
Understanding the Potential Role of Sirtuin 2 on Aging: Consequences of SIRT2.3 Overexpression in Senescence

 , and
, and
Abstract
1. Introduction
2. Results
2.1. SIRT2 Expression in Aging
2.2. Potential Role of SIRT2 on Neurodegenerative Diseases
2.2.1. Is SIRT2 Beneficial for Age-Related Neurodegenerative Diseases?
2.2.2. Is SIRT2 Detrimental for Age-Related Neurodegenerative Diseases?
2.3. Potential Role of SIRT2 on Inflammation
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Adeno-Associated Virus Production and Purification
4.3. AAV Injection
4.4. Behavioral Tests: Morris Water Maze
4.5. Microglia and Astrocytes Isolation
4.6. RNA Extraction and Quantitative PCR
4.7. Western Blot
4.8. Immunofluorescence
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Kennedy, B.K.; Berger, S.L.; Brunet, A.; Campisi, J.; Cuervo, A.M.; Epel, E.S.; Franceschi, C.; Lithgow, G.J.; Morimoto, R.I.; Pessin, J.E.; et al. Aging: A common driver of chronic diseases and a target for novel interventions. Cell 2014, 159, 709–713. [Google Scholar] [CrossRef]
- United Nations, Department of Economic and Social Affairs, Population Division. World Population Prospects 2019: Highlights; United Nations: New York, NY, USA, 2019; ISBN 978-92-1-148316-1. [Google Scholar]
- López-Otín, C.; Galluzzi, L.; Freije, J.M.P.; Madeo, F.; Kroemer, G. Metabolic Control of Longevity. Cell 2016, 166, 802–821. [Google Scholar] [CrossRef]
- Pagiatakis, C.; Musolino, E.; Gornati, R.; Bernardini, G.; Papait, R. Epigenetics of aging and disease: A brief overview. Aging Clin. Exp. Res. 2019, 1–9. [Google Scholar] [CrossRef]
- Martinez-Zamudio, R.; Ha, H.C. Histone ADP-Ribosylation Facilitates Gene Transcription by Directly Remodeling Nucleosomes. Mol. Cell. Biol. 2012, 32, 2490–2502. [Google Scholar] [CrossRef]
- Thompson, P.R.; Fast, W. Histone Citrullination by Protein Arginine Deiminase: Is Arginine Methylation a Green Light or a Roadblock? ACS Chem. Biol. 2006, 1, 433–441. [Google Scholar] [CrossRef]
- Sola-Sevilla, N.; Solas, M.; Puerta, E. Sirtuin 2: A Potential Target for Age-Related Neurodegenerative Diseases? In Horizons in Neuroscience Research; Costa, A., Villalba, E., Eds.; Nova Science Publishers: Hauppauge, NY, USA, 2020; pp. 97–132. ISBN 978-1-53618-480-8. [Google Scholar]
- Lee, D.Y.; Hayes, J.J.; Pruss, D.; Wolffe, A.P. A positive role for histone acetylation in transcription factor access to nucleosomal DNA. Cell 1993, 72, 73–84. [Google Scholar] [CrossRef]
- Kurdistani, S.K.; Grunstein, M. Histone acetylation and deacetylation in yeast. Nat. Rev. Mol. Cell Biol. 2003, 4, 276–284. [Google Scholar] [CrossRef]
- Lazo-Gómez, R.; Ramírez-Jarquín, U.N.; Tovar-y-Romo, L.B.; Tapia, R. Histone deacetylases and their role in motor neuron degeneration. Front. Cell. Neurosci. 2013, 7, 243. [Google Scholar] [CrossRef] [PubMed]
- Jayasena, T.; Poljak, A.; Braidy, N.; Zhong, L.; Rowlands, B.; Muenchhoff, J.; Grant, R.; Smythe, G.; Teo, C.; Raftery, M.; et al. Application of Targeted Mass Spectrometry for the Quantification of Sirtuins in the Central Nervous System. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Jęśko, H.; Wencel, P.; Strosznajder, R.P.; Strosznajder, J.B. Sirtuins and Their Roles in Brain Aging and Neurodegenerative Disorders. Neurochem. Res. 2017, 42, 876–890. [Google Scholar] [CrossRef] [PubMed]
- Polito, L.; Kehoe, P.G.; Forloni, G.; Albani, D. The molecular genetics of sirtuins: Association with human longevity and age-related diseases. Int. J. Mol. Epidemiol. Genet. 2010, 1, 214–225. [Google Scholar]
- Satoh, A.; Imai, S.I.; Guarente, L. The brain, sirtuins, and ageing. Nat. Rev. Neurosci. 2017, 18, 362–374. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, J.; Hong, T.; Chen, X.; Cui, L. SIRT2: Controversy and multiple roles in disease and physiology. Ageing Res. Rev. 2019, 55, 100961. [Google Scholar] [CrossRef]
- Wątroba, M.; Dudek, I.; Skoda, M.; Stangret, A.; Rzodkiewicz, P.; Szukiewicz, D. Sirtuins, epigenetics and longevity. Ageing Res. Rev. 2017, 40, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Guarente, L. Sir2 links chromatin silencing, metabolism, and aging. Genes Dev. 2000, 14, 1021–1026. [Google Scholar] [PubMed]
- Michan, S.; Sinclair, D. Sirtuins in mammals: Insights into their biological function. Biochem. J. 2007, 404, 1–13. [Google Scholar] [CrossRef]
- Buler, M.; Andersson, U.; Hakkola, J. Who watches the watchmen? Regulation of the expression and activity of sirtuins. FASEB J. 2016, 30, 3942–3960. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, N.; Schwer, B.; Carobbio, S.; Waltregny, D.; North, B.J.; Castronovo, V.; Maechler, P.; Verdin, E. Regulation of Insulin Secretion by SIRT4, a Mitochondrial ADP-ribosyltransferase. J. Biol. Chem. 2007, 282, 33583–33592. [Google Scholar] [CrossRef] [PubMed]
- Liszt, G.; Ford, E.; Kurtev, M.; Guarente, L. Mouse Sir2 Homolog SIRT6 Is a Nuclear ADP-ribosyltransferase. J. Biol. Chem. 2005, 280, 21313–21320. [Google Scholar] [CrossRef]
- Michishita, E.; Park, J.Y.; Burneskis, J.M.; Barrett, J.C.; Horikawa, I. Evolutionarily Conserved and Nonconserved Cellular Localizations and Functions of Human SIRT Proteins. Mol. Biol. Cell 2005, 16, 4623–4635. [Google Scholar] [CrossRef]
- Liu, G.; Park, S.-H.; Imbesi, M.; Nathan, W.J.; Zou, X.; Zhu, Y.; Jiang, H.; Parisiadou, L.; Gius, D. Loss of NAD-Dependent Protein Deacetylase Sirtuin-2 Alters Mitochondrial Protein Acetylation and Dysregulates Mitophagy. Antioxid. Redox Signal. 2017, 26, 849–863. [Google Scholar] [CrossRef]
- Anwar, T.; Khosla, S.; Ramakrishna, G. Increased expression of SIRT2 is a novel marker of cellular senescence and is dependent on wild type p53 status. Cell Cycle 2016, 15, 1883–1897. [Google Scholar] [CrossRef]
- Esteves, A.R.; Palma, A.M.; Gomes, R.; Santos, D.; Silva, D.F.; Cardoso, S.M. Acetylation as a major determinant to microtubule-dependent autophagy: Relevance to Alzheimer’s and Parkinson disease pathology. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2008–2023. [Google Scholar] [CrossRef] [PubMed]
- Harting, K.; Knöll, B. SIRT2-mediated protein deacetylation: An emerging key regulator in brain physiology and pathology. Eur. J. Cell Biol. 2010, 89, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, M.M.; Tomkinson, E.M.; Nobles, J.; Wizeman, J.W.; Amore, A.M.; Quinti, L.; Chopra, V.; Hersch, S.M.; Kazantsev, A.G. The Sirtuin 2 microtubule deacetylase is an abundant neuronal protein that accumulates in the aging CNS. Hum. Mol. Genet. 2011, 20, 3986–3996. [Google Scholar] [CrossRef]
- North, B.J.; Marshall, B.L.; Borra, M.T.; Denu, J.M.; Verdin, E. The Human Sir2 Ortholog, SIRT2, Is an NAD+-Dependent Tubulin Deacetylase. Mol. Cell 2003, 11, 437–444. [Google Scholar] [CrossRef]
- Silva, D.F.; Esteves, A.R.; Oliveira, C.R.; Cardoso, S.M. Mitochondrial Metabolism Power SIRT2-Dependent Deficient Traffic Causing Alzheimer’s-Disease Related Pathology. Mol. Neurobiol. 2017, 54, 4021–4040. [Google Scholar] [CrossRef]
- Beirowski, B.; Gustin, J.; Armour, S.M.; Yamamoto, H.; Viader, A.; North, B.J.; Michan, S.; Baloh, R.H.; Golden, J.P.; Schmidt, R.E.; et al. Sir-two-homolog 2 (Sirt2) modulates peripheral myelination through polarity protein Par-3/atypical protein kinase C (aPKC) signaling. Proc. Natl. Acad. Sci. USA 2011, 108, E952–E961. [Google Scholar] [CrossRef]
- Tang, B.L.; Chua, C.E.L. SIRT2, tubulin deacetylation, and oligodendroglia differentiation. Cell Motil. Cytoskelet. 2008, 65, 179–182. [Google Scholar] [CrossRef] [PubMed]
- Gal, J.; Bang, Y.; Choi, H.J. SIRT2 interferes with autophagy-mediated degradation of protein aggregates in neuronal cells under proteasome inhibition. Neurochem. Int. 2012, 61, 992–1000. [Google Scholar] [CrossRef]
- Inoue, T.; Nakayama, Y.; Li, Y.; Matsumori, H.; Takahashi, H.; Kojima, H.; Wanibuchi, H.; Katoh, M.; Oshimura, M. SIRT2 knockdown increases basal autophagy and prevents postslippage death by abnormally prolonging the mitotic arrest that is induced by microtubule inhibitors. FEBS J. 2014, 281, 2623–2637. [Google Scholar] [CrossRef]
- Mendes, K.L.; de Farias Lelis, D.; Santos, S.H.S. Nuclear sirtuins and inflammatory signaling pathways. Cytokine Growth Factor Rev. 2017, 38, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Chambers, S.M.; Shaw, C.A.; Gatza, C.; Fisk, C.J.; Donehower, L.A.; Goodell, M.A. Aging Hematopoietic Stem Cells Decline in Function and Exhibit Epigenetic Dysregulation. PLoS Biol. 2007, 5, e201. [Google Scholar] [CrossRef]
- Luo, H.; Mu, W.C.; Karki, R.; Chiang, H.H.; Mohrin, M.; Shin, J.J.; Ohkubo, R.; Ito, K.; Kanneganti, T.D.; Chen, D. Mitochondrial Stress-Initiated Aberrant Activation of the NLRP3 Inflammasome Regulates the Functional Deterioration of Hematopoietic Stem Cell Aging. Cell Rep. 2019, 26, 945–954.e4. [Google Scholar] [CrossRef]
- He, M.; Chiang, H.-H.; Luo, H.; Zheng, Z.; Qiao, Q.; Wang, L.; Tan, M.; Ohkubo, R.; Mu, W.-C.; Zhao, S.; et al. An Acetylation Switch of the NLRP3 Inflammasome Regulates Aging-Associated Chronic Inflammation and Insulin Resistance. Cell Metab. 2020, 31, 580–591.e5. [Google Scholar] [CrossRef] [PubMed]
- Yudoh, K.; Karasawa, R.; Ishikawa, J. Age-related Decrease of Sirtuin 2 Protein in Human Peripheral Blood Mononuclear Cells. Curr. Aging Sci. 2015, 8, 256–258. [Google Scholar] [CrossRef] [PubMed]
- Wongchitrat, P.; Pakpian, N.; Kitidee, K.; Phopin, K.; Dharmasaroja, P.A.; Govitrapong, P. Alterations in the Expression of Amyloid Precursor Protein Cleaving Enzymes mRNA in Alzheimer Peripheral Blood. Curr. Alzheimer Res. 2018, 16, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, B.; Tang, J.; Cao, Q.; Wu, Y.; Wu, C.; Guo, J.; Ling, E.-A.; Liang, F. Sirtuin 2, a Mammalian Homolog of Yeast Silent Information Regulator-2 Longevity Regulator, Is an Oligodendroglial Protein That Decelerates Cell Differentiation through Deacetylating alfa-Tubulin. J. Neurosci. 2007, 27, 2606–2616. [Google Scholar] [CrossRef]
- Vaquero, A. SirT2 is a histone deacetylase with preference for histone H4 Lys 16 during mitosis. Genes Dev. 2006, 20, 1256–1261. [Google Scholar] [CrossRef]
- Rothgiesser, K.M.; Erener, S.; Waibel, S.; Luscher, B.; Hottiger, M.O. SIRT2 regulates NF- B-dependent gene expression through deacetylation of p65 Lys310. J. Cell Sci. 2010, 123, 4251–4258. [Google Scholar] [CrossRef]
- Wang, X.; Buechler, N.L.; Martin, A.; Wells, J.; Yoza, B.; McCall, C.E.; Vachharajani, V. Sirtuin-2 Regulates Sepsis Inflammation in ob/ob Mice. PLoS ONE 2016, 11, e0160431. [Google Scholar] [CrossRef]
- Yuan, F.; Xu, Z.M.; Lu, L.Y.; Nie, H.; Ding, J.; Ying, W.H.; Tian, H.L. SIRT2 inhibition exacerbates neuroinflammation and blood-brain barrier disruption in experimental traumatic brain injury by enhancing NF-κB p65 acetylation and activation. J. Neurochem. 2016, 136, 581–593. [Google Scholar] [CrossRef]
- Zhang, Y.; Chi, D. Overexpression of SIRT2 Alleviates Neuropathic Pain and Neuroinflammation through Deacetylation of Transcription Factor Nuclear Factor-Kappa, B. Inflammation 2018, 41, 569–578. [Google Scholar] [CrossRef]
- Diaz-Perdigon, T.; Belloch, F.B.; Ricobaraza, A.; Elboray, E.E.; Suzuki, T.; Tordera, R.M.; Puerta, E. Early sirtuin 2 inhibition prevents age-related cognitive decline in a senescence-accelerated mouse model. Neuropsychopharmacology 2020, 45, 347–357. [Google Scholar] [CrossRef]
- Kireev, R.A.; Vara, E.; Tresguerres, J.A.F. Growth hormone and melatonin prevent age-related alteration in apoptosis processes in the dentate gyrus of male rats. Biogerontology 2013, 14, 431–442. [Google Scholar] [CrossRef]
- Braidy, N.; Poljak, A.; Grant, R.; Jayasena, T.; Mansour, H.; Chan-Ling, T.; Smythe, G.; Sachdev, P.; Guillemin, G.J. Differential expression of sirtuins in the aging rat brain. Front. Cell. Neurosci. 2015, 9, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Garg, G.; Singh, S.; Singh, A.K.; Rizvi, S.I. Antiaging Effect of Metformin on Brain in Naturally Aged and Accelerated Senescence Model of Rat. Rejuvenation Res. 2017, 20, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Hanson, P.S.; Morris, C.M. Sirtuin-2 Protects Neural Cells from Oxidative Stress and Is Elevated in Neurodegeneration. Parkinson’s Dis. 2017, 2017, 1–17. [Google Scholar] [CrossRef]
- Fourcade, S.; Morató, L.; Parameswaran, J.; Ruiz, M.; Ruiz-Cortés, T.; Jové, M.; Naudí, A.; Martínez-Redondo, P.; Dierssen, M.; Ferrer, I.; et al. Loss of SIRT2 leads to axonal degeneration and locomotor disability associated with redox and energy imbalance. Aging Cell 2017, 16, 1404–1413. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Li, S.; Gilbert, J.; Gritton, H.J.; Wang, Z.; Li, Z.; Han, X.; Selkoe, D.J.; Man, H.-Y. Crucial Roles for SIRT2 and AMPA Receptor Acetylation in Synaptic Plasticity and Memory. Cell Rep. 2017, 20, 1335–1347. [Google Scholar] [CrossRef] [PubMed]
- Martin, B.; Mattson, M.P.; Maudsley, S. Caloric restriction and intermittent fasting: Two potential diets for successful brain aging. Ageing Res. Rev. 2006, 5, 332–353. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Nguyen, M.; Qin, F.X.F.; Tong, Q. SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell 2007, 6, 505–514. [Google Scholar] [CrossRef] [PubMed]
- North, B.J.; Rosenberg, M.A.; Jeganathan, K.B.; Hafner, A.V.; Michan, S.; Dai, J.; Baker, D.J.; Cen, Y.; Wu, L.E.; Sauve, A.A.; et al. SIRT2 induces the checkpoint kinase BubR1 to increase lifespan. EMBO J. 2014, 33, 1438–1453. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yang, J.; Liao, W.; Liu, X.; Zhang, H.; Wang, S.; Wang, D.; Feng, J.; Yu, L.; Zhu, W.-G. Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat. Cell Biol. 2010, 12, 665–675. [Google Scholar] [CrossRef]
- Wong, E.; Cuervo, A.M. Autophagy gone awry in neurodegenerative diseases. Nat. Neurosci. 2010, 13, 805–811. [Google Scholar] [CrossRef] [PubMed]
- Van Leeuwen, I.M.M.; Higgins, M.; Campbell, J.; McCarthy, A.R.; Sachweh, M.C.C.; Navarro, A.M.; Laín, S. Modulation of p53 C-Terminal Acetylation by mdm2, p14 ARF, and Cytoplasmic SirT2. Mol. Cancer Ther. 2013, 12, 471–480. [Google Scholar] [CrossRef]
- Sun, S.; Han, X.; Li, X.; Song, Q.; Lu, M.; Jia, M.; Ding, J.; Hu, G. MicroRNA-212-5p Prevents Dopaminergic Neuron Death by Inhibiting SIRT2 in MPTP-Induced Mouse Model of Parkinson’s Disease. Front. Mol. Neurosci. 2018, 11, 1–14. [Google Scholar] [CrossRef]
- Guan, Q.; Wang, M.; Chen, H.; Yang, L.; Yan, Z.; Wang, X. Aging-related 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurochemial and behavioral deficits and redox dysfunction: Improvement by AK-7. Exp. Gerontol. 2016, 82, 19–29. [Google Scholar] [CrossRef]
- Chen, X.; Mai, H.; Chen, X.; Cai, Y.; Cheng, Q.; Chen, X.; Li, X.; Fan, W.; Tang, P.; Ou, M.; et al. Rs2015 Polymorphism in miRNA Target Site of Sirtuin2 Gene Is Associated with the Risk of Parkinson’s Disease in Chinese Han Population. Biomed. Res. Int. 2019, 2019, 1–8. [Google Scholar] [CrossRef]
- Baldo, B.; Gabery, S.; Soylu-Kucharz, R.; Cheong, R.Y.; Henningsen, J.B.; Englund, E.; McLean, C.; Kirik, D.; Halliday, G.; Petersén, Å. SIRT1 is increased in affected brain regions and hypothalamic metabolic pathways are altered in Huntington disease. Neuropathol. Appl. Neurobiol. 2019, 45, 361–379. [Google Scholar] [CrossRef]
- Yang, J.W.; Czech, T.; Felizardo, M.; Baumgartner, C.; Lubec, G. Aberrant expression of cytoskeleton proteins in hippocampus from patients with mesial temporal lobe epilepsy. Amino Acids 2006, 30, 477–493. [Google Scholar] [CrossRef]
- Erburu, M.; Muñoz-Cobo, I.; Domínguez-Andrés, J.; Beltran, E.; Suzuki, T.; Mai, A.; Valente, S.; Puerta, E.; Tordera, R.M. Chronic stress and antidepressant induced changes in Hdac5 and Sirt2 affect synaptic plasticity. Eur. Neuropsychopharmacol. 2015, 25, 2036–2048. [Google Scholar] [CrossRef]
- Guerreiro, P.S.; Coelho, J.E.; Sousa-Lima, I.; Macedo, P.; Lopes, L.V.; Outeiro, T.F.; Pais, T.F. Mutant A53T α-Synuclein Improves Rotarod Performance Before Motor Deficits and Affects Metabolic Pathways. Neuromol. Med. 2017, 19, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Harrison, I.F.; Smith, A.D.; Dexter, D.T. Pathological histone acetylation in Parkinson’s disease: Neuroprotection and inhibition of microglial activation through SIRT 2 inhibition. Neurosci. Lett. 2018, 666, 48–57. [Google Scholar] [CrossRef]
- Outeiro, T.F.; Kontopoulos, E.; Altmann, S.M.; Kufareva, I.; Strathearn, K.E.; Amore, A.M.; Volk, C.B.; Maxwell, M.M.; Rochet, J.-C.; McLean, P.J.; et al. Sirtuin 2 Inhibitors Rescue -Synuclein-Mediated Toxicity in Models of Parkinson’s Disease. Science 2007, 317, 516–519. [Google Scholar] [CrossRef]
- Di Fruscia, P.; Zacharioudakis, E.; Liu, C.; Moniot, S.; Laohasinnarong, S.; Khongkow, M.; Harrison, I.F.; Koltsida, K.; Reynolds, C.R.; Schmidtkunz, K.; et al. The Discovery of a Highly Selective 5,6,7,8-Tetrahydrobenzo[4,5]thieno[2,3-d ]pyrimidin-4(3H)-one SIRT2 Inhibitor that is Neuroprotective in an in vitro Parkinson’s Disease Model. ChemMedChem 2015, 10, 69–82. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, R.M.; Vicente Miranda, H.; Francelle, L.; Pinho, R.; Szegö, É.M.; Martinho, R.; Munari, F.; Lázaro, D.F.; Moniot, S.; Guerreiro, P.; et al. The mechanism of sirtuin 2–mediated exacerbation of alpha-synuclein toxicity in models of Parkinson disease. PLoS Biol. 2017, 15, e2000374. [Google Scholar] [CrossRef]
- Esteves, A.R.; Arduíno, D.M.; Silva, D.F.; Viana, S.D.; Pereira, F.C.; Cardoso, S.M. Mitochondrial Metabolism Regulates Microtubule Acetylome and Autophagy Trough Sirtuin-2: Impact for Parkinson’s Disease. Mol. Neurobiol. 2018, 55, 1440–1462. [Google Scholar] [CrossRef]
- Bobrowska, A.; Donmez, G.; Weiss, A.; Guarente, L.; Bates, G. SIRT2 Ablation Has No Effect on Tubulin Acetylation in Brain, Cholesterol Biosynthesis or the Progression of Huntington’s Disease Phenotypes In Vivo. PLoS ONE 2012, 7, e34805. [Google Scholar] [CrossRef]
- Luthi-Carter, R.; Taylor, D.M.; Pallos, J.; Lambert, E.; Amore, A.; Parker, A.; Moffitt, H.; Smith, D.L.; Runne, H.; Gokce, O.; et al. SIRT2 inhibition achieves neuroprotection by decreasing sterol biosynthesis. Proc. Natl. Acad. Sci. USA 2010, 107, 7927–7932. [Google Scholar] [CrossRef] [PubMed]
- Chopra, V.; Quinti, L.; Kim, J.; Vollor, L.; Narayanan, K.L.; Edgerly, C.; Cipicchio, P.M.; Lauver, M.A.; Choi, S.H.; Silverman, R.B.; et al. The Sirtuin 2 Inhibitor AK-7 Is Neuroprotective in Huntington’s Disease Mouse Models. Cell Rep. 2012, 2, 1492–1497. [Google Scholar] [CrossRef] [PubMed]
- Pallos, J.; Bodai, L.; Lukacsovich, T.; Purcell, J.M.; Steffan, J.S.; Thompson, L.M.; Marsh, J.L. Inhibition of specific HDACs and sirtuins suppresses pathogenesis in a Drosophila model of Huntington’s disease. Hum. Mol. Genet. 2008, 17, 3767–3775. [Google Scholar] [CrossRef]
- Quinti, L.; Casale, M.; Moniot, S.; Pais, T.F.; Van Kanegan, M.J.; Kaltenbach, L.S.; Pallos, J.; Lim, R.G.; Naidu, S.D.; Runne, H.; et al. SIRT2- and NRF2-Targeting Thiazole-Containing Compound with Therapeutic Activity in Huntington’s Disease Models. Cell Chem. Biol. 2016, 23, 849–861. [Google Scholar] [CrossRef] [PubMed]
- Scuderi, C.; Stecca, C.; Bronzuoli, R.M.R.; Rotili, D.; Valente, S.; Mai, A.; Steardo, L. Sirtuin modulators control reactive gliosis in an in vitro model of alzheimer’s disease. Front. Pharmacol. 2014, 5, 1–8. [Google Scholar] [CrossRef]
- Spires-Jones, T.L.; Fox, L.M.; Rozkalne, A.; Pitstick, R.; Carlson, G.A.; Kazantsev, A.G. Inhibition of sirtuin 2 with sulfobenzoic acid derivative AK1 is non-toxic and potentially neuroprotective in a mouse model of frontotemporal dementia. Front. Pharmacol. 2012, 3, 1–7. [Google Scholar] [CrossRef]
- Biella, G.; Fusco, F.; Nardo, E.; Bernocchi, O.; Colombo, A.; Lichtenthaler, S.F.; Forloni, G.; Albani, D. Sirtuin 2 Inhibition Improves Cognitive Performance and Acts on Amyloid-β Protein Precursor Processing in Two Alzheimer’s Disease Mouse Models. J. Alzheimer’s Dis. 2016, 53, 1193–1207. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, J.; Hong, T.-T.; Sun, Y.; Huang, H.; Chen, F.; Chen, X.; Chen, H.; Dong, S.; Cui, L.; et al. RTN4B-mediated suppression of Sirtuin 2 activity ameliorates β-amyloid pathology and cognitive impairment in Alzheimer’s disease mouse model. Aging Cell 2020, 19, 1–13. [Google Scholar] [CrossRef]
- Pallas, M.; Camins, A.; Smith, M.A.; Perry, G.; Lee, H.; Casadesus, G. From Aging to Alzheimer’s Disease: Unveiling “The Switch” with the Senescence-Accelerated Mouse Model (SAMP8). J. Alzheimer’s Dis. 2008, 15, 615–624. [Google Scholar] [CrossRef]
- Akiguchi, I.; Pallàs, M.; Budka, H.; Akiyama, H.; Ueno, M.; Han, J.; Yagi, H.; Nishikawa, T.; Chiba, Y.; Sugiyama, H.; et al. SAMP8 mice as a neuropathological model of accelerated brain aging and dementia: Toshio Takeda’s legacy and future directions. Neuropathology 2017, 37, 293–305. [Google Scholar] [CrossRef]
- Morrisette-Thomas, V.; Cohen, A.A.; Fülöp, T.; Riesco, É.; Legault, V.; Li, Q.; Milot, E.; Dusseault-Bélanger, F.; Ferrucci, L. Inflamm-aging does not simply reflect increases in pro-inflammatory markers. Mech. Ageing Dev. 2014, 139, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Bektas, A.; Schurman, S.H.; Sen, R.; Ferrucci, L. Aging, inflammation and the environment. Exp. Gerontol. 2018, 105, 10–18. [Google Scholar] [CrossRef]
- Lo Sasso, G.; Menzies, K.J.; Mottis, A.; Piersigilli, A.; Perino, A.; Yamamoto, H.; Schoonjans, K.; Auwerx, J. SIRT2 Deficiency Modulates Macrophage Polarization and Susceptibility to Experimental Colitis. PLoS ONE 2014, 9, e103573. [Google Scholar] [CrossRef]
- Lin, J.; Sun, B.; Jiang, C.; Hong, H.; Zheng, Y. Sirt2 suppresses inflammatory responses in collagen-induced arthritis. Biochem. Biophys. Res. Commun. 2013, 441, 897–903. [Google Scholar] [CrossRef]
- Kara, M.; Yolbas, S.; Sahin, C.; Koca, S.S. Changes in sirtuin 2 and sirtuin 3 mRNA expressions in rheumatoid arthritis. Eur. J. Rheumatol. 2017, 4, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Pais, T.F.; Szego, É.M.; Marques, O.; Miller-Fleming, L.; Antas, P.; Guerreiro, P.; De Oliveira, R.M.H.; Kasapoglu, B.; Outeiro, T.F. The NAD-dependent deacetylase sirtuin 2 is a suppressor of microglial activation and brain inflammation. EMBO J. 2013, 32, 2603–2616. [Google Scholar] [CrossRef]
- Kim, M.J.; Kim, D.W.; Park, J.H.; Kim, S.J.; Lee, C.H.; Yong, J.I.; Ryu, E.J.; Cho, S.B.; Yeo, H.J.; Hyeon, J.; et al. PEP-1-SIRT2 inhibits inflammatory response and oxidative stress-induced cell death via expression of antioxidant enzymes in murine macrophages. Free Radic. Biol. Med. 2013, 63, 432–445. [Google Scholar] [CrossRef]
- Sun, K.; Wang, X.; Fang, N.; Xu, A.; Lin, Y.; Zhao, X.; Nazarali, A.J.; Ji, S. SIRT2 suppresses expression of inflammatory factors via Hsp90-glucocorticoid receptor signalling. J. Cell. Mol. Med. 2020, 24, 7439–7450. [Google Scholar] [CrossRef]
- Lee, A.S.; Jung, Y.J.; Kim, D.; Nguyen-Thanh, T.; Kang, K.P.; Lee, S.; Park, S.K.; Kim, W. SIRT2 ameliorates lipopolysaccharide-induced inflammation in macrophages. Biochem. Biophys. Res. Commun. 2014, 450, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wu, D.; Ding, X.; Ying, W. SIRT2 is required for lipopolysaccharide-induced activation of BV2 microglia. Neuroreport 2015, 26, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Jiao, F.; Wang, Y.; Zhang, W.; Zhang, H.; Chen, Q.; Wang, L.; Shi, C.; Gong, Z. AGK2 Alleviates Lipopolysaccharide Induced Neuroinflammation through Regulation of Mitogen-Activated Protein Kinase Phosphatase-1. J. Neuroimmune Pharmacol. 2019, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Alam, H.B.; Liu, B.; Bronson, R.T.; Nikolian, V.C.; Wu, E.; Chong, W.; Li, Y. Selective Inhibition of SIRT2 Improves Outcomes in a Lethal Septic Model. Curr. Mol. Med. 2015, 15, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Ciarlo, E.; Heinonen, T.; Théroude, C.; Herderschee, J.; Mombelli, M.; Lugrin, J.; Pfefferlé, M.; Tyrrell, B.; Lensch, S.; Acha-Orbea, H.; et al. Sirtuin 2 Deficiency Increases Bacterial Phagocytosis by Macrophages and Protects from Chronic Staphylococcal Infection. Front. Immunol. 2017, 8, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhang, Y.; Cao, W.; Wei, X.; Chen, J.; Ying, W. SIRT2 Plays Significant Roles in Lipopolysaccharides-Induced Neuroinflammation and Brain Injury in Mice. Neurochem. Res. 2016, 41, 2490–2500. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Zhang, Y.; Zhang, Y.; Zhao, P. Sirtuin 2 Inhibition Attenuates Sevoflurane-Induced Learning and Memory Deficits in Developing Rats via Modulating Microglial Activation. Cell. Mol. Neurobiol. 2020, 40, 437–446. [Google Scholar] [CrossRef]
- Abellanas, M.A.; Zamarbide, M.; Basurco, L.; Luquin, E.; Garcia-Granero, M.; Clavero, P.; San Martin-Uriz, P.; Vilas, A.; Mengual, E.; Hervas-Stubbs, S.; et al. Midbrain microglia mediate a specific immunosuppressive response under inflammatory conditions. J. Neuroinflamm. 2019, 16, 233. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
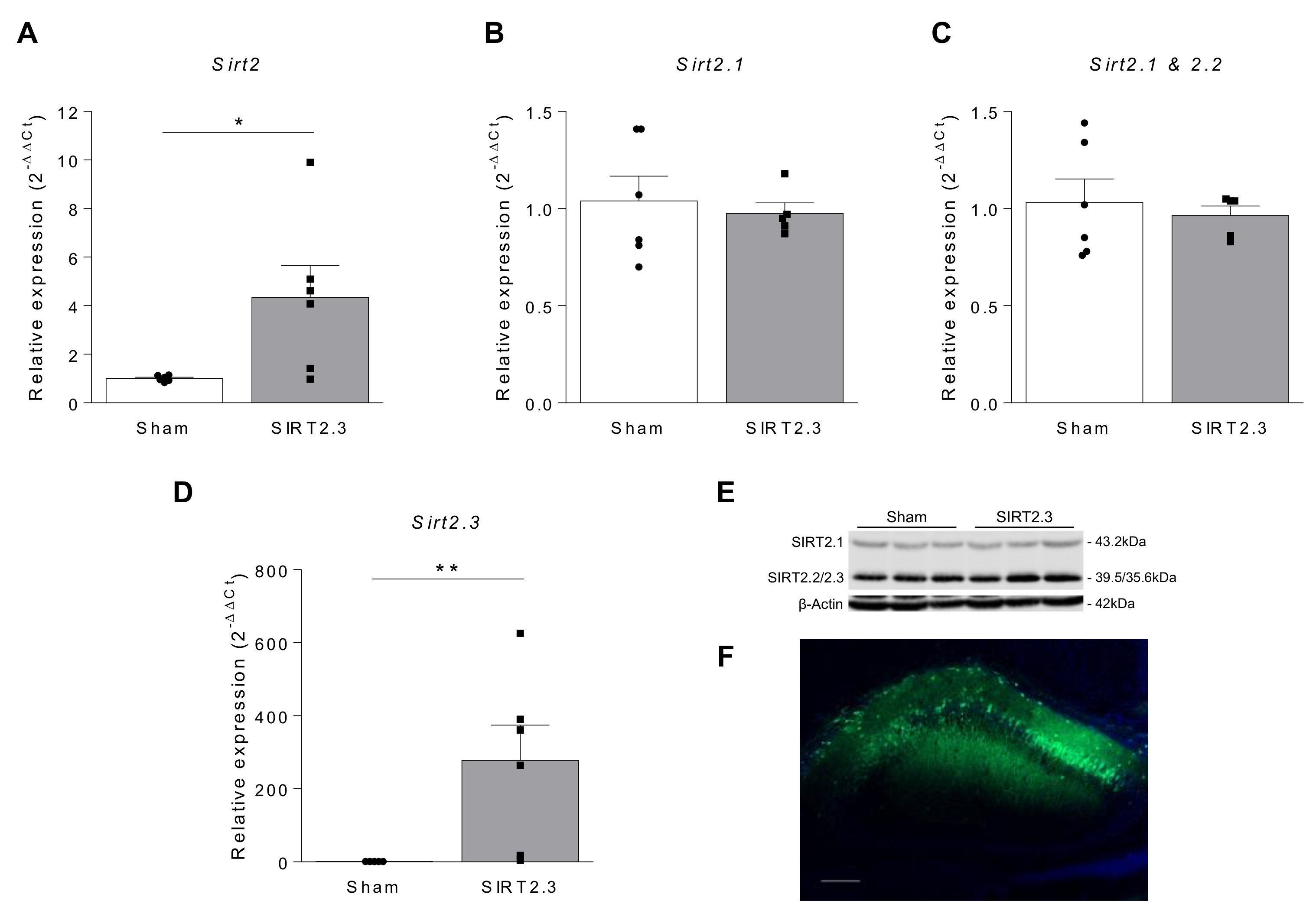{kind=link}
{kind=link}
{kind=link}
{kind=link}
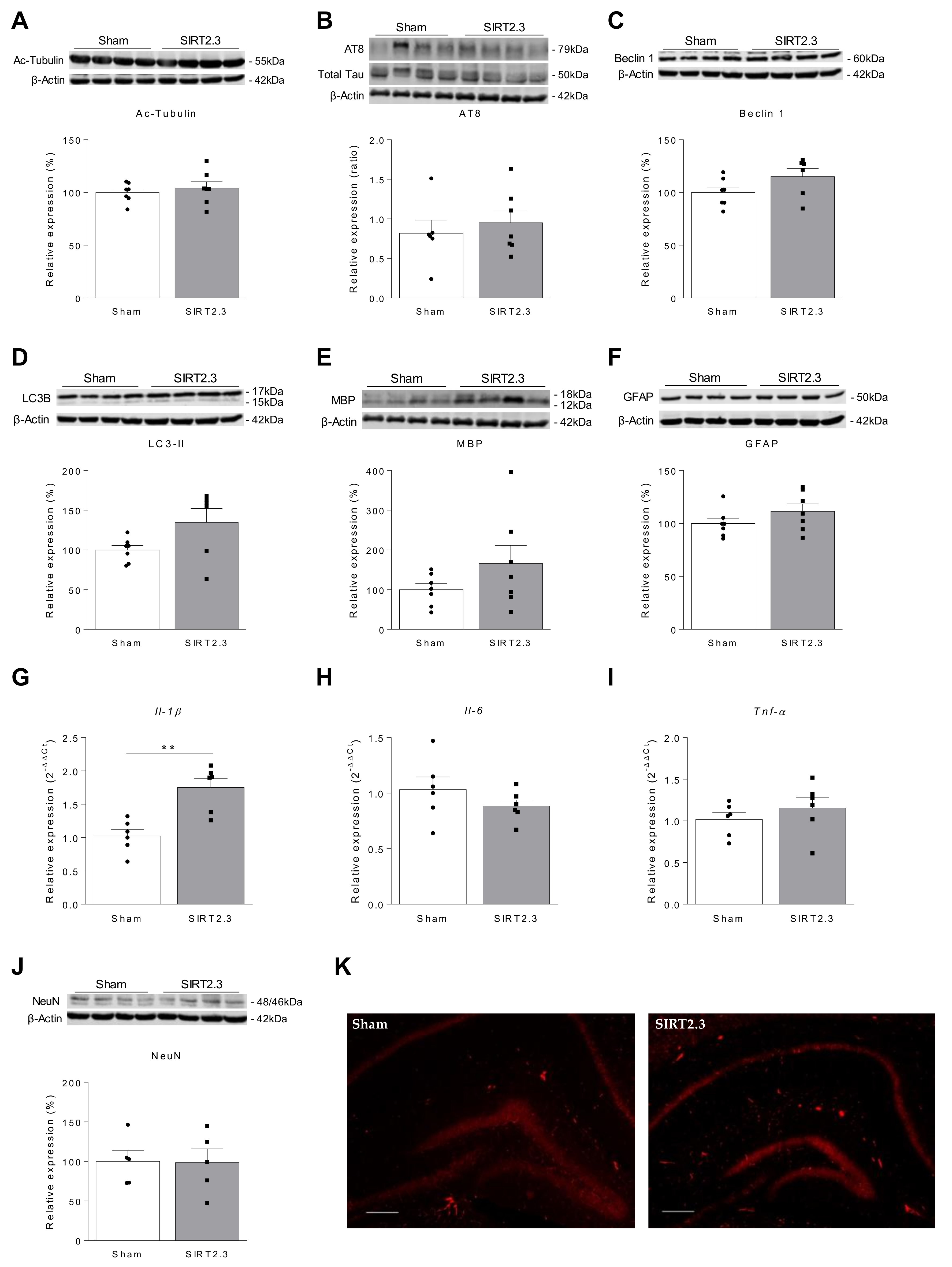{kind=link}
| Author, Year | Model-Species | Sample | SIRT2 Expression | Reference | ||
|---|---|---|---|---|---|---|
| Model | Ages Compared | Effect with Aging | Gene or Protein | |||
| Chambers et al., 2007 | Mouse C57BL/6 | 2 vs. 21 months old | Isolated mouse hematopoietic stem cells from bone marrow | Decrease | mRNA | [35] |
| Maxwell et al., 2011 | Mouse (both sexes) (1) C57BL/6J (2) B6CBA | (1) 4 vs. 19 months old (2) 3 vs. 18 months old | (1) Spinal cord (2) Cortex | Increase | Protein | [27] |
| Luo et al., 2019 | Mouse (both sexes) C57BL/6 | 3 vs. 24 months old | Isolated mouse hematopoietic stem cells from bone marrow | Decrease | mRNA | [36] |
| Diaz-Perdigon et al., 2020 | Mouse (male) SAMR1 and SAMP8 | 2 vs. 9 months old | Hippocampus | Increase | Protein | [46] |
| He et al., 2020 | Mouse (male) C57BL/6 | 3 vs. 24 months old | Isolated macrophages | Decrease | mRNA | [37] |
| Kireev et al., 2013 | Rat (male) Wistar | 2 vs. 22 months old | Dentate gyrus (brain) | Decrease | mRNA | [47] |
| Braidy et al., 2015 | Rat (female) Wistar | 3, 12 and 24 months old | Occipital lobe (brain) | Increase | mRNA and protein | [48] |
| Garg et al., 2017 | Rat (male) Wistar | (1) 4 vs. 24 months old (2) young controls vs. D-galactose induced accelerated senescence rat model | (1) Whole brain (2) Whole brain | (1) Increase (2) Increase | mRNA | [49] |
| Yudoh et al., 2015 | Human (both sexes) | 20–58 years old vs. old subjects | Peripheral blood mononuclear cells | Decrease | Protein | [38] |
| Wongchitrat et al., 2018 | Human (both sexes) | 25–35 years old vs. old subjects (≥ 65 years) | Plasma (peripheral blood) | Increase | mRNA | [39] |
| Author, Year | Model-Species | Sample | SIRT2 Expression | Reference | ||
|---|---|---|---|---|---|---|
| Model | Groups Compared | Expression in the Disease | Gene or Protein | |||
| Yang et al., 2006 | Human samples Mesial temporal lobe epilepsy | Control vs. patients | Hippocampus | Decrease | Protein | [63] |
| Erburu et al., 2015 | (1) Mouse C57BL/6J (2) Postmortem samples depressed patients | (1) Control vs. Chronic social defeat stress model (2) Control vs. patients | (1) Prefrontal cortex (2) Prefrontal cortex | (1) Increase (2) Increase | mRNA | [64] |
| Guan et al., 2016 | Mouse C57BL/6 MPTP model of PD | (1) 12 week old control vs. MPTP mice (2) 72 week old control vs. MPTP mice | Substantia nigra | (1) No changes (2) Increase | Protein | [60] |
| Guerreiro et al., 2017 | Mouse (male) a-SynA53T model of PD | 17 months old control vs. transgenic mice | Whole Brain | Decrease | mRNA | [65] |
| Silva et al., 2017 | Postmortem human brains of AD | Control vs. AD patients | Temporal cortex | Increase | Protein | [29] |
| Singh et al., 2017 | Postmortem human samples of PD, PDD, DLB, AD | Control vs. patients | Frontal and temporal cortex | (1) Increase in PD and DLB (2) Increase activity in all diseases | (1) Protein (2) Activity | [50] |
| Harrison et al., 2018 | Postmortem human brains of PD | Control vs. PD patients | Substantia nigra pars compacta | No changes | mRNA | [66] |
| Sun et al., 2018 | Mouse C57BL/6 MPTP model of PD | 12 weeks old control vs. MPTP mice | Midbrain | Increase | Protein (no changes in mRNA) | [59] |
| Wongchitrat et al., 2018 | Human samples of AD (≥65 years old) | Control vs. AD patients | Plasma | No changes | mRNA | [39] |
| Baldo et al., 2019 | Postmortem human brains of HD | Control vs. HD patients | Striatum (brain) | Increase | mRNA | [62] |
| Chen et al., 2019 | Human samples of PD | Control vs. HD patients | Peripheral blood leukocytes | Increase | mRNA | [61] |
| Forward Primer (5′-3′) | Reverse Primer (5′-3′) | |
|---|---|---|
| Sirt2.1 | TCAGGATTCAGACTCGGACAC | TGTAGCGTGTCACTCCTTCG |
| Sirt2.1&2.2 | AGCGAGCGCTGCCGCAAG | AAGATGGCCTCTGGGTAAGGAAGGTG |
| Sirt2.3 | AGCCGGACCGCCGCAAGG | AAGATGGCCTCTGGGTAAGGAAGGTG |
| Gapdh | CCAAGGTCATCCATGACAAC | TGTCATACCAGGAAATGAGC |
| 36b4 | AACAATCTCCCCCTTCTCCTT | GAAGGCCTTGACCTTTTCAG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sola-Sevilla, N.; Ricobaraza, A.; Hernandez-Alcoceba, R.; Aymerich, M.S.; Tordera, R.M.; Puerta, E. Understanding the Potential Role of Sirtuin 2 on Aging: Consequences of SIRT2.3 Overexpression in Senescence. Int. J. Mol. Sci. 2021, 22, 3107. https://doi.org/10.3390/ijms22063107
Sola-Sevilla N, Ricobaraza A, Hernandez-Alcoceba R, Aymerich MS, Tordera RM, Puerta E. Understanding the Potential Role of Sirtuin 2 on Aging: Consequences of SIRT2.3 Overexpression in Senescence. International Journal of Molecular Sciences. 2021; 22(6):3107. https://doi.org/10.3390/ijms22063107
Chicago/Turabian StyleSola-Sevilla, Noemi, Ana Ricobaraza, Ruben Hernandez-Alcoceba, Maria S. Aymerich, Rosa M. Tordera, and Elena Puerta. 2021. "Understanding the Potential Role of Sirtuin 2 on Aging: Consequences of SIRT2.3 Overexpression in Senescence" International Journal of Molecular Sciences 22, no. 6: 3107. https://doi.org/10.3390/ijms22063107
APA StyleSola-Sevilla, N., Ricobaraza, A., Hernandez-Alcoceba, R., Aymerich, M. S., Tordera, R. M., & Puerta, E. (2021). Understanding the Potential Role of Sirtuin 2 on Aging: Consequences of SIRT2.3 Overexpression in Senescence. International Journal of Molecular Sciences, 22(6), 3107. https://doi.org/10.3390/ijms22063107