
Butyrate and the Fine-Tuning of Colonic Homeostasis: Implication for Inflammatory Bowel Diseases



Abstract
1. Introduction
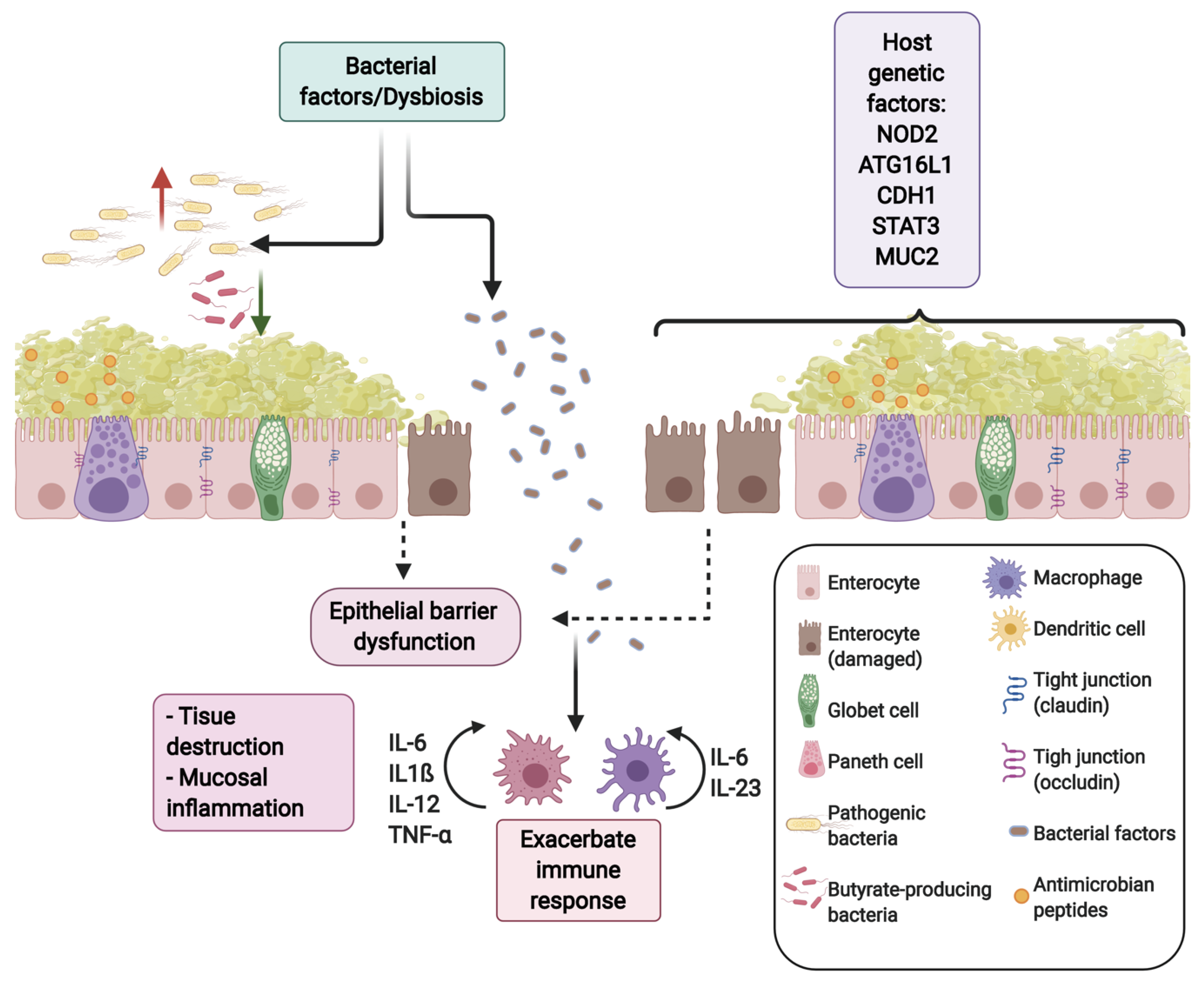
2. The Gut Microbiota in Health and IBDs
3. Butyrate Production
4. Butyrate Uptake by Epithelial Cells
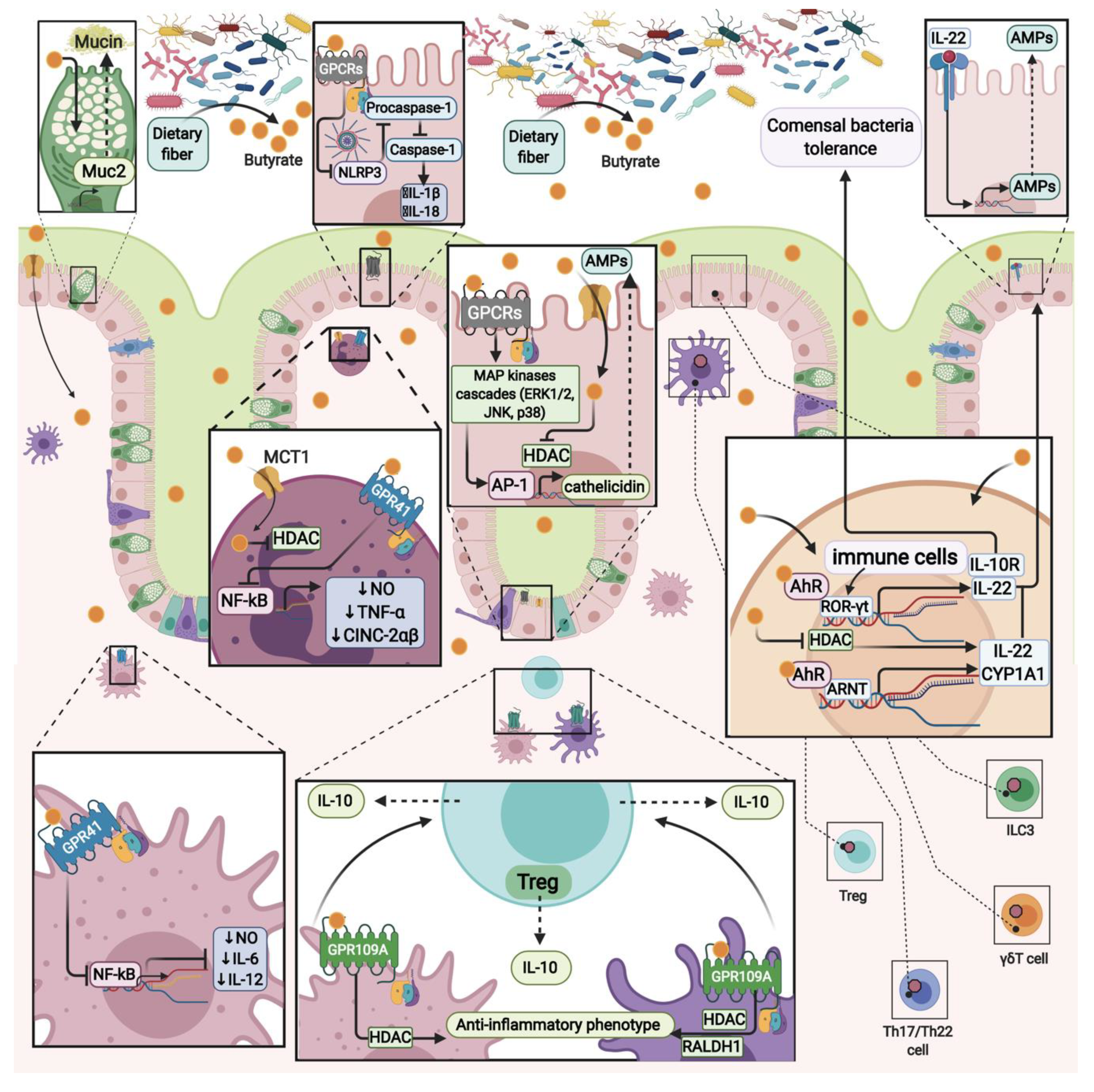
5. Butyrate Effects on Epithelial Cells
6. Butyrate in the Colonocyte
6.1. The Paradoxical Effect of Butyrate
6.2. Butyrate and the Generation of Oxygen Gradient
7. Butyrate as a Regulator of Epigenetic Processes
8. Anti-inflammatory Butyrate Effects
9. Butyrate as a Ligand for the Aryl Hydrocarbon Receptor (AhR)
10. Butyrate and Intestinal Barrier Function
11. Role of Butyrate in the Development of Colorectal Cancer
12. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| GM | Gut Microbiota |
| SCFA | Short-Chain Fatty Acids |
| IBD | Inflammatory Bowel Diseases |
| CD | Crohn’s Disease |
| UC | Ulcerative Colitis |
| GALT | Gastrointestinal Associated Lymphoid Tissue |
| MCT1 | Proton-coupled Monocarboxylate Transporter-1 |
| SMCT1 | Sodium-coupled Monocarboxylate Transporter-1 |
| GPR41 | G-protein coupled Receptor 41 |
| GRP43 | G-protein coupled Receptor 43 |
| GPR109A | G-protein coupled Receptor 109A |
| NF-kB | Nuclear Factor kappa Beta |
| HAT | Histone Acetyl Transferase |
| HDAC | Histone Deacetylase |
| DSS | Dextran Sodium Sulfate |
| TCA | Tricarboxylic acid |
| HIF | Hypoxia-inducible Factor |
| NOS2 | Nitric Oxide Synthase 2 |
| PPAR-γ | Peroxisome Proliferator-activated Receptor-γ |
| NLRP3 | Pyrin Domain Containing 3 |
| LPS | Lipopolysaccharide |
| AhR | Aryl Hydrocarbon Receptor |
| TEER | Transepithelial Electrical Resistance |
| MUC2 | Mucin 2 |
| ZO-1 | Zonulin 1 |
References
- Edelblum, K.L.; Turner, J.R. The tight junction in inflammatory disease: Communication breakdown. Curr. Opin. Pharmacol. 2009, 9, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Vindigni, S.M.; Zisman, T.L.; Suskind, D.L.; Damman, C.J. The intestinal microbiome, barrier function, and immune system in inflammatory bowel disease: A tripartite pathophysiological circuit with implications for new therapeutic directions. Ther. Adv. Gastroenterol. 2016, 9, 606–625. [Google Scholar] [CrossRef] [PubMed]
- Hooper, L.V.; Littman, D.R.; Macpherson, A.J. Interactions between the microbiota and the immune system. Science 2012, 336, 1268–1273. [Google Scholar] [CrossRef] [PubMed]
- Littman, D.R.; Pamer, E.G. Role of the commensal microbiota in normal and pathogenic host immune responses. Cell Host Microbe 2011, 10, 311–323. [Google Scholar] [CrossRef]
- Lozupone, C.A.; Stombaugh, J.I.; Gordon, J.I.; Jansson, J.K.; Knight, R. Diversity, stability and resilience of the human gut microbiota. Nature 2012, 489, 220–230. [Google Scholar] [CrossRef]
- Ballester Ferré, M.P.; Boscá-Watts, M.M.; Mínguez Pérez, M. Enfermedad de Crohn. Med. Clín. 2018, 151, 26–33. [Google Scholar] [CrossRef]
- Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef]
- Kuo, S.-M. The Interplay Between Fiber and the Intestinal Microbiome in the Inflammatory Response. Adv. Nutr. 2013, 4, 16–28. [Google Scholar] [CrossRef]
- Macfarlane, S.; Macfarlane, G.T. Regulation of short-chain fatty acid production. Proc. Nutr. Soc. 2003, 62, 67–72. [Google Scholar] [CrossRef]
- Vinolo, M.A.R.; Rodrigues, H.G.; Nachbar, R.T.; Curi, R. Regulation of Inflammation by Short Chain Fatty Acids. Nutrients 2011, 3, 858–876. [Google Scholar] [CrossRef]
- Carding, S.; Verbeke, K.; Vipond, D.T.; Corfe, B.M.; Owen, L.J. Dysbiosis of the gut microbiota in disease. Microb. Ecol. Health Dis. 2015, 26, 26191. [Google Scholar] [CrossRef] [PubMed]
- Gentile, C.L.; Weir, T.L. The gut microbiota at the intersection of diet and human health. Science 2018, 362, 776–780. [Google Scholar] [CrossRef]
- Amre, D.K.; Seidman, G. Imbalances in Dietary Consumption of Fatty Acids, Vegetables, and Fruits Are Associated with Risk for Crohn’s Disease in Children. Am. J. Gastroenterol. 2007, 102, 2016–2025. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.K.; Abraham, B.; El-Serag, H. Dietary Intake and Risk of Developing Inflammatory Bowel Disease: A Systematic Review of the Literature. Am. J. Gastroenterol. 2011, 106, 563–573. [Google Scholar] [CrossRef]
- Hansen, L.; Skeie, G.; Landberg, R.; Lund, E.; Palmqvist, R.; Johansson, I.; Dragsted, L.O.; Egeberg, R.; Johnsen, N.F.; Christensen, J.; et al. Intake of dietary fiber, especially from cereal foods, is associated with lower incidence of colon cancer in the HELGA cohort. Int. J. Cancer 2012, 131, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Ben, Q.; Sun, Y.; Chai, R.; Qian, A.; Xu, B.; Yuan, Y. Dietary fiber intake reduces risk for colorectal adenoma: A meta-analysis. Gastroenterology 2014, 146, 689–699.e6. [Google Scholar] [CrossRef]
- Roediger, W.E.W. The starved colon—Diminished mucosal nutrition, diminished absorption, and colitis. Dis. Colon Rectum 1990, 33, 858–862. [Google Scholar] [CrossRef] [PubMed]
- Scheppach, W.; Christl, S.U.; Bartram, H.P.; Richter, F.; Kasper, H. Effects of short-chain fatty acids on the inflamed colonic mucosa. Scand. J. Gastroenterol. Suppl. 1997, 222, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Guillemot, F.; Colombel, J.F.; Neut, C.; Verplanck, N.; Lecomte, M.; Romond, C.; Paris, J.C.; Cortot, A. Treatment of diversion colitis by short-chain fatty acids. Prospective and double-blind study. Dis. Colon Rectum 1991, 34, 861–864. [Google Scholar] [CrossRef]
- Kaczmarek, N.; Kokot, M.; Makarewicz, A.; Glapa-Nowak, A.; Nowak, J.; Jamka, M.; Walkowiak, J. The therapeutic potential of short-chain fatty acids enemas in inflammatory bowel diseases: A systematic review. Farm. Pol. 2020, 76, 297–304. [Google Scholar] [CrossRef]
- Sokol, H.; Pigneur, B.; Watterlot, L.; Lakhdari, O.; Bermudez-Humaran, L.G.; Gratadoux, J.-J.; Blugeon, S.; Bridonneau, C.; Furet, J.-P.; Corthier, G.; et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc. Natl. Acad. Sci. USA 2008, 105, 16731–16736. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Nishida, A.; Fujimoto, T.; Fujii, M.; Shioya, M.; Imaeda, H.; Inatomi, O.; Bamba, S.; Sugimoto, M.; Andoh, A. Reduced Abundance of Butyrate-Producing Bacteria Species in the Fecal Microbial Community in Crohn’s Disease. Digestion 2016, 93, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Chen, L.; Zhou, R.; Wang, X.; Huang, S.; Wang, G.; Xia, B. Increased Proportions of Bifidobacterium and the Lactobacillus Group and Loss of Butyrate-Producing Bacteria in Inflammatory Bowel Disease. J. Clin. Microbiol. 2014, 52, 10. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.N.; St Amand, A.L.; Feldman, R.A.; Boedeker, E.C.; Harpaz, N.; Pace, N.R. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 13780–13785. [Google Scholar] [CrossRef]
- Sartor, R.B. The Intestinal Microbiota in Inflammatory Bowel Diseases. In Nestlé Nutrition Institute Workshop Series; Lewis, J.D., Ruemmele, F.M., Wu, G.D., Eds.; S. KARGER AG: Basel, Switzerland, 2014; Volume 79, pp. 29–39. ISBN 978-3-318-02669-6. [Google Scholar]
- Vidal, R.; Ginard, D.; Khorrami, S.; Mora-Ruiz, M.; Munoz, R.; Hermoso, M.; Díaz, S.; Cifuentes, A.; Orfila, A.; Rosselló-Móra, R. Crohn associated microbial communities associated to colonic mucosal biopsies in patients of the western Mediterranean. Syst. Appl. Microbiol. 2015, 38, 442–452. [Google Scholar] [CrossRef]
- Olaisen, M.; Flatberg, A.; Granlund, A.V.B.; Røyset, E.S.; Martinsen, T.C.; Sandvik, A.K.; Fossmark, R. Bacterial Mucosa-associated Microbiome in Inflamed and Proximal Noninflamed Ileum of Patients with Crohn’s Disease. Inflamm. Bowel Dis. 2020. [Google Scholar] [CrossRef]
- Rolhion, N.; Darfeuille-Michaud, A. Adherent-invasive Escherichia coli in inflammatory bowel disease. Inflamm. Bowel Dis. 2007, 13, 7. [Google Scholar] [CrossRef]
- De la Fuente, M.; Franchi, L.; Araya, D.; Díaz-Jiménez, D.; Olivares, M.; Álvarez-Lobos, M.; Golenbock, D.; González, M.-J.; López-Kostner, F.; Quera, R.; et al. Escherichia coli isolates from inflammatory bowel diseases patients survive in macrophages and activate NLRP3 inflammasome. Int. J. Med. Microbiol. IJMM 2014, 304, 384–392. [Google Scholar] [CrossRef]
- Liguori, G.; Lamas, B.; Richard, M.L.; Brandi, G.; da Costa, G.; Hoffmann, T.W.; Di Simone, M.P.; Calabrese, C.; Poggioli, G.; Langella, P.; et al. Fungal Dysbiosis in Mucosa-associated Microbiota of Crohn’s Disease Patients. J. Crohns Colitis 2016, 10, 296–305. [Google Scholar] [CrossRef]
- Honda, K.; Littman, D.R. The Microbiome in Infectious Disease and Inflammation. Annu. Rev. Immunol. 2012, 30, 759–795. [Google Scholar] [CrossRef]
- Hamer, H.M.; Jonkers, D.; Venema, K.; Vanhoutvin, S.; Troost, F.J.; Brummer, R.-J. Review article: The role of butyrate on colonic function: REVIEW: ROLE OF BUTYRATE ON COLONIC FUNCTION. Aliment. Pharmacol. Ther. 2007, 27, 104–119. [Google Scholar] [CrossRef] [PubMed]
- Vital, M.; Karch, A.; Pieper, D.H. Colonic Butyrate-Producing Communities in Humans: An Overview Using Omics Data. mSystems 2017, 2, e00130-17. [Google Scholar] [CrossRef]
- Thomson, P.; Medina, D.A.; Ortúzar, M.V.; Gotteland, M.; Garrido, D. Anti-inflammatory effect of microbial consortia during the utilization of dietary. Food Res. Int. 2018, 109, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, D.S.G.; Jensen, B.B.; Theil, P.K.; Nielsen, T.S.; Knudsen, K.E.B.; Purup, S. Effect of butyrate and fermentation products on epithelial integrity in a mucus-secreting human colon cell line. J. Funct. Foods 2018, 40, 9–17. [Google Scholar] [CrossRef]
- Iwanaga, T.; Takebe, K.; Kato, I.; Karaki, S.-I.; Kuwahara, A. Cellular expression of monocarboxylate transporters (MCT) in the digestive tract of the mouse, rat, and humans, with special reference to slc5a8. Biomed. Res. Tokyo Jpn. 2006, 27, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; van Esch, B.C.A.M.; Wagenaar, G.T.M.; Garssen, J.; Folkerts, G.; Henricks, P.A.J. Pro- and anti-inflammatory effects of short chain fatty acids on immune and endothelial cells. Eur. J. Pharmacol. 2018, 831, 52–59. [Google Scholar] [CrossRef]
- Borthakur, A.; Priyamvada, S.; Kumar, A.; Natarajan, A.A.; Gill, R.K.; Alrefai, W.A.; Dudeja, P.K. A novel nutrient sensing mechanism underlies substrate-induced regulation of monocarboxylate transporter-1. Am. J. Physiol.-Gastrointest. Liver Physiol. 2012, 303, G1126–G1133. [Google Scholar] [CrossRef]
- Ramakrishna, B.S.; Young, G.P. Amylase-Resistant Starch plus Oral Rehydration Solution for Cholera. N. Engl. J. Med. 2000, 342, 308–313. [Google Scholar] [CrossRef]
- De Preter, V.; Geboes, K.P.; Bulteel, V.; Vandermeulen, G.; Suenaert, P.; Rutgeerts, P.; Verbeke, K. Kinetics of butyrate metabolism in the normal colon and in ulcerative colitis: The effects of substrate concentration and carnitine on the β-oxidation pathway: Kinetics of butyrate metabolism in ulcerative colitis. Aliment. Pharmacol. Ther. 2011, 34, 526–532. [Google Scholar] [CrossRef]
- Gonçalves, P.; Martel, F. Regulation of colonic epithelial butyrate transport: Focus on colorectal cancer. Porto Biomed. J. 2016, 1, 83–91. [Google Scholar] [CrossRef]
- Thibault, R.; De Coppet, P.; Daly, K.; Bourreille, A.; Cuff, M.; Bonnet, C.; Mosnier, J.; Galmiche, J.; Shirazi–Beechey, S.; Segain, J. Down-Regulation of the Monocarboxylate Transporter 1 Is Involved in Butyrate Deficiency During Intestinal Inflammation. Gastroenterology 2007, 133, 1916–1927. [Google Scholar] [CrossRef] [PubMed]
- Karaki, S.; Mitsui, R.; Hayashi, H.; Kato, I.; Sugiya, H.; Iwanaga, T.; Furness, J.B.; Kuwahara, A. Short-chain fatty acid receptor, GPR43, is expressed by enteroendocrine cells and mucosal mast cells in rat intestine. Cell Tissue Res. 2006, 324, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Thangaraju, M.; Cresci, G.A.; Liu, K.; Ananth, S.; Gnanaprakasam, J.P.; Browning, D.D.; Mellinger, J.D.; Smith, S.B.; Digby, G.J.; Lambert, N.A.; et al. GPR109A is a G-protein-coupled receptor for the bacterial fermentation product butyrate and functions as a tumor suppressor in colon. Cancer Res. 2009, 69, 2826–2832. [Google Scholar] [CrossRef] [PubMed]
- DeGruttola, A.K.; Low, D.; Mizoguchi, A.; Mizoguchi, E. Current Understanding of Dysbiosis in Disease in Human and Animal Models. Inflamm. Bowel Dis. 2016, 22, 1137–1150. [Google Scholar] [CrossRef] [PubMed]
- Donohoe, D.R.; Garge, N.; Zhang, X.; Sun, W.; O’Connell, T.M.; Bunger, M.K.; Bultman, S.J. The Microbiome and Butyrate Regulate Energy Metabolism and Autophagy in the Mammalian Colon. Cell Metab. 2011, 13, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.A.; Ogawa, S.A.; Chau, L.; Whelan, K.A.; Hamilton, K.E.; Chen, J.; Tan, L.; Chen, E.Z.; Keilbaugh, S.; Fogt, F.; et al. Mitochondrial dysfunction in inflammatory bowel disease alters intestinal epithelial metabolism of hepatic acylcarnitines. J. Clin. Investig. 2020. [Google Scholar] [CrossRef]
- Peltekova, V.D.; Wintle, R.F.; Rubin, L.A.; Amos, C.I.; Huang, Q.; Gu, X.; Newman, B.; Van Oene, M.; Cescon, D.; Greenberg, G.; et al. Functional variants of OCTN cation transporter genes are associated with Crohn disease. Nat. Genet. 2004, 36, 471–475. [Google Scholar] [CrossRef]
- Kaiko, G.E.; Ryu, S.H.; Koues, O.I.; Collins, P.L.; Solnica-Krezel, L.; Pearce, E.J.; Pearce, E.L.; Oltz, E.M.; Stappenbeck, T.S. The Colonic Crypt Protects Stem Cells from Microbiota-Derived Metabolites. Cell 2016, 165, 1708–1720. [Google Scholar] [CrossRef]
- Lin, J.; Peng, L.; Itzkowitz, S.; Holzman, I.R.; Babyatsky, M.W. Short-Chain Fatty Acid Induces Intestinal Mucosal Injury in Newborn Rats and Down-Regulates Intestinal Trefoil Factor Gene Expression In Vivo and In Vitro. J. Pediatr. Gastroenterol. Nutr. 2005, 41, 607–611. [Google Scholar] [CrossRef]
- Cummins, A.G. Effect of breast milk and weaning on epithelial growth of the small intestine in humans. Gut 2002, 51, 748–754. [Google Scholar] [CrossRef]
- Appert, O.; Garcia, A.R.; Frei, R.; Roduit, C.; Constancias, F.; Neuzil-Bunesova, V.; Ferstl, R.; Zhang, J.; Akdis, C.; Lauener, R.; et al. Initial butyrate producers during infant gut microbiota development are endospore formers. Environ. Microbiol. 2020, 22, 3909–3921. [Google Scholar] [CrossRef] [PubMed]
- Midtvedt, A.C.; Midtvedt, T. Production of short chain fatty acids by the intestinal microflora during the first 2 years of human life. J. Pediatr. Gastroenterol. Nutr. 1992, 15, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Kelly, C.J. Crosstalk between Microbiota-Derived Short-Chain Fatty Acids and Intestinal Epithelial HIF Augments Tissue Barrier Function. Cell Host Microbe 2015, 17, 662–671. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Chávez, F. Depletion of Butyrate-Producing Clostridia from the Gut Microbiota Drives an Aerobic Luminal Expansion of Salmonella. Cell Host Microbe 2016, 19, 443–454. [Google Scholar] [CrossRef]
- Winter, S.E.; Winter, M.G.; Xavier, M.N.; Thiennimitr, P.; Poon, V.; Keestra, A.M.; Laughlin, R.C.; Gomez, G.; Wu, J.; Lawhon, S.D.; et al. Host-Derived Nitrate Boosts Growth of E. coli in the Inflamed Gut. Science 2013, 339, 708–711. [Google Scholar] [CrossRef]
- Byndloss, M.X.; Olsan, E.E.; Rivera-Chávez, F.; Tiffany, C.R.; Cevallos, S.A.; Lokken, K.L.; Torres, T.P.; Byndloss, A.J.; Faber, F.; Gao, Y.; et al. Microbiota-activated PPAR-g signaling inhibits dysbiotic Enterobacteriaceae expansion. Science 2017, 357, 570–575. [Google Scholar] [CrossRef]
- Rigottier-Gois, L. Dysbiosis in inflammatory bowel diseases: The oxygen hypothesis. ISME J. 2013, 7, 1256–1261. [Google Scholar] [CrossRef]
- Theochari, N.A.; Stefanopoulos, A.; Mylonas, K.S.; Economopoulos, K.P. Antibiotics exposure and risk of inflammatory bowel disease: A systematic review. Scand. J. Gastroenterol. 2018, 53, 1–7. [Google Scholar] [CrossRef]
- Dubuquoy, L.; Jansson, E.Å.; Deeb, S.; Rakotobe, S.; Karoui, M.; Colombel, J.-F.; Auwerx, J.; Pettersson, S.; Desreumaux, P. Impaired expression of peroxisome proliferator-activated receptor γ in ulcerative colitis. Gastroenterology 2003, 124, 1265–1276. [Google Scholar] [CrossRef]
- Donohoe, D.R. The Warburg Effect Dictates the Mechanism of Butyrate-Mediated Histone Acetylation and Cell Proliferation. Mol. Cell 2012, 48, 612–626. [Google Scholar] [CrossRef]
- Furusawa, Y. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Kaisar, M.M.M.; Pelgrom, L.R.; van der Ham, A.J.; Yazdanbakhsh, M.; Everts, B. Butyrate Conditions Human Dendritic Cells to Prime Type 1 Regulatory T Cells via both Histone Deacetylase Inhibition and G Protein-Coupled Receptor 109A Signaling. Front. Immunol. 2017, 8, 1429. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Gurav, A.; Sivaprakasam, S.; Brady, E.; Padia, R.; Shi, H.; Thangaraju, M.; Prasad, P.D.; Manicassamy, S.; Munn, D.H.; et al. Activation of Gpr109a, Receptor for Niacin and the Commensal Metabolite Butyrate, Suppresses Colonic Inflammation and Carcinogenesis. Immunity 2014, 40, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Nastasi, C.; Fredholm, S.; Willerslev-Olsen, A.; Hansen, M.; Bonefeld, C.M.; Geisler, C.; Andersen, M.H.; Ødum, N.; Woetmann, A. Butyrate and propionate inhibit antigen-specific CD8+ T cell activation by suppressing IL-12 production by antigen-presenting cells. Sci. Rep. 2017, 7, 14516. [Google Scholar] [CrossRef] [PubMed]
- Petersen, C.; Round, J.L. Defining dysbiosis and its influence on host immunity and disease. Cell. Microbiol. 2014, 16, 1024–1033. [Google Scholar] [CrossRef]
- Andoh, A.; Fujiyama, Y.; Hata, K.; Araki, Y.; Takaya, H.; Shimada, M.; Bamba, T. Counter-regulatory effect of sodium butyrate on tumour necrosis factor-alpha (TNF-a)-induced complement C3 and factor B biosynthesis in human intestinal epithelial cells. Clin. Exp. Immunol. 1999, 118, 23. [Google Scholar] [CrossRef]
- Chang, P.V.; Hao, L.; Offermanns, S.; Medzhitov, R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc. Natl. Acad. Sci. USA 2014, 111, 2247–2252. [Google Scholar] [CrossRef]
- Vinolo, M.A.R. Suppressive effect of short-chain fatty acids on production of proinflammatory mediators by neutrophils. J. Nutr. Biochem. 2011, 22, 849–855. [Google Scholar] [CrossRef]
- Kellow, N.J.; Coughlan, M.T.; Reid, C.M. Metabolic benefits of dietary prebiotics in human subjects: A systematic review of randomised controlled trials. Br. J. Nutr. 2014, 111, 1147–1161. [Google Scholar] [CrossRef]
- Fusunyan, R.D.; Quinn, J.J.; Fujimoto, M.; MacDermott, R.P.; Sanderson, I.R. Butyrate Switches the Pattern of Chemokine Secretion by Intestinal Epithelial Cells through Histone Acetylation. Mol. Med. 1999, 5, 631–640. [Google Scholar] [CrossRef]
- Böcker, U.; Nebe, T.; Herweck, F.; Holt, L.; Panja, A.; Jobin, C.; Rossol, S.; Sartor, R.B.; Singer, M.V. Butyrate modulates intestinal epithelial cell-mediated neutrophil migration: Immunomodulatory effects of butyrate on IEC. Clin. Exp. Immunol. 2003, 131, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Asarat, M.; Vasiljevic, T.; Apostolopoulos, V.; Donkor, O. Short-Chain Fatty Acids Regulate Secretion of IL-8 from Human Intestinal Epithelial Cell Lines in vitro. Immunol. Investig. 2015, 44, 678–693. [Google Scholar] [CrossRef] [PubMed]
- Hung, T.V.; Suzuki, T. Short-Chain Fatty Acids Suppress Inflammatory Reactions in Caco-2 Cells and Mouse Colons. J. Agric. Food Chem. 2018, 66, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Shao, B.-Z.; Xu, Z.-Q.; Han, B.-Z.; Su, D.-F.; Liu, C. NLRP3 inflammasome and its inhibitors: A review. Front. Pharmacol. 2015, 6, 262. [Google Scholar] [CrossRef]
- Malago, J.J.; Tooten, P.C.J.; van Liere, E.A.; van Dijk, J.E. Anti-inflammatory properties of heat shock protein 70 and butyrate on Salmonella-induced interleukin-8 secretion in enterocyte-like Caco. Clin. Exp. Immunol. 2005, 141, 62–71. [Google Scholar] [CrossRef]
- Yang, W.; Yu, T.; Huang, X.; Bilotta, A.J.; Xu, L.; Lu, Y.; Sun, J.; Pan, F.; Zhou, J.; Zhang, W.; et al. Intestinal microbiota-derived short-chain fatty acids regulation of immune cell IL-22 production and gut immunity. Nat. Commun. 2020, 11, 4457. [Google Scholar] [CrossRef]
- Liu, H.; Wang, J.; He, T.; Becker, S.; Zhang, G.; Li, D.; Ma, X. Butyrate: A Double-Edged Sword for Health? Adv. Nutr. 2018, 9, 21–29. [Google Scholar] [CrossRef]
- Lührs, H.; Gerke, T.; Müller, J.G.; Melcher, R.; Schauber, J.; Boxberger, F.; Scheppach, W.; Menzel, T. Butyrate Inhibits NF-κB Activation in Lamina Propria Macrophages of Patients with Ulcerative Colitis. Scand. J. Gastroenterol. 2002, 37, 458–466. [Google Scholar] [CrossRef]
- Hamer, H.M.; Jonkers, D.M.A.E.; Vanhoutvin, S.A.L.W.; Troost, F.J.; Rijkers, G.; de Bruïne, A.; Bast, A.; Venema, K.; Brummer, R.-J.M. Effect of butyrate enemas on inflammation and antioxidant status in the colonic mucosa of patients with ulcerative colitis in remission. Clin. Nutr. 2010, 29, 738–744. [Google Scholar] [CrossRef]
- Breuer, R.I.; Soergel, K.H.; Lashner, B.A.; Christ, M.L.; Hanauer, S.B.; Vanagunas, A.; Harig, J.M.; Keshavarzian, A.; Robinson, M.; Sellin, J.H.; et al. Short chain fatty acid rectal irrigation for left-sided ulcerative colitis: A randomised, placebo controlled trial. Gut 1997, 40, 485–491. [Google Scholar] [CrossRef]
- van der Does, A.M.; Bergman, P.; Agerberth, B.; Lindbom, L. Induction of the human cathelicidin LL-37 as a novel treatment against bacterial infections. J. Leukoc. Biol. 2012, 92, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Kida, Y.; Shimizu, T.; Kuwano, K. Sodium butyrate up-regulates cathelicidin gene expression via activator protein-1 and histone acetylation at the promoter region in a human lung epithelial cell line, EBC-1. Mol. Immunol. 2006, 43, 1972–1981. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Xu, K.; Liu, H.; Liu, G.; Bai, M.; Peng, C.; Li, T.; Yin, Y. Impact of the Gut Microbiota on Intestinal Immunity Mediated by Tryptophan Metabolism. Front. Cell. Infect. Microbiol. 2018, 8, 13. [Google Scholar] [CrossRef] [PubMed]
- Hankinson, O. The Aryl Hydrocarbon Receptor Complex. Annu. Rev. Pharmacol. Toxicol. 1995, 35, 307–340. [Google Scholar] [CrossRef] [PubMed]
- Quintana, F.J.; Sherr, D.H. Aryl hydrocarbon receptor control of adaptive immunity. Pharmacol. Rev. 2013, 65, 1148–1161. [Google Scholar] [CrossRef]
- Veldhoen, M.; Ferreira, C. Influence of nutrient-derived metabolites on lymphocyte immunity. Nat. Med. 2015, 21, 709–718. [Google Scholar] [CrossRef]
- Barouki, R.; Aggerbeck, M.; Aggerbeck, L.; Coumoul, X. The aryl hydrocarbon receptor system. Drug Metabol. Drug Interact. 2012, 27, 3–8. [Google Scholar] [CrossRef]
- Hubbard, T.D.; Murray, I.A.; Bisson, W.H.; Lahoti, T.S.; Gowda, K.; Amin, S.G.; Patterson, A.D.; Perdew, G.H. Adaptation of the human aryl hydrocarbon receptor to sense microbiota-derived indoles. Sci. Rep. 2015, 5, 12689. [Google Scholar] [CrossRef]
- Qiu, J.; Guo, X.; Chen, Z.-M.E.; He, L.; Sonnenberg, G.F.; Artis, D.; Fu, Y.-X.; Zhou, L. Group 3 innate lymphoid cells inhibit T-cell-mediated intestinal inflammation through aryl hydrocarbon receptor signaling and regulation of microflora. Immunity 2013, 39, 386–399. [Google Scholar] [CrossRef]
- Qiu, J.; Zhou, L. Aryl hydrocarbon receptor promotes RORγt+ group 3 ILCs and controls intestinal immunity and inflammation. Semin. Immunopathol. 2013, 35, 657–670. [Google Scholar] [CrossRef]
- Monteleone, I.; Rizzo, A.; Sarra, M.; Sica, G.; Sileri, P.; Biancone, L.; MacDonald, T.T.; Pallone, F.; Monteleone, G. Aryl hydrocarbon receptor-induced signals up-regulate IL-22 production and inhibit inflammation in the gastrointestinal tract. Gastroenterology 2011, 141, 237–248.e1. [Google Scholar] [CrossRef] [PubMed]
- Lamas, B.; Richard, M.L.; Leducq, V.; Pham, H.-P.; Michel, M.-L.; Da Costa, G.; Bridonneau, C.; Jegou, S.; Hoffmann, T.W.; Natividad, J.M.; et al. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat. Med. 2016, 22, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Marinelli, L.; Martin-Gallausiaux, C.; Bourhis, J.-M.; Béguet-Crespel, F.; Blottière, H.M.; Lapaque, N. Identification of the novel role of butyrate as AhR ligand in human intestinal epithelial cells. Sci. Rep. 2019, 9, 643. [Google Scholar] [CrossRef] [PubMed]
- Gitter, A.H.; Wullstein, F.; Fromm, M.; Schulzke, J.R.D. Epithelial Barrier Defects in Ulcerative Colitis: Characterization and Quantification by Electrophysiological Imaging. Gastroenterology 2001, 121, 1320–1328. [Google Scholar] [CrossRef]
- Caviglia, G.P.; Dughera, F.; Ribaldone, D.G.; Rosso, C.; Abate, M.L.; Pellicano, R.; Bresso, F.; Smedile, A.; Saracco, G.M.; Astegiano, M. Serum zonulin in patients with inflammatory bowel disease: A pilot study. Minerva Med. 2019, 110, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Li, Z.-R.; Green, R.S.; Holzman, I.R.; Lin, J. Butyrate Enhances the Intestinal Barrier by Facilitating Tight Junction Assembly via Activation of AMP-Activated Protein Kinase in Caco-2 Cell Monolayers. J. Nutr. 2009, 139, 1619–1625. [Google Scholar] [CrossRef]
- Peng, L.; He, Z.; Chen, W.; Holzman, I.R.; Lin, J. Effects of Butyrate on Intestinal Barrier Function in a Caco-2 Cell Monolayer Model of Intestinal Barrier. Pediatric Res. 2007, 61, 37–41. [Google Scholar] [CrossRef]
- Finnie, I.A.; Dwarakanath, A.D.; Taylor, B.A.; Rhodes, J.M. Colonic mucin synthesis is increased by sodium butyrate. Gut 1995, 36, 93–99. [Google Scholar] [CrossRef]
- Jiminez, J.A.; Uwiera, T.C.; Abbott, D.W.; Uwiera, R.R.E.; Inglis, G.D. Butyrate Supplementation at High Concentrations Alters Enteric Bacterial Communities and Reduces Intestinal Inflammation in Mice Infected with Citrobacter rodentium. mSphere 2017, 2, e00243-17. [Google Scholar] [CrossRef]
- Chen, G.; Ran, X.; Li, B.; Li, Y.; He, D.; Huang, B.; Fu, S.; Liu, J.; Wang, W. Sodium Butyrate Inhibits Inflammation and Maintains Epithelium Barrier Integrity in a TNBS-induced Inflammatory Bowel Disease Mice Model. EBioMedicine 2018, 30, 317–325. [Google Scholar] [CrossRef]
- Takakuwa, A.; Nakamura, K.; Kikuchi, M.; Sugimoto, R.; Ohira, S.; Yokoi, Y.; Ayabe, T. Butyric Acid and Leucine Induce α-Defensin Secretion from Small Intestinal Paneth Cells. Nutrients 2019, 11, 2817. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Guo, B.; Gan, Z.; Song, D.; Lu, Z.; Yi, H.; Wu, Y.; Wang, Y.; Du, H. Butyrate upregulates endogenous host defense peptides to enhance disease resistance in piglets via histone deacetylase inhibition. Sci. Rep. 2016, 6, 27070. [Google Scholar] [CrossRef]
- Wong, S.H.; Zhao, L.; Zhang, X.; Nakatsu, G.; Han, J.; Xu, W.; Xiao, X.; Kwong, T.N.Y.; Tsoi, H.; Wu, W.K.K.; et al. Gavage of Fecal Samples from Patients with Colorectal Cancer Promotes Intestinal Carcinogenesis in Germ-Free and Conventional Mice. Gastroenterology 2017, 153, 1621–1633.e6. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, G.W.; Begley, M.; Cotter, P.D.; Guinane, C.M. Potential Use of Biotherapeutic Bacteria to Target Colorectal Cancer-Associated Taxa. Int. J. Mol. Sci. 2020, 21, 924. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.-C.; Shen, M.-H.; Liu, C.-Y.; Pu, C.-M.; Hu, J.-M.; Huang, C.-J. A gut butyrate-producing bacterium Butyricicoccus pullicaecorum regulates short-chain fatty acid transporter and receptor to reduce the progression of 1,2-dimethylhydrazine-associated colorectal cancer. Oncol. Lett. 2020, 20, 327. [Google Scholar] [CrossRef]
- McNabney, S.; Henagan, T. Short Chain Fatty Acids in the Colon and Peripheral Tissues: A Focus on Butyrate, Colon Cancer, Obesity and Insulin Resistance. Nutrients 2017, 9, 1348. [Google Scholar] [CrossRef] [PubMed]
- Zagato, E.; Pozzi, C.; Bertocchi, A.; Schioppa, T.; Saccheri, F.; Guglietta, S.; Fosso, B.; Melocchi, L.; Nizzoli, G.; Troisi, J.; et al. Endogenous murine microbiota member Faecalibaculum rodentium and its human homologue protect from intestinal tumour growth. Nat. Microbiol. 2020, 5, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Belcheva, A.; Irrazabal, T.; Robertson, S.J.; Streutker, C.; Maughan, H.; Rubino, S.; Moriyama, E.H.; Copeland, J.K.; Surendra, A.; Kumar, S.; et al. Gut microbial metabolism drives transformation of MSH2-deficient colon epithelial cells. Cell 2014, 158, 288–299. [Google Scholar] [CrossRef]
- Bultman, S.J.; Jobin, C. Microbial-derived butyrate: An oncometabolite or tumor-suppressive metabolite? Cell Host Microbe 2014, 16, 143–145. [Google Scholar] [CrossRef]
- Schmitt, M.G.; Soergel, K.H.; Wood, C.M. Absorption of Short Chain Fatty Acids from the Human Jejunum. Gastroenterology 1976, 70, 211–215. [Google Scholar] [CrossRef]
- Lama, A.; Annunziata, C.; Coretti, L.; Pirozzi, C.; Di Guida, F.; Nitrato Izzo, A.; Cristiano, C.; Mollica, M.P.; Chiariotti, L.; Pelagalli, A.; et al. N-(1-carbamoyl-2-phenylethyl) butyramide reduces antibiotic-induced intestinal injury, innate immune activation and modulates microbiota composition. Sci. Rep. 2019, 9, 4832. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Butyrate Pathway | Firmicutes | Bacteroidetes | GM Abundance of the Bacterial Populations Expressing Each Pathway | |
|---|---|---|---|---|
| Lachnospiraceae | Rumicococcaceae | |||
| Acetyl-CoA | Eubacterium Butyrivibrio Clostridium XIVa Ruminococcus Anaerostipes Coprococcus | Subdoligranulum Butyricicoccus Roseburia Pseudoflavonifractor Flavonifractor Oscillibacter Faecalibacterium | Odoribacter Butyricimonas Porphyromonas | 24.2% |
| Glutarate | Clostridium XIVa | Pseudoflavonifractor Oscillibacter | 1.8% | |
| 4-aminobutyrate | Flavonifractor | Odoribacter Butyricimonas | 1.7% | |
| Lysine | Flavonifractor | Odoribacter Alistipes Butyricimonas | 4.4% | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gasaly, N.; Hermoso, M.A.; Gotteland, M. Butyrate and the Fine-Tuning of Colonic Homeostasis: Implication for Inflammatory Bowel Diseases. Int. J. Mol. Sci. 2021, 22, 3061. https://doi.org/10.3390/ijms22063061
Gasaly N, Hermoso MA, Gotteland M. Butyrate and the Fine-Tuning of Colonic Homeostasis: Implication for Inflammatory Bowel Diseases. International Journal of Molecular Sciences. 2021; 22(6):3061. https://doi.org/10.3390/ijms22063061
Chicago/Turabian StyleGasaly, Naschla, Marcela A. Hermoso, and Martín Gotteland. 2021. "Butyrate and the Fine-Tuning of Colonic Homeostasis: Implication for Inflammatory Bowel Diseases" International Journal of Molecular Sciences 22, no. 6: 3061. https://doi.org/10.3390/ijms22063061
APA StyleGasaly, N., Hermoso, M. A., & Gotteland, M. (2021). Butyrate and the Fine-Tuning of Colonic Homeostasis: Implication for Inflammatory Bowel Diseases. International Journal of Molecular Sciences, 22(6), 3061. https://doi.org/10.3390/ijms22063061