudomonas aeruginosa in human mucus. Mol. Microbiol. 2007, 63, 1026–1038. [Google Scholar] [CrossRef] [PubMed]
- Floyd, M.; Winn, M.; Cullen, C.; Sil, P.; Chassaing, B.; Yoo, D.G.; Gewirtz, A.T.; Goldberg, J.B.; McCarter, L.L.; Rada, B. Swimming Motility Mediates the Formation of Neutrophil Extracellular Traps Induced by Flagellated Pseudomonas aeruginosa. PLoS Pathog. 2016, 12, e1005987. [Google Scholar] [CrossRef] [PubMed]
- Amiel, E.; Lovewell, R.R.; O’Toole, G.A.; Hogan, D.A.; Berwin, B. Pseudomonas aeruginosa evasion of phagocytosis is mediated by loss of swimming motility and is independent of flagellum expression. Infect. Immun. 2010, 78, 2937–2945. [Google Scholar] [CrossRef]
- Patankar, Y.R.; Lovewell, R.R.; Poynter, M.E.; Jyot, J.; Kazmierczak, B.I.; Berwin, B. Flagellar motility is a key determinant of the magnitude of the inflammasome response to Pseudomonas aeruginosa. Infect. Immun. 2013, 81, 2043–2052. [Google Scholar] [CrossRef]
- Chang, Y.S.; Klockgether, J.; Tümmler, B. An intragenic deletion in pilQ leads to nonpiliation of a Pseudomonas aeruginosa strain isolated from cystic fibrosis lung. FEMS Microbiol. Lett. 2007, 270, 201–206. [Google Scholar] [CrossRef]
- Hogardt, M.; Heesemann, J. Adaptation of Pseudomonas aeruginosa during persistence in the cystic fibrosis lung. Int. J. Med. Microbiol. 2010, 300, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Xu, A.; Zhang, M.; Du, W.; Wang, D.; Ma, L.Z. A molecular mechanism for how sigma factor AlgT and transcriptional regulator AmrZ inhibit twitching motility in Pseudomonas aeruginosa. Environ. Microbiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Badrane, H.; Arora, S.; Baker, H.V.; Jin, S. MucA-mediated coordination of type III secretion and alginate synthesis in Pseudomonas aeruginosa. J. Bacteriol. 2004, 186, 7575–7585. [Google Scholar] [CrossRef] [PubMed]
- Intile, P.J.; Diaz, M.R.; Urbanowski, M.L.; Wolfgang, M.C.; Yahr, T.L. The AlgZR two-component system recalibrates the RsmAYZ posttranscriptional regulatory system to inhibit expression of the Pseudomonas aeruginosa type III secretion system. J. Bacteriol. 2014, 196, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Ryall, B.; Carrara, M.; Zlosnik, J.E.; Behrends, V.; Lee, X.; Wong, Z.; Lougheed, K.E.; Williams, H.D. The mucoid switch in Pseudomonas aeruginosa represses quorum sensing systems and leads to complex changes to stationary phase virulence factor regulation. PLoS ONE 2014, 9, e96166. [Google Scholar] [CrossRef]
- Mehta, H.H.; Prater, A.G.; Beabout, K.; Elworth, R.A.L.; Karavis, M.; Gibbons, H.S.; Shamoo, Y. The Essential Role of Hypermutation in Rapid Adaptation to Antibiotic Stress. Antimicrob. Agents Chemother. 2019, 63, e00744-19. [Google Scholar] [CrossRef]
- D’Argenio, D.A.; Wu, M.; Hoffman, L.R.; Kulasekara, H.D.; Déziel, E.; Smith, E.E.; Nguyen, H.; Ernst, R.K.; Larson Freeman, T.J.; Spencer, D.H.; et al. Growth phenotypes of Pseudomonas aeruginosa lasR mutants adapted to the airways of cystic fibrosis patients. Mol. Microbiol. 2007, 64, 512–533. [Google Scholar] [CrossRef]
- Hoffman, L.R.; Kulasekara, H.D.; Emerson, J.; Houston, L.S.; Burns, J.L.; Ramsey, B.W.; Miller, S.I. Pseudomonas aeruginosa lasR mutants are associated with cystic fibrosis lung disease progression. J. Cyst. Fibros. 2009, 8, 66–70. [Google Scholar] [CrossRef]
- Jiricny, N.; Molin, S.; Foster, K.; Diggle, S.P.; Scanlan, P.D.; Ghoul, M.; Johansen, H.K.; Santorelli, L.A.; Popat, R.; West, S.A.; et al. Loss of social behaviours in populations of Pseudomonas aeruginosa infecting lungs of patients with cystic fibrosis. PLoS ONE 2014, 9, e83124. [Google Scholar] [CrossRef]
- LaFayette, S.L.; Houle, D.; Beaudoin, T.; Wojewodka, G.; Radzioch, D.; Hoffman, L.R.; Burns, J.L.; Dandekar, A.A.; Smalley, N.E.; Chandler, J.R.; et al. Cystic fibrosis–adapted Pseudomonas aeruginosa quorum sensing lasR mutants cause hyperinflammatory responses. Sci. Adv. 2015, 1, e1500199. [Google Scholar] [CrossRef]
- La Rosa, R.; Johansen, H.K.; Molin, S. Adapting to the Airways: Metabolic Requirements of Pseudomonas aeruginosa during the Infection of Cystic Fibrosis Patients. Metabolites 2019, 9, 234. [Google Scholar] [CrossRef]
- Kumar, S.S.; Penesyan, A.; Elbourne, L.D.H.; Gillings, M.R.; Paulsen, I.T. Catabolism of Nucleic Acids by a Cystic Fibrosis Pseudomonas aeruginosa Isolate: An adaptive pathway to cystic fibrosis sputum environment. Front. Microbiol. 2019, 10, 1199. [Google Scholar] [CrossRef]
- Marvig, R.L.; Damkiær, S.; Khademi, S.M.; Markussen, T.M.; Molin, S.; Jelsbak, L. Within-host evolution of Pseudomonas aeruginosa reveals adaptation toward iron acquisition from hemoglobin. mBio 2014, 5, e00966-14. [Google Scholar] [CrossRef]
- Dingemans, J.; Ye, L.; Hildebrand, F.; Tontodonati, F.; Craggs, M.; Bilocq, F.; De Vos, D.; Crabbé, A.; Van Houdt, R.; Malfroot, A.; et al. The deletion of TonB-dependent receptor genes is part of the genome reduction process that occurs during adaptation of Pseudomonas aeruginosa to the cystic fibrosis lung. Pathog. Dis. 2014, 71, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Bianconi, I.; D’Arcangelo, S.; Esposito, A.; Benedet, M.; Piffer, E.; Dinnella, G.; Gualdi, P.; Schinella, M.; Baldo, E.; Donati, C.; et al. Persistence and Microevolution of Pseudomonas aeruginosa in the Cystic Fibrosis Lung: A Single-Patient Longitudinal Genomic Study. Front. Microbiol. 2018, 9, 3242. [Google Scholar] [CrossRef] [PubMed]
- Llanes, C.; Pourcel, C.; Richardot, C.; Plésiat, P.; Fichant, G.; Cavallo, J.D.; Mérens, A.; Group, G.S. Diversity of β-lactam resistance mechanisms in cystic fibrosis isolates of Pseudomonas aeruginosa: A French multicentre study. J. Antimicrob. Chemother. 2013, 68, 1763–17671. [Google Scholar] [CrossRef]
- Castanheira, M.; Mills, J.C.; Farrell, D.J.; Jones, R.N. Mutation-driven β-lactam resistance mechanisms among contemporary ceftazidime-nonsusceptible Pseudomonas aeruginosa isolates from U.S. hospitals. Antimicrob. Agents Chemother. 2014, 58, 6844–6850. [Google Scholar] [CrossRef] [PubMed]
- Varga, J.J.; Barbier, M.; Mulet, X.; Bielecki, P.; Bartell, J.A.; Owings, J.P.; Martinez-Ramos, I.; Hittle, L.E.; Davis, M.R.; Damron, F.H.; et al. Genotypic and phenotypic analyses of a Pseudomonas aeruginosa chronic bronchiectasis isolate reveal differences from cystic fibrosis and laboratory strains. BMC Genom. 2015, 16, 883. [Google Scholar] [CrossRef]
- Schmidt, K.D.; Tümmler, B.; Römling, U. Comparative genome mapping of Pseudomonas aeruginosa PAO with P. aeruginosa C, which belongs to a major clone in cystic fibrosis patients and aquatic habitats. J. Bacteriol. 1996, 178, 85–93. [Google Scholar] [CrossRef]
- Mielko, K.A.; Jabłoński, S.J.; Milczewska, J.; Sands, D.; Łukaszewicz, M.; Młynarz, P. Metabolomic studies of Pseudomonas aeruginosa. World J. Microbiol. Biotechnol. 2019, 35, 178. [Google Scholar] [CrossRef] [PubMed]
- Parkins, M.D.; Somayaji, R.; Waters, V.J. Epidemiology, Biology, and Impact of Clonal Pseudomonas aeruginosa Infections in Cystic Fibrosis. Clin. Microbiol. Rev. 2018, 31. [Google Scholar] [CrossRef] [PubMed]
- Winsor, G.L.; Griffiths, E.J.; Lo, R.; Dhillon, B.K.; Shay, J.A.; Brinkman, F.S. Enhanced annotations and features for comparing thousands of Pseudomonas genomes in the Pseudomonas genome database. Nucleic Acids Res. 2016, 44, D646–D653. [Google Scholar] [CrossRef]
- Romero, P.; Karp, P. PseudoCyc, a pathway-genome database for Pseudomonas aeruginosa. J. Mol. Microbiol. Biotechnol. 2003, 5, 230–239. [Google Scholar] [CrossRef]
- Choi, C.; Münch, R.; Leupold, S.; Klein, J.; Siegel, I.; Thielen, B.; Benkert, B.; Kucklick, M.; Schobert, M.; Barthelmes, J.; et al. SYSTOMONAS--an integrated database for systems biology analysis of Pseudomonas. Nucleic Acids Res. 2007, 35, D533–D537. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem Substance and Compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef]
- Wishart, D.S.; Jewison, T.; Guo, A.C.; Wilson, M.; Knox, C.; Liu, Y.; Djoumbou, Y.; Mandal, R.; Aziat, F.; Dong, E.; et al. HMDB 3.0—The Human Metabolome Database in 2013. Nucleic Acids Res. 2013, 41, D801–D807. [Google Scholar] [CrossRef]
- Marvig, R.L.; Sommer, L.M.; Molin, S.; Johansen, H.K. Convergent evolution and adaptation of Pseudomonas aeruginosa within patients with cystic fibrosis. Nat. Genet. 2015, 47, 57–64. [Google Scholar] [CrossRef]
- Greipel, L.; Fischer, S.; Klockgether, J.; Dorda, M.; Mielke, S.; Wiehlmann, L.; Cramer, N.; Tümmler, B. Molecular Epidemiology of Mutations in Antimicrobial Resistance Loci of Pseudomonas aeruginosa Isolates from Airways of Cystic Fibrosis Patients. Antimicrob. Agents Chemother. 2016, 60, 6726–6734. [Google Scholar] [CrossRef]
- Ahmed, M.N.; Abdelsamad, A.; Wassermann, T.; Porse, A.; Becker, J.; Sommer, M.O.A.; Høiby, N.; Ciofu, O. The evolutionary trajectories of P. aeruginosa in biofilm and planktonic growth modes exposed to ciprofloxacin: Beyond selection of antibiotic resistance. NPJ Biofilms Microbiomes 2020, 6, 28. [Google Scholar] [CrossRef]
- La Rosa, R.; Johansen, H.K.; Molin, S. Convergent Metabolic Specialization through Distinct Evolutionary Paths in Pseudomonas aeruginosa. mBio 2018, 9. [Google Scholar] [CrossRef]
- Martínez-Carranza, E.; García-Reyes, S.; González-Valdez, A.; Soberón-Chávez, G. Tracking the genome of four Pseudomonas aeruginosa isolates that have a defective Las quorum-sensing system, but are still virulent. Access Microbiol. 2020, 2, acmi000132. [Google Scholar] [CrossRef]
- Bartell, J.A.; Sommer, L.M.; Haagensen, J.A.J.; Loch, A.; Espinosa, R.; Molin, S.; Johansen, H.K. Evolutionary highways to persistent bacterial infection. Nat. Commun. 2019, 10, 629. [Google Scholar] [CrossRef]
- Williams, D.; Evans, B.; Haldenby, S.; Walshaw, M.J.; Brockhurst, M.A.; Winstanley, C.; Paterson, S. Divergent, coexisting Pseudomonas aeruginosa lineages in chronic cystic fibrosis lung infections. Am. J. Respir. Crit. Care Med. 2015, 191, 775–785. [Google Scholar] [CrossRef]
- Cramer, N.; Klockgether, J.; Wrasman, K.; Schmidt, M.; Davenport, C.F.; Tümmler, B. Microevolution of the major common Pseudomonas aeruginosa clones C and PA14 in cystic fibrosis lungs. Environ. Microbiol. 2011, 13, 1690–1704. [Google Scholar] [CrossRef]
- Grothues, D.; Koopmann, U.; von der Hardt, H.; Tümmler, B. Genome fingerprinting of Pseudomonas aeruginosa indicates colonization of cystic fibrosis siblings with closely related strains. J. Clin. Microbiol. 1988, 26, 1973–1977. [Google Scholar] [CrossRef]
- Thomassen, M.J.; Demko, C.A.; Doershuk, C.F.; Root, J.M. Pseudomonas aeruginosa isolates: Comparisons of isolates from campers and from sibling pairs with cystic fibrosis. Pediatr Pulmonol. 1985, 1, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Wolz, C.; Kiosz, G.; Ogle, J.W.; Vasil, M.L.; Schaad, U.; Botzenhart, K.; Döring, G. Pseudomonas aeruginosa cross-colonization and persistence in patients with cystic fibrosis. Use of a DNA probe. Epidemiol. Infect. 1989, 102, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, S.S.; Jensen, T.; Pressler, T.; Høiby, N.; Rosendal, K. Does centralized treatment of cystic fibrosis increase the risk of Pseudomonas aeruginosa infection? Acta Paediatr Scand. 1986, 75, 840–845. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, S.S.; Koch, C.; Høiby, N.; Rosendal, K. An epidemic spread of multiresistant Pseudomonas aeruginosa in a cystic fibrosis centre. J. Antimicrob. Chemother. 1986, 17, 505–516. [Google Scholar] [CrossRef]
- Cheng, K.; Smyth, R.L.; Govan, J.R.; Doherty, C.; Winstanley, C.; Denning, N.; Heaf, D.P.; van Saene, H.; Hart, C.A. Spread of beta-lactam-resistant Pseudomonas aeruginosa in a cystic fibrosis clinic. Lancet 1996, 348, 639–642. [Google Scholar] [CrossRef]
- Hu, H.; Harmer, C.; Anuj, S.; Wainwright, C.E.; Manos, J.; Cheney, J.; Harbour, C.; Zablotska, I.; Turnbull, L.; Whitchurch, C.B.; et al. Type 3 secretion system effector genotype and secretion phenotype of longitudinally collected Pseudomonas aeruginosa isolates from young children diagnosed with cystic fibrosis following newborn screening. Clin. Microbiol. Infect. 2013, 19, 266–272. [Google Scholar] [CrossRef]
- Kidd, T.J.; Ramsay, K.A.; Vidmar, S.; Carlin, J.B.; Bell, S.C.; Wainwright, C.E.; Grimwood, K.; Investigators, A.S. Pseudomonas aeruginosa genotypes acquired by children with cystic fibrosis by age 5-years. J. Cyst. Fibros. 2015, 14, 361–369. [Google Scholar] [CrossRef]
- Ranganathan, S.C.; Skoric, B.; Ramsay, K.A.; Carzino, R.; Gibson, A.M.; Hart, E.; Harrison, J.; Bell, S.C.; Kidd, T.J. Geographical differences in first acquisition of Pseudomonas aeruginosa in cystic fibrosis. Ann. Am. Thorac. Soc. 2013, 10, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Fluge, G.; Ojeniyi, B.; Høiby, N.; Digranes, A.; Ciofu, O.; Hunstad, E.; Haanaes, O.C.; Storrøsten, O.T. Typing of Pseudomonas aeruginosa strains in Norwegian cystic fibrosis patients. Clin. Microbiol. Infect. 2001, 7, 238–243. [Google Scholar] [CrossRef] [PubMed]
- Schmid, J.; Ling, L.J.; Leung, J.L.; Zhang, N.; Kolbe, J.; Wesley, A.W.; Mills, G.D.; Brown, P.J.; Jones, D.T.; Laing, R.T.; et al. Pseudomonas aeruginosa transmission is infrequent in New Zealand cystic fibrosis clinics. Eur. Respir. J. 2008, 32, 1583–1590. [Google Scholar] [CrossRef] [PubMed]
- Van Daele, S.; Vaneechoutte, M.; De Boeck, K.; Knoop, C.; Malfroot, A.; Lebecque, P.; Leclercq-Foucart, J.; Van Schil, L.; Desager, K.; De Baets, F. Survey of Pseudomonas aeruginosa genotypes in colonised cystic fibrosis patients. Eur. Respir. J. 2006, 28, 740–747. [Google Scholar] [CrossRef] [PubMed]
- Ashish, A.; Paterson, S.; Mowat, E.; Fothergill, J.L.; Walshaw, M.J.; Winstanley, C. Extensive diversification is a common feature of Pseudomonas aeruginosa populations during respiratory infections in cystic fibrosis. J. Cyst. Fibros. 2013, 12, 790–793. [Google Scholar] [CrossRef] [PubMed]
- Tingpej, P.; Smith, L.; Rose, B.; Zhu, H.; Conibear, T.; Al Nassafi, K.; Manos, J.; Elkins, M.; Bye, P.; Willcox, M.; et al. Phenotypic characterization of clonal and nonclonal Pseudomonas aeruginosa strains isolated from lungs of adults with cystic fibrosis. J. Clin. Microbiol. 2007, 45, 1697–1704. [Google Scholar] [CrossRef]
- Ciofu, O.; Mandsberg, L.F.; Bjarnsholt, T.; Wassermann, T.; Høiby, N. Genetic adaptation of Pseudomonas aeruginosa during chronic lung infection of patients with cystic fibrosis: Strong and weak mutators with heterogeneous genetic backgrounds emerge in mucA and/or lasR mutants. Microbiology 2010, 156, 1108–1119. [Google Scholar] [CrossRef]
- Sokurenko, E.V.; Hasty, D.L.; Dykhuizen, D.E. Pathoadaptive mutations: Gene loss and variation in bacterial pathogens. Trends Microbiol. 1999, 7, 191–195. [Google Scholar] [CrossRef]
- Sanz-García, F.; Hernando-Amado, S.; Martínez, J.L. Mutation-Driven Evolution of Pseudomonas aeruginosa in the Presence of either. Ceftazidime or Ceftazidime-Avibactam. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Rojas, A.; Mena, A.; Martín, S.; Borrell, N.; Oliver, A.; Blázquez, J. Inactivation of the hmgA gene of Pseudomonas aeruginosa leads to pyomelanin hyperproduction, stress resistance and increased persistence in chronic lung infection. Microbiology 2009, 155, 1050–1057. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Ortega, C.; Wiegand, I.; Olivares, J.; Hancock, R.E.; Martínez, J.L. Genetic determinants involved in the susceptibility of Pseudomonas aeruginosa to beta-lactam antibiotics. Antimicrob. Agents Chemother. 2010, 54, 4159–4167. [Google Scholar] [CrossRef]
- Masuda, N.; Sakagawa, E.; Ohya, S.; Gotoh, N.; Tsujimoto, H.; Nishino, T. Contribution of the MexX-MexY-oprM efflux system to intrinsic resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2000, 44, 2242–2246. [Google Scholar] [CrossRef]
- Oakley, J.L.; Weiser, R.; Powell, L.C.; Forton, J.; Mahenthiralingam, E.; Rye, P.D.; Hill, K.E.; Thomas, D.W.; Pritchard, M.F. Phenotypic and Genotypic Adaptations in Pseudomonas aeruginosa Biofilms following Long-Term Exposure to an Alginate Oligomer Therapy. mSphere 2021, 6. [Google Scholar] [CrossRef]
- Flynn, K.M.; Dowell, G.; Johnson, T.M.; Koestler, B.J.; Waters, C.M.; Cooper, V.S. Evolution of Ecological Diversity in Biofilms of Pseudomonas aeruginosa by Altered Cyclic Diguanylate Signaling. J. Bacteriol. 2016, 198, 2608–2618. [Google Scholar] [CrossRef] [PubMed]
- Vanderwoude, J.; Fleming, D.; Azimi, S.; Trivedi, U.; Rumbaugh, K.P.; Diggle, S.P. The evolution of virulence in Pseudomonas aeruginosa during chronic wound infection. Proc. Biol. Sci. 2020, 287, 20202272. [Google Scholar] [CrossRef]



{kind=link}
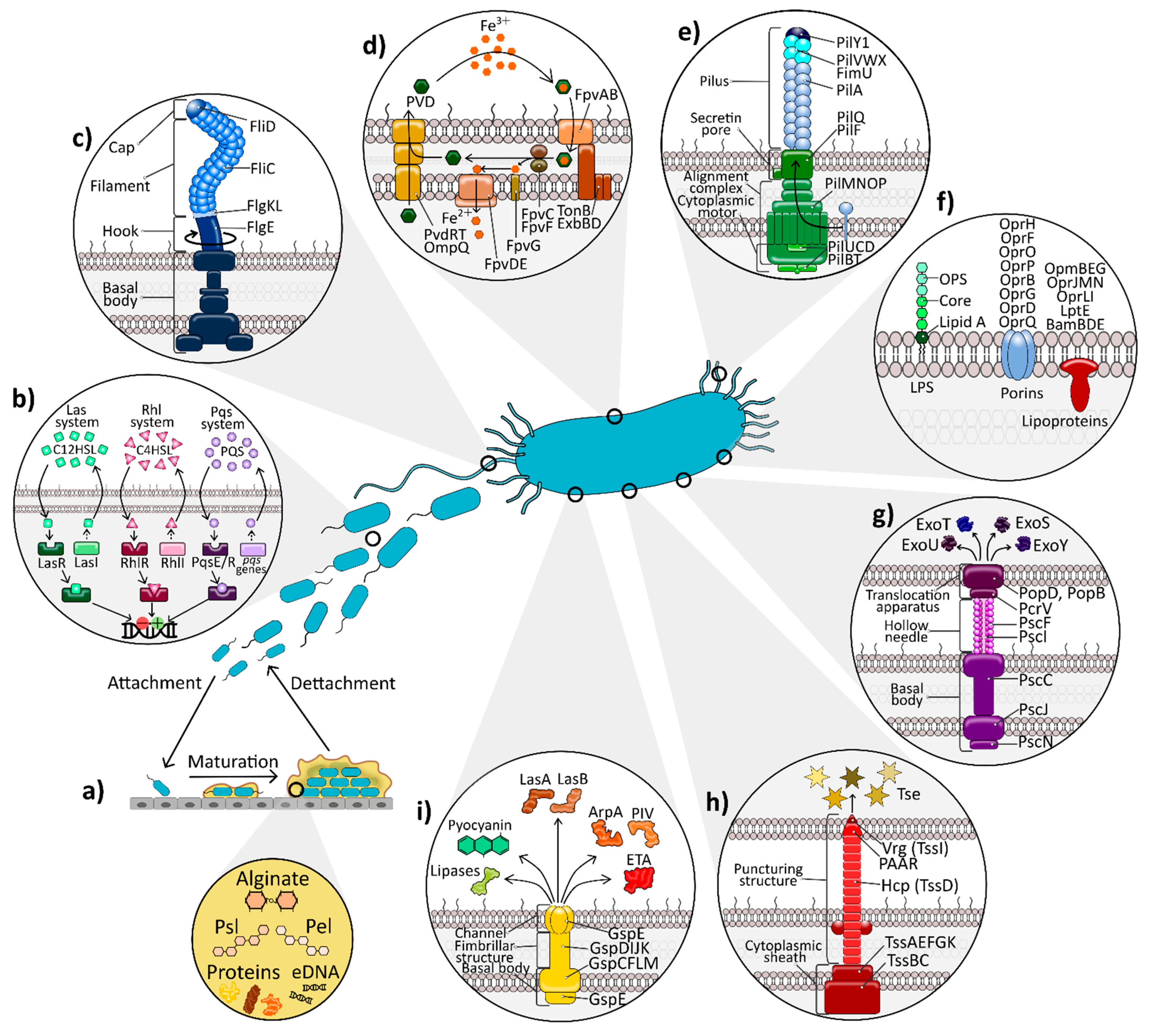{kind=link}
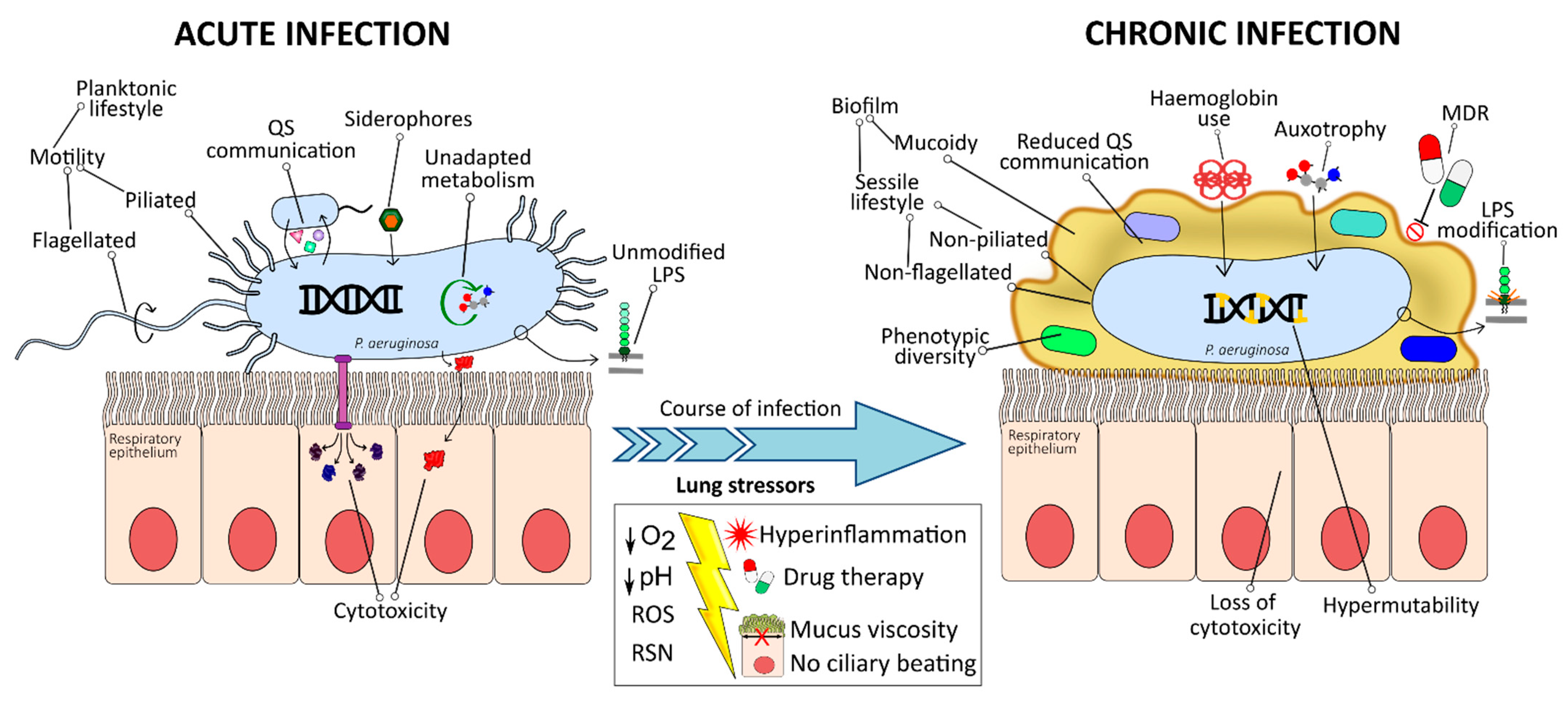{kind=link}
| OMP in P. aeruginosa | Homolog in E. coli | Function | Ref. |
|---|---|---|---|
| OprF | OmpA | Cell integrity maintenance | [42,43,44] |
| Ion and saccharide acquisition | [26] | ||
| Peptidoglycan binding | [42,44] | ||
| Diffusion channel (toluene, siderophores, nitrates, nitrites) | [42] | ||
| Adhesion (alveolar epithelial cells and other bacteria) | [42,43] | ||
| Regulation of other virulence factors | [42,45,46,47,50] | ||
| Immune system sensor | [43,47,51,52] | ||
| OprH | OmpW family | Protein binding (SP-A and laminin) | [53,54] |
| Aminoglycoside and polymyxin resistance | [42] | ||
| Transport (hydrophobic molecules, amino acids, iron, and cations) | [42,50] | ||
| OprD (OccD1) | OmpF | Laminin binding | [42,54] |
| Carbapenem resistance | [42] | ||
| Molecule transport (amino acids, peptides, gluconate) | [42] | ||
| OprG | OmpW family | Laminin binding | [42,54] |
| OprQ (OccD6) | Fibronectin binding | [55] | |
| Adhesion (epithelial cells) | [55] | ||
| OprL | Pal | Cell integrity maintenance | [24] |
| Protection against oxidative stress | [56] | ||
| OprI | Lpp | Cell integrity maintenance | [24] |
| BamBDE | OM biogenesis | [24] | |
| LptE | OM biogenesis | [24] | |
| OprJMN | Antibiotic resistance | [24,57] | |
| OmpBEG | Antibiotic resistance | [24,57] |
| Type of Study | Source of Isolates | Main Findings | Frequently Mutated Genes | Function of Identified Mutated Genes | Ref. |
|---|---|---|---|---|---|
| In vivo evolution study using whole genome sequencing | 474 longitudinal CF clinical isolates from 34 children and young individuals. | 36 lineages with convergent evolution in 52 genes | asR, mexA, mexS, nex, yecS, algU, gyrA, gyrB, mexB, oprD, pela, and rbdA | Host adaptation, AMR, and loss of extracellular virulence factors | [302] |
| In vivo evolution study of 17 AMR loci | 361, independent CF isolates collected from 30 CF centres. | 1112 sequence variants not present in the 20 most common PA clones | spuE, mexA, gyrA, rpoB, fusA1, mexZ, mexY, oprD, ampD, parR, parS, and envZ (amgS), and pagL | Unrelated. Translation, transport, LPS modification, and AMR | [303] |
| In vivo longitudinal and evolution analysis | 14 isolates from the same clonal lineage of a CF patient (20 years of the infection). | Evolution towards purifying selection. Different evolutionary pathways affecting genes of the same functional categories | ampC, ftsI | Codification of β-lactamase and penicillin-binding protein 3 (AMR) | [243] |
| In vitro biofilm and stationary-phase planktonic culture evolution study | 57 CIP-evolved populations and 35 control. | CIP-resistance development depends on bacterial lifestyle | ftsZ, murG, sdhA | Cell-wall recycling, TCA cycle, and arginine catabolism | [304] |
| Real-time in vivo evolution, metabolic and genomic study. | 26 from a single CF patient (8 years of infection). | Convergence at the phenotypic level but different mutational patterns | Not specified (functional grouping) | Amino acid transport and metabolism, defense, signal transduction and translation | [305] |
| In vivo genome analysis (wgMLST) | 2 environmental, 1 veterinary and a CF clinical isolates with a defective Las QS system | Identification of ten highly discriminatory loci between the studied strains and the PAO1 and PA14 strains | exsA, rsmN, and hopJ | T3SS and QS-regulated virulence traits. | [306] |
| Screening of 8 infection-relevant phenotypes (In vivo evolution) | 443 longitudinal isolates from 39 young cystic fibrosis patients over 10 years | Identification of phenotypic changes that deviate from expected evolutionary trajectories | mexZ, nfxB, nalDmucA, algU, retS/gacAS/rsmA gyrA and gyrB47 | Drug efflux pumps, mucoidity regulators, ciprofloxacin resistance | [307] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jurado-Martín, I.; Sainz-Mejías, M.; McClean, S. Pseudomonas aeruginosa: An Audacious Pathogen with an Adaptable Arsenal of Virulence Factors. Int. J. Mol. Sci. 2021, 22, 3128. https://doi.org/10.3390/ijms22063128
Jurado-Martín I, Sainz-Mejías M, McClean S. Pseudomonas aeruginosa: An Audacious Pathogen with an Adaptable Arsenal of Virulence Factors. International Journal of Molecular Sciences. 2021; 22(6):3128. https://doi.org/10.3390/ijms22063128
Chicago/Turabian StyleJurado-Martín, Irene, Maite Sainz-Mejías, and Siobhán McClean. 2021. "Pseudomonas aeruginosa: An Audacious Pathogen with an Adaptable Arsenal of Virulence Factors" International Journal of Molecular Sciences 22, no. 6: 3128. https://doi.org/10.3390/ijms22063128
APA StyleJurado-Martín, I., Sainz-Mejías, M., & McClean, S. (2021). Pseudomonas aeruginosa: An Audacious Pathogen with an Adaptable Arsenal of Virulence Factors. International Journal of Molecular Sciences, 22(6), 3128. https://doi.org/10.3390/ijms22063128