
A Novel Position-Specific Encoding Algorithm (SeqPose) of Nucleotide Sequences and Its Application for Detecting Enhancers


Abstract
1. Introduction
2. Results and Discussion
2.1. Evaluating the Length of K-mers
2.2. Selecting the Best SeqPose Features
2.3. Symbolic Interpretation of SeqPoseDict
2.4. Optimizing the Best Choices of the Three Parameters
2.5. Comparing spEnhancer with the Existing Models
2.6. Evaluating the Three-Class Classification Model
2.7. Evaluating Different Word Vector Dimensions
3. Materials and Methods
3.1. Datasets and Performance Metrics
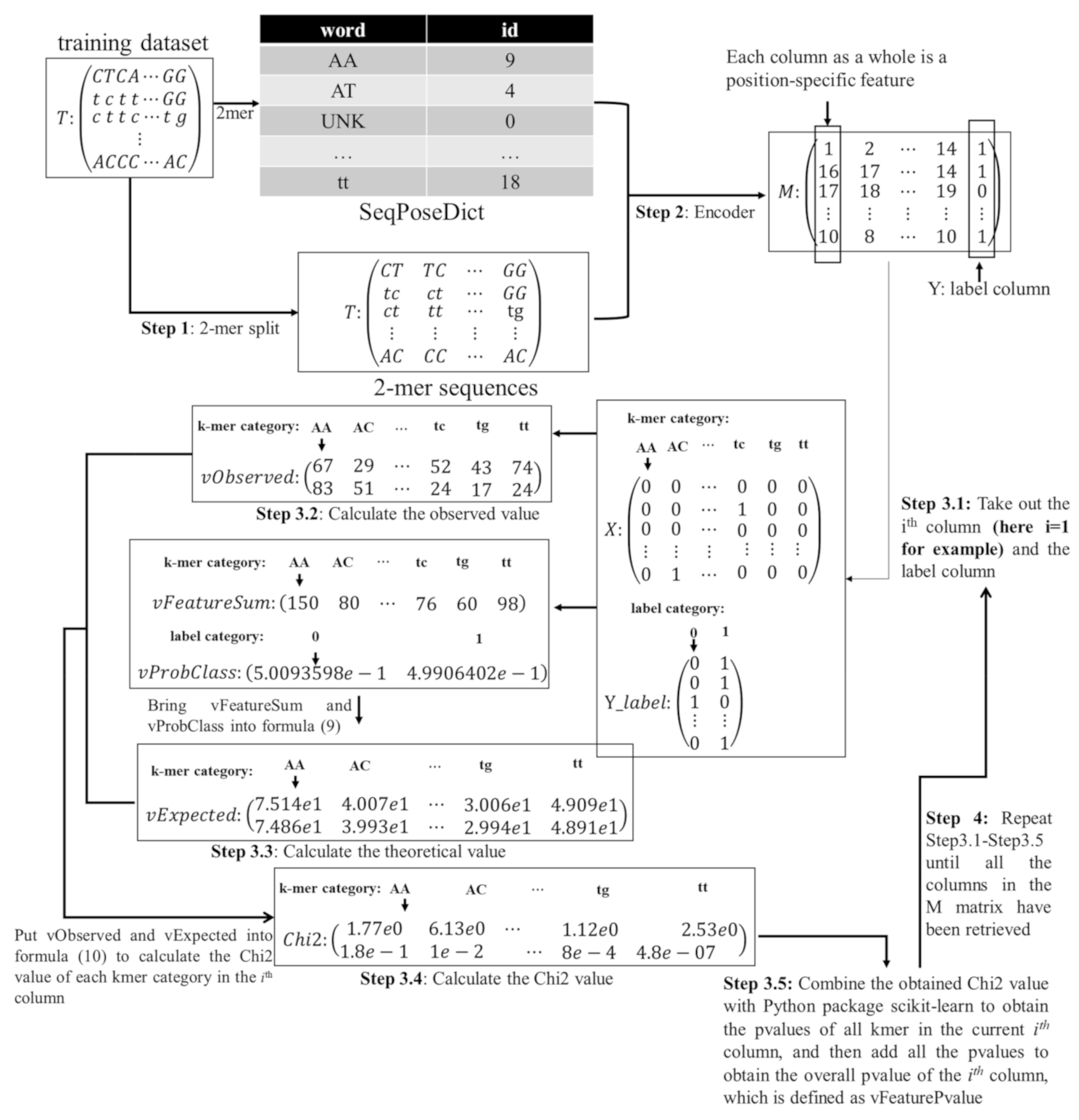
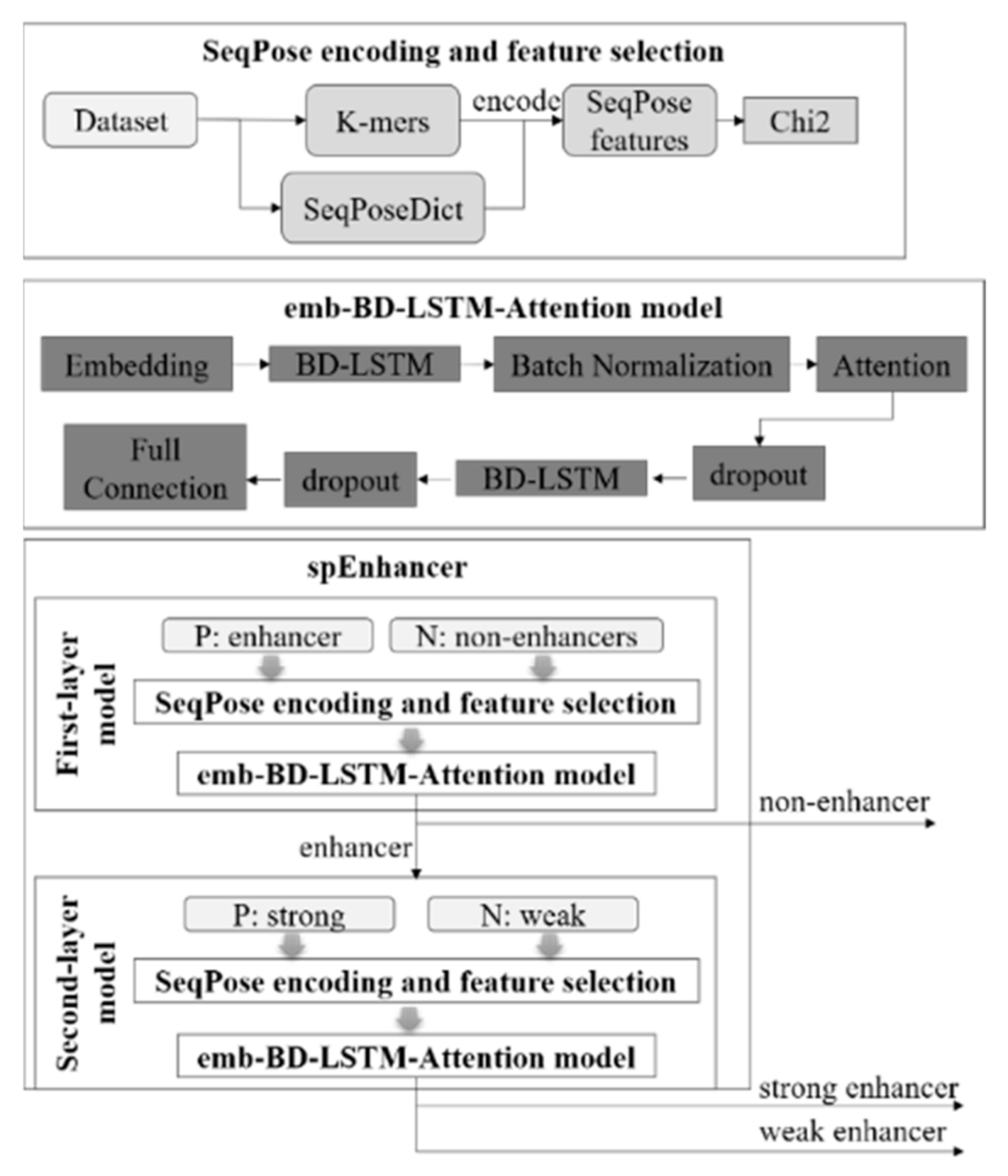
3.2. K-mer Indexing and SeqPose Feature Extraction
3.3. Selecting the Subset of Best Features
3.4. Vectorization
3.5. Structural Description of the Classifier SpEnhancer
3.6. Training Procedure of SpEnhancer
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wierzbicki, A.T. The role of long non-coding RNA in transcriptional gene silencing. Curr. Opin. Plant Biol. 2012, 15, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Ramji, D.P.; Foka, P. CCAAT/enhancer-binding proteins: Structure, function and regulation. Biochem. J. 2002, 365, 561–575. [Google Scholar] [CrossRef] [PubMed]
- Erwin, G.D.; Oksenberg, N.; Truty, R.M.; Kostka, D.; Murphy, K.K.; Ahituv, N.; Pollard, K.S.; Capra, J.A. Integrating Diverse Datasets Improves Developmental Enhancer Prediction. PLoS Comput. Biol. 2014, 10, e1003677. [Google Scholar] [CrossRef]
- Gillies, S.D.; Morrison, S.L.; Oi, V.T.; Tonegawa, S. A tissue-specific transcription enhancer element is located in the major intron of a rearranged immunoglobulin heavy chain gene. Cell 1983, 33, 717–728. [Google Scholar] [CrossRef]
- Larsson, A.J.M.; Johnsson, P.; Hagemann-Jensen, M.; Hartmanis, L.; Faridani, O.R.; Reinius, B.; Segerstolpe, Å.; Rivera, C.M.; Ren, B.; Sandberg, R. Genomic encoding of transcriptional burst kinetics. Nat. Cell Biol. 2019, 565, 251–254. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.-K.; Hemberg, M.; Gray, J.M.; Costa, A.M.; Bear, D.M.; Wu, J.; Harmin, D.A.; Laptewicz, M.; Barbara-Haley, K.; Kuersten, S.; et al. Widespread transcription at neuronal activity-regulated enhancers. Nat. Cell Biol. 2010, 465, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Heintzman, N.D.; Ren, B. Finding distal regulatory elements in the human genome. Curr. Opin. Genet. Dev. 2009, 19, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Boyle, A.P.; Song, L.; Lee, B.-K.; London, D.; Keefe, D.; Birney, E.; Iyer, V.R.; Crawford, G.E.; Furey, T.S. High-resolution genome-wide in vivo footprinting of diverse transcription factors in human cells. Genome Res. 2010, 21, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Davis, H.L.; Weeratna, R.; Waldschmidt, T.J.; Tygrett, L.; Schorr, J.; Krieg, A.M. CpG DNA is a potent enhancer of specific immunity in mice immunized with recombinant hepatitis B surface antigen. J. Immunol. 1998, 160, 870–876. [Google Scholar] [PubMed]
- Firpi, H.A.; Ucar, D.; Tan, K. Discover regulatory DNA elements using chromatin signatures and artificial neural network. Bioinformatics 2010, 26, 1579–1586. [Google Scholar] [CrossRef] [PubMed]
- Rajagopal, N.; Xie, W.; Li, Y.; Wagner, U.; Wang, W.; Stamatoyannopoulos, J.; Ernst, J.; Kellis, M.; Ren, B. RFECS: A Random-Forest Based Algorithm for Enhancer Identification from Chromatin State. PLoS Comput. Biol. 2013, 9, e1002968. [Google Scholar] [CrossRef] [PubMed]
- Bu, H.; Gan, Y.; Wang, Y.; Zhou, S.; Guan, J. A new method for enhancer prediction based on deep belief network. BMC Bioinform. 2017, 18, 418. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Liu, H.; Yang, J.; Chou, K.-C. Prediction of linear B-cell epitopes using amino acid pair antigenicity scale. Amino Acids 2007, 33, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Fang, L.; Longyun, F.; Lan, X.; Chou, K.-C. iEnhancer-2L: A two-layer predictor for identifying enhancers and their strength by pseudok-tuple nucleotide composition. Bioinformatics 2016, 32, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.; He, W. EnhancerPred: A predictor for discovering enhancers based on the combination and selection of multiple features. Sci. Rep. 2016, 6, 38741. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, Q.H.; Nguyen-Vo, T.-H.; Le, N.Q.K.; Do, T.T.; Rahardja, S.; Nguyen, B.P. iEnhancer-ECNN: Identifying enhancers and their strength using ensembles of convolutional neural networks. BMC Genom. 2019, 20, 951. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Li, K.; Huang, D.-S.; Chou, K.-C. iEnhancer-EL: Identifying enhancers and their strength with ensemble learning approach. Bioinformatics 2018, 34, 3835–3842. [Google Scholar] [CrossRef] [PubMed]
- Chou, K. A vectorized sequence-coupling model for predicting HIV protease cleavage sites in proteins. J. Biol. Chem. 1993, 268, 16938–16948. [Google Scholar] [CrossRef]
- Fawcett, T. An introduction to ROC analysis. Pattern Recognit. Lett. 2006, 27, 861–874. [Google Scholar] [CrossRef]
- Devlin, J.; Chang, M.-W.; Lee, K.; Toutanova, K. BERT: Pre-training of Deep Bidirectional Transformers for Language Understanding. In Proceedings of the North American Association for Computational Linguistics-Human Language Technologies 2019, Minneapolis, MN, USA, 2–7 June 2019; pp. 4171–4186. [Google Scholar]
- Kingma, D.P.; Ba, J. Adam: A method for stochastic optimization. In Proceedings of the International Conference on Learnning Representations (ICLR), San Diego, CA, USA, 5–8 May 2015. [Google Scholar]
- Chen, Z.; Pang, M.; Zhao, Z.; Li, S.; Miao, R.; Zhang, Y.; Feng, X.; Feng, X.; Zhang, Y.; Duan, M.; et al. Feature selection may improve deep neural networks for the bioinformatics problems. Bioinformatics 2019, 36, 1542–1552. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| k-mer | Acc | Sn | Sp | MCC | AUC |
|---|---|---|---|---|---|
| 1-mer | 0.7811 | 0.7183 | 0.8387 | 0.5319 | 0.8783 |
| 2-mer | 0.8047 | 0.7254 | 0.8774 | 0.5673 | 0.8781 |
| 3-mer | 0.7912 | 0.7042 | 0.8710 | 0.5383 | 0.8693 |
| 4-mer | 0.8013 | 0.7113 | 0.8839 | 0.5554 | 0.8717 |
| 5-mer | 0.7677 | 0.7887 | 0.7484 | 0.5534 | 0.8637 |
| 6-mer | 0.7778 | 0.6338 | 0.9097 | 0.4870 | 0.8652 |
| 7-mer | 0.7306 | 0.6972 | 0.7613 | 0.4498 | 0.8322 |
| Acc | Sn | Sp | MCC | AUC | |
|---|---|---|---|---|---|
| Res1 | 0.7946 | 0.6620 | 0.9161 | 0.5221 | 0.8827 |
| Res2 | 0.7912 | 0.8028 | 0.7806 | 0.5951 | 0.8797 |
| Res3 | 0.8114 | 0.7394 | 0.8774 | 0.8774 | 0.8920 |
| Res4 | 0.7845 | 0.8099 | 0.7613 | 0.5905 | 0.8827 |
| Res5 | 0.7845 | 0.8380 | 0.7355 | 0.6100 | 0.8885 |
| Sn | Sp | MCC | Acc | AUC | Sn | Sp | MCC | Acc | AUC | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 64,0.1 | 0.7180 | 0.8219 | 0.5161 | 0.7692 | 0.8441 | 64,0.1 | 0.7057 | 0.8429 | 0.5173 | 0.7729 | 0.8476 |
| 64,0.2 | 0.7489 | 0.8032 | 0.5384 | 0.7759 | 0.8496 | 64,0.2 | 0.7468 | 0.7993 | 0.5327 | 0.7729 | 0.8471 |
| 64,0.3 | 0.7135 | 0.8395 | 0.5224 | 0.7773 | 0.8516 | 64,0.3 | 0.7301 | 0.7895 | 0.5061 | 0.7571 | 0.8484 |
| 64,0.4 | 0.7309 | 0.8207 | 0.5307 | 0.7756 | 0.8504 | 64,0.4 | 0.7145 | 0.8317 | 0.5172 | 0.7712 | 0.8508 |
| 64,0.5 | 0.7408 | 0.7976 | 0.5252 | 0.7685 | 0.8475 | 64,0.5 | 0.7001 | 0.8597 | 0.5205 | 0.7800 | 0.8430 |
| 64,0.6 | 0.7393 | 0.8102 | 0.5307 | 0.7743 | 0.8485 | 64,0.6 | 0.7242 | 0.8075 | 0.5129 | 0.7652 | 0.8519 |
| 64,0.7 | 0.7319 | 0.8286 | 0.5366 | 0.7810 | 0.8514 | 64,0.7 | 0.7263 | 0.8254 | 0.5282 | 0.7756 | 0.8478 |
| 128,0.1 | 0.6746 | 0.8602 | 0.4908 | 0.7662 | 0.8427 | 128,0.1 | 0.7295 | 0.8147 | 0.5250 | 0.7729 | 0.8433 |
| 128,0.2 | 0.7256 | 0.7758 | 0.4894 | 0.7507 | 0.8347 | 128,0.2 | 0.6743 | 0.8589 | 0.4970 | 0.7642 | 0.8459 |
| 128,0.3 | 0.6919 | 0.8426 | 0.4983 | 0.7665 | 0.8441 | 128,0.3 | 0.6894 | 0.8304 | 0.4946 | 0.7642 | 0.8361 |
| 128,0.4 | 0.6854 | 0.8534 | 0.5019 | 0.7709 | 0.8479 | 128,0.4 | 0.7620 | 0.7570 | 0.5219 | 0.7571 | 0.8418 |
| 128,0.5 | 0.6947 | 0.8337 | 0.4982 | 0.7638 | 0.8438 | 128,0.5 | 0.7322 | 0.7987 | 0.5187 | 0.7655 | 0.8452 |
| 128,0.6 | 0.7289 | 0.8028 | 0.5154 | 0.7655 | 0.8441 | 128,0.6 | 0.7539 | 0.7820 | 0.5302 | 0.7682 | 0.8384 |
| 128,0.7 | 0.6866 | 0.8509 | 0.4990 | 0.7672 | 0.8373 | 128,0.7 | 0.7255 | 0.7894 | 0.5015 | 0.7551 | 0.8416 |
| 192,0.1 | 0.6969 | 0.7745 | 0.4674 | 0.7385 | 0.8364 | 192,0.1 | 0.6825 | 0.8220 | 0.4753 | 0.7520 | 0.8276 |
| 192,0.2 | 0.6868 | 0.8228 | 0.4821 | 0.7537 | 0.8291 | 192,0.2 | 0.6480 | 0.8739 | 0.4752 | 0.7618 | 0.8364 |
| 192,0.3 | 0.7047 | 0.8098 | 0.4913 | 0.7574 | 0.8357 | 192,0.3 | 0.7250 | 0.8237 | 0.5255 | 0.7756 | 0.8467 |
| 192,0.4 | 0.6406 | 0.8555 | 0.4547 | 0.7470 | 0.8329 | 192,0.4 | 0.6430 | 0.8482 | 0.4576 | 0.7439 | 0.8352 |
| 192,0.5 | 0.6286 | 0.8500 | 0.4427 | 0.7358 | 0.8146 | 192,0.5 | 0.6854 | 0.8527 | 0.5000 | 0.7692 | 0.8484 |
| 192,0.6 | 0.7567 | 0.7800 | 0.5312 | 0.7695 | 0.8420 | 192,0.6 | 0.7468 | 0.7604 | 0.5105 | 0.7564 | 0.8383 |
| 192,0.7 | 0.7089 | 0.8322 | 0.5186 | 0.7746 | 0.8438 | 192,0.7 | 0.7373 | 0.7608 | 0.4924 | 0.7456 | 0.8377 |
| Sn | Sp | MCC | Acc | AUC | Sn | Sp | MCC | Acc | AUC | ||
| 64,0.1 | 0.7199 | 0.8250 | 0.5185 | 0.7709 | 0.8497 | 64,0.1 | 0.7152 | 0.8372 | 0.5233 | 0.7773 | 0.8527 |
| 64,0.2 | 0.7377 | 0.8393 | 0.5509 | 0.7877 | 0.8567 | 64,0.2 | 0.7191 | 0.8276 | 0.5188 | 0.7729 | 0.8507 |
| 64,0.3 | 0.7427 | 0.8106 | 0.5358 | 0.7756 | 0.8521 | 64,0.3 | 0.6868 | 0.8618 | 0.5074 | 0.7732 | 0.8530 |
| 64,0.4 | 0.6896 | 0.8444 | 0.4987 | 0.7665 | 0.8464 | 64,0.4 | 0.6722 | 0.8288 | 0.4664 | 0.7483 | 0.8316 |
| 64,0.5 | 0.7403 | 0.8361 | 0.5521 | 0.7884 | 0.8506 | 64,0.5 | 0.7152 | 0.8233 | 0.5133 | 0.7689 | 0.8428 |
| 64,0.6 | 0.6898 | 0.8411 | 0.4968 | 0.7631 | 0.8507 | 64,0.6 | 0.6995 | 0.8154 | 0.4874 | 0.7577 | 0.8401 |
| 64,0.7 | 0.7265 | 0.8049 | 0.5136 | 0.7642 | 0.8486 | 64,0.7 | 0.7136 | 0.8191 | 0.5081 | 0.7665 | 0.8497 |
| 128,0.1 | 0.6909 | 0.8390 | 0.4946 | 0.7648 | 0.8340 | 128,0.1 | 0.6920 | 0.8464 | 0.5029 | 0.7709 | 0.8466 |
| 128,0.2 | 0.7260 | 0.8166 | 0.5238 | 0.7722 | 0.8479 | 128,0.2 | 0.6973 | 0.8428 | 0.5080 | 0.7692 | 0.8469 |
| 128,0.3 | 0.6754 | 0.8299 | 0.4764 | 0.7544 | 0.8393 | 128,0.3 | 0.6719 | 0.8415 | 0.4755 | 0.7547 | 0.8447 |
| 128,0.4 | 0.6765 | 0.8409 | 0.4863 | 0.7540 | 0.8443 | 128,0.4 | 0.6916 | 0.8168 | 0.4858 | 0.7534 | 0.8444 |
| 128,0.5 | 0.7060 | 0.8300 | 0.5075 | 0.7675 | 0.8470 | 128,0.5 | 0.6994 | 0.8343 | 0.5048 | 0.7668 | 0.8392 |
| 128,0.6 | 0.6852 | 0.8611 | 0.5064 | 0.7732 | 0.8459 | 128,0.6 | 0.7085 | 0.8366 | 0.5152 | 0.7712 | 0.8498 |
| 128,0.7 | 0.6918 | 0.8507 | 0.5091 | 0.7702 | 0.8430 | 128,0.7 | 0.6962 | 0.8189 | 0.4929 | 0.7598 | 0.8518 |
| 192,0.1 | 0.6767 | 0.8462 | 0.4860 | 0.7631 | 0.8378 | 192,0.1 | 0.6529 | 0.8787 | 0.4824 | 0.7679 | 0.8488 |
| 192,0.2 | 0.6848 | 0.8393 | 0.4939 | 0.7618 | 0.8480 | 192,0.2 | 0.6793 | 0.8445 | 0.4884 | 0.7625 | 0.8345 |
| 192,0.3 | 0.6654 | 0.8538 | 0.4768 | 0.7584 | 0.8283 | 192,0.3 | 0.7350 | 0.8156 | 0.5325 | 0.7749 | 0.8528 |
| 192,0.4 | 0.6989 | 0.8349 | 0.5050 | 0.7658 | 0.8437 | 192,0.4 | 0.6592 | 0.8623 | 0.4781 | 0.7594 | 0.8444 |
| 192,0.5 | 0.6812 | 0.8499 | 0.4933 | 0.7638 | 0.8472 | 192,0.5 | 0.6046 | 0.8926 | 0.4439 | 0.7480 | 0.8441 |
| 192,0.6 | 0.6801 | 0.8374 | 0.4836 | 0.7574 | 0.8313 | 192,0.6 | 0.7090 | 0.7949 | 0.4944 | 0.7480 | 0.8405 |
| 192,0.7 | 0.7044 | 0.8305 | 0.5065 | 0.7665 | 0.8422 | 192,0.7 | 0.7177 | 0.8210 | 0.5145 | 0.7675 | 0.8467 |
| Sn | Sp | MCC | Acc | AUC | Sn | Sp | MCC | Acc | AUC | ||
| 64,0.1 | 0.6857 | 0.8343 | 0.4910 | 0.7598 | 0.8363 | 64,0.1 | 0.6857 | 0.7948 | 0.4581 | 0.7433 | 0.8242 |
| 64,0.2 | 0.6979 | 0.8412 | 0.5052 | 0.7702 | 0.8410 | 64,0.2 | 0.6514 | 0.8488 | 0.4591 | 0.7483 | 0.8217 |
| 64,0.3 | 0.6923 | 0.8483 | 0.5027 | 0.7706 | 0.8422 | 64,0.3 | 0.6614 | 0.8005 | 0.4319 | 0.7338 | 0.8102 |
| 64,0.4 | 0.6840 | 0.8524 | 0.4975 | 0.7682 | 0.8436 | 64,0.4 | 0.6781 | 0.7835 | 0.4398 | 0.7315 | 0.8109 |
| 64,0.5 | 0.7304 | 0.8148 | 0.5252 | 0.7736 | 0.8460 | 64,0.5 | 0.6705 | 0.8206 | 0.4593 | 0.7456 | 0.8203 |
| 64,0.6 | 0.7113 | 0.8147 | 0.5040 | 0.7618 | 0.8433 | 64,0.6 | 0.6490 | 0.7762 | 0.3986 | 0.7113 | 0.8037 |
| 64,0.7 | 0.7132 | 0.8421 | 0.5245 | 0.7746 | 0.8538 | 64,0.7 | 0.6253 | 0.8211 | 0.4091 | 0.7217 | 0.8011 |
| 128,0.1 | 0.6982 | 0.8451 | 0.5123 | 0.7702 | 0.8494 | 128,0.1 | 0.6601 | 0.8469 | 0.4701 | 0.7534 | 0.8383 |
| 128,0.2 | 0.7124 | 0.8149 | 0.5069 | 0.7675 | 0.8423 | 128,0.2 | 0.6412 | 0.8730 | 0.4655 | 0.7564 | 0.8237 |
| 128,0.3 | 0.6600 | 0.8739 | 0.4869 | 0.7689 | 0.8400 | 128,0.3 | 0.6770 | 0.8292 | 0.4736 | 0.7527 | 0.8303 |
| 128,0.4 | 0.6407 | 0.8799 | 0.4708 | 0.7581 | 0.8456 | 128,0.4 | 0.7036 | 0.8361 | 0.5097 | 0.7675 | 0.8444 |
| 128,0.5 | 0.6726 | 0.8494 | 0.4857 | 0.7581 | 0.8469 | 128,0.5 | 0.7253 | 0.8013 | 0.5114 | 0.7621 | 0.8374 |
| 128,0.6 | 0.6995 | 0.8190 | 0.4900 | 0.7591 | 0.8343 | 128,0.6 | 0.6967 | 0.8230 | 0.4887 | 0.7601 | 0.8281 |
| 128,0.7 | 0.7194 | 0.8276 | 0.5228 | 0.7732 | 0.8418 | 128,0.7 | 0.6680 | 0.8668 | 0.4912 | 0.7679 | 0.8455 |
| 192,0.1 | 0.7208 | 0.7946 | 0.4964 | 0.7567 | 0.8375 | 192,0.1 | 0.7118 | 0.8042 | 0.4961 | 0.7564 | 0.8404 |
| 192,0.2 | 0.6794 | 0.8020 | 0.4556 | 0.7419 | 0.8239 | 192,0.2 | 0.7025 | 0.7904 | 0.4719 | 0.7473 | 0.8363 |
| 192,0.3 | 0.6893 | 0.7819 | 0.4526 | 0.7355 | 0.8287 | 192,0.3 | 0.6949 | 0.8085 | 0.4779 | 0.7520 | 0.8358 |
| 192,0.4 | 0.6651 | 0.8435 | 0.4715 | 0.7537 | 0.8392 | 192,0.4 | 0.7163 | 0.7901 | 0.4908 | 0.7524 | 0.8314 |
| 192,0.5 | 0.6942 | 0.8122 | 0.4836 | 0.7527 | 0.8317 | 192,0.5 | 0.6893 | 0.8321 | 0.4885 | 0.7621 | 0.8426 |
| 192,0.6 | 0.6847 | 0.8196 | 0.4739 | 0.7476 | 0.8278 | 192,0.6 | 0.6970 | 0.8009 | 0.4750 | 0.7507 | 0.8334 |
| 192,0.7 | 0.6840 | 0.8309 | 0.4816 | 0.7567 | 0.8436 | 192,0.7 | 0.6688 | 0.8532 | 0.4808 | 0.7625 | 0.8395 |
| Methods | Acc | Sn | Sp | MCC | AUC | |
|---|---|---|---|---|---|---|
| enhancers vs non-enhancers | spEnhancer | 0.7793 | 0.7082 | 0.8504 | 0.5227 | 0.8468 |
| iEnhancer-EL | 0.7803 | 0.7567 | 0.8039 | 0.5613 | 0.8547 | |
| iEnhancer-2L | 0.7689 | 0.7809 | 0.7588 | 0.5400 | 0.8500 | |
| EnhancerPred | 0.7318 | 0.7257 | 0.7379 | 0.4636 | 0.8082 | |
| strong enhancers vs weak enhancers | spEnhancer | 0.6413 | 0.8503 | 0.3052 | 0.2105 | 0.6148 |
| iEnhancer-EL | 0.6503 | 0.6900 | 0.6105 | 0.3149 | 0.6957 | |
| iEnhancer-2L | 0.6193 | 0.6221 | 0.6182 | 0.2400 | 0.6600 | |
| EnhancerPred | 0.6206 | 0.6267 | 0.6146 | 0.2413 | 0.6601 |
| Methods | Acc | Sn | Sp | MCC | AUC | |
|---|---|---|---|---|---|---|
| enhancers vs non-enhancers | spEnhancer | 0.7725 | 0.8300 | 0.7150 | 0.5793 | 0.8235 |
| iEnhancer-ECNN | 0.7690 | 0.7850 | 0.7520 | 0.5370 | 0.8320 | |
| iEnhancer-EL | 0.7475 | 0.7100 | 0.7850 | 0.4964 | 0.8173 | |
| iEnhancer-2L | 0.7300 | 0.7100 | 0.7500 | 0.4604 | 0.8062 | |
| EnhancerPred | 0.7400 | 0.7350 | 0.7450 | 0.4800 | 0.8013 | |
| strong enhancers vs weak enhancers | spEnhancer | 0.6200 | 0.9100 | 0.3300 | 0.3703 | 0.6253 |
| iEnhancer-ECNN | 0.6780 | 0.7910 | 0.7480 | 0.3680 | 0.7480 | |
| iEnhancer-EL | 0.6100 | 0.5400 | 0.6800 | 0.2222 | 0.6801 | |
| iEnhancer-2L | 0.6050 | 0.4700 | 0.7400 | 0.2181 | 0.6678 | |
| EnhancerPred | 0.5500 | 0.4500 | 0.6500 | 0.1021 | 0.5790 |
| Word Vector Dimension | Acc | Sn | Sp | MCC | AUC | |
|---|---|---|---|---|---|---|
| enhancers vs. non-enhancers | 12 | 0.7085 | 0.8550 | 0.5606 | 0.4943 | 0.8177 |
| 24 | 0.6658 | 0.9150 | 0.4141 | 0.4655 | 0.8094 | |
| 48 | 0.7538 | 0.8150 | 0.6919 | 0.5408 | 0.8167 | |
| 96 | 0.7060 | 0.8650 | 0.5455 | 0.4971 | 0.7359 | |
| 192 | 0.7186 | 0.7650 | 0.6717 | 0.4580 | 0.8078 | |
| 394 | 0.7337 | 0.8400 | 0.6263 | 0.5253 | 0.8172 | |
| 768 (this study) | 0.7725 | 0.8300 | 0.7150 | 0.5793 | 0.8235 | |
| 1536 | 0.7764 | 0.7550 | 0.7980 | 0.5408 | 0.8281 | |
| strong enhancers vs. weak enhancers | 12 | 0.5550 | 0.4300 | 0.6800 | 0.0984 | 0.6351 |
| 24 | 0.5000 | 1.0000 | 0.0000 | 0.0000 | 0.6324 | |
| 48 | 0.6000 | 0.7100 | 0.4900 | 0.2265 | 0.6342 | |
| 96 | 0.6250 | 0.8000 | 0.4500 | 0.3101 | 0.6275 | |
| 192 | 0.5700 | 0.6700 | 0.4700 | 0.1565 | 0.6279 | |
| 394 | 0.5950 | 0.7100 | 0.4800 | 0.2165 | 0.5987 | |
| 768 (this study) | 0.6200 | 0.9100 | 0.3300 | 0.3703 | 0.6253 | |
| 1536 | 0.5800 | 0.8700 | 0.2900 | 0.2469 | 0.5972 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mu, X.; Wang, Y.; Duan, M.; Liu, S.; Li, F.; Wang, X.; Zhang, K.; Huang, L.; Zhou, F. A Novel Position-Specific Encoding Algorithm (SeqPose) of Nucleotide Sequences and Its Application for Detecting Enhancers. Int. J. Mol. Sci. 2021, 22, 3079. https://doi.org/10.3390/ijms22063079
Mu X, Wang Y, Duan M, Liu S, Li F, Wang X, Zhang K, Huang L, Zhou F. A Novel Position-Specific Encoding Algorithm (SeqPose) of Nucleotide Sequences and Its Application for Detecting Enhancers. International Journal of Molecular Sciences. 2021; 22(6):3079. https://doi.org/10.3390/ijms22063079
Chicago/Turabian StyleMu, Xuechen, Yueying Wang, Meiyu Duan, Shuai Liu, Fei Li, Xiuli Wang, Kai Zhang, Lan Huang, and Fengfeng Zhou. 2021. "A Novel Position-Specific Encoding Algorithm (SeqPose) of Nucleotide Sequences and Its Application for Detecting Enhancers" International Journal of Molecular Sciences 22, no. 6: 3079. https://doi.org/10.3390/ijms22063079
APA StyleMu, X., Wang, Y., Duan, M., Liu, S., Li, F., Wang, X., Zhang, K., Huang, L., & Zhou, F. (2021). A Novel Position-Specific Encoding Algorithm (SeqPose) of Nucleotide Sequences and Its Application for Detecting Enhancers. International Journal of Molecular Sciences, 22(6), 3079. https://doi.org/10.3390/ijms22063079