
Therapeutic Potential of AAV1-Rheb(S16H) Transduction against Neurodegenerative Diseases



Abstract
1. Introduction
2. Importance of Supporting Neurotrophic Factors as a Therapeutic Strategy for Neurodegenerative Diseases
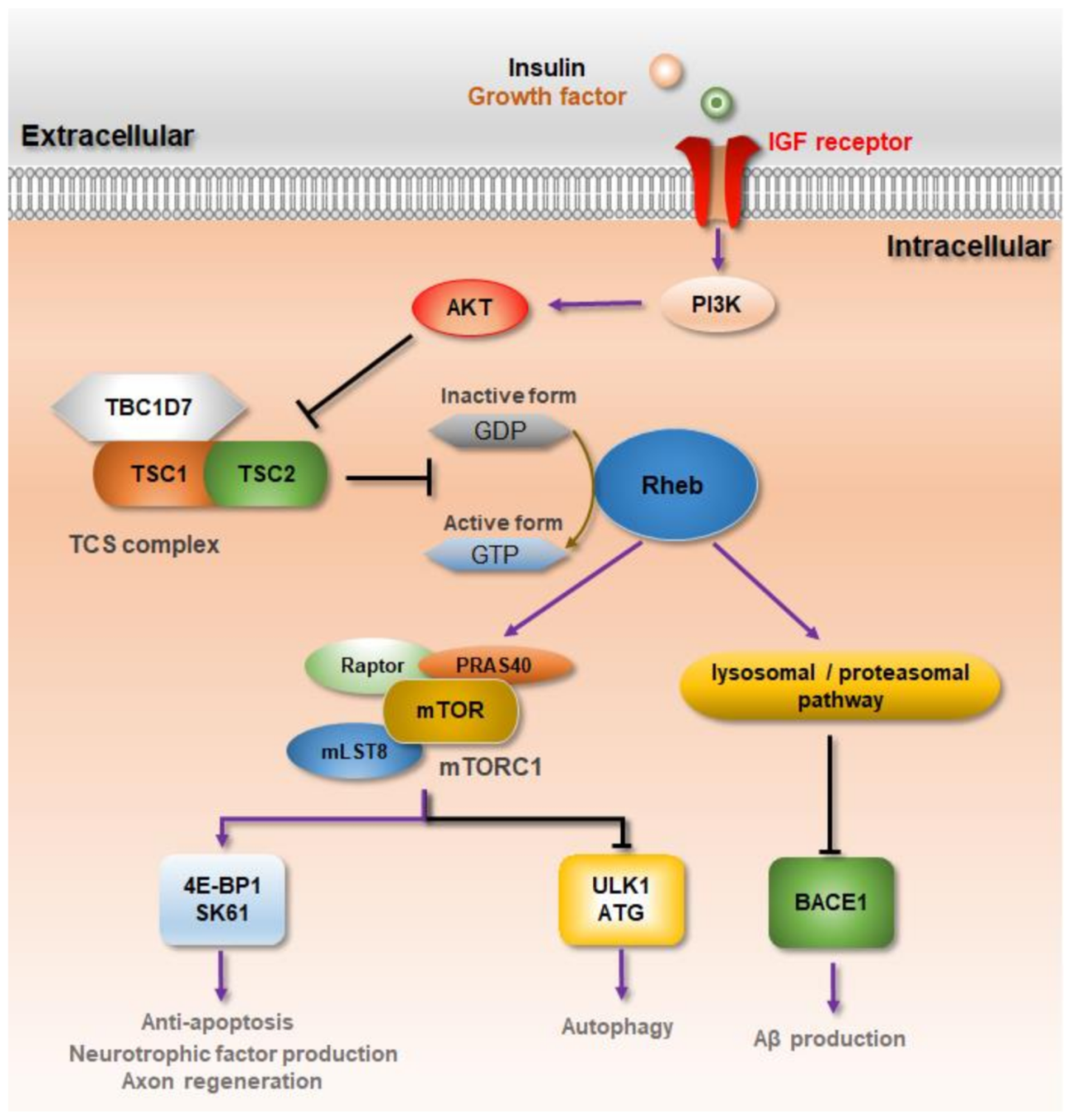
3. Rheb-mTORC1 Signaling against Neurodegenerative Diseases
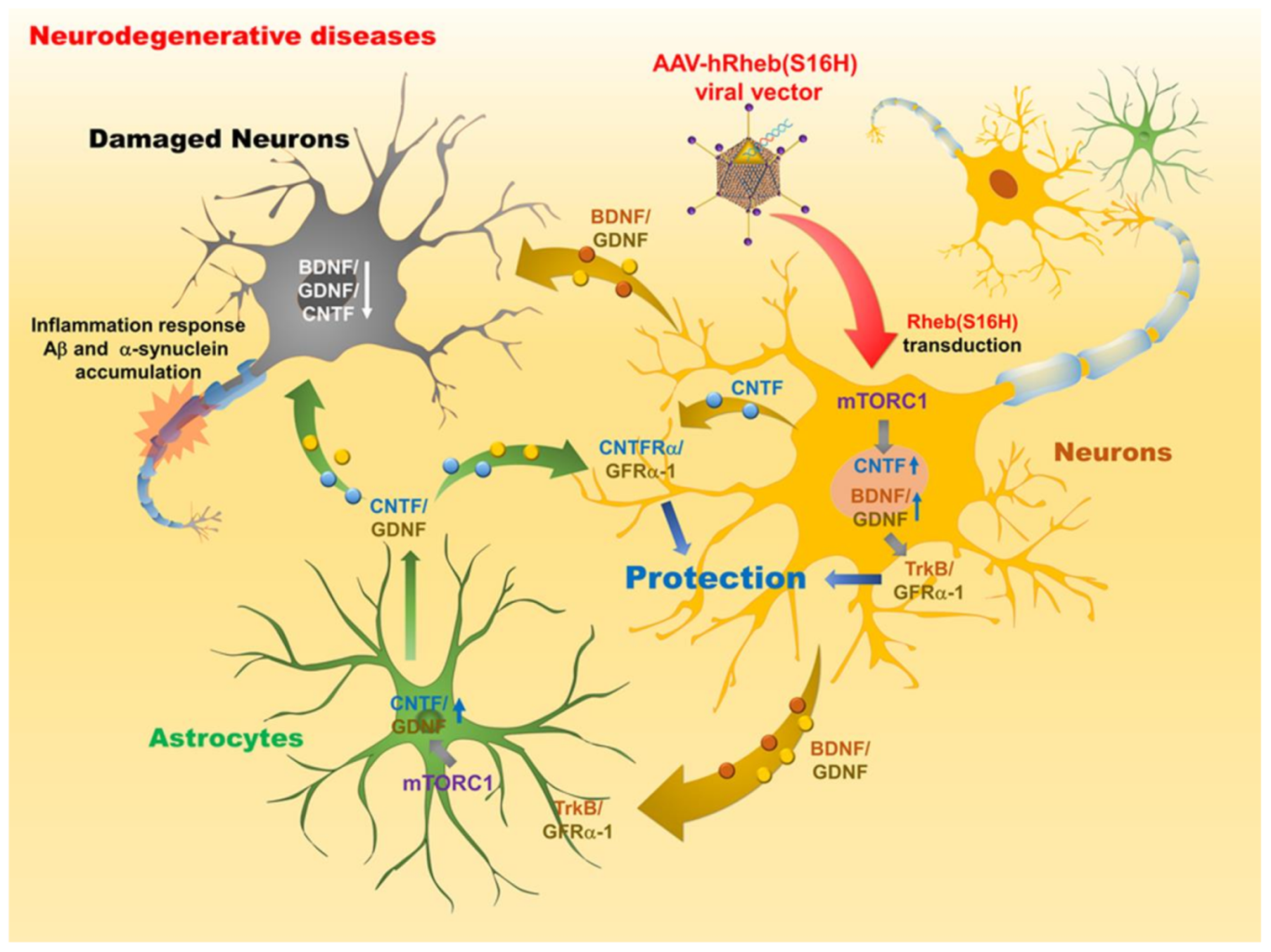
4. Therapeutic Potential of Rheb(S16H) Transduction via AAV1 against Neurodegenerative Diseases
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Dugger, B.N.; Dickson, D.W. Pathology of Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef]
- Erkkinen, M.G.; Kim, M.O.; Geschwind, M.D. Clinical Neurology and Epidemiology of the Major Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2018, 10. [Google Scholar] [CrossRef]
- Fu, H.; Hardy, J.; Duff, K.E. Selective vulnerability in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1350–1358. [Google Scholar] [CrossRef] [PubMed]
- Gitler, A.D.; Dhillon, P.; Shorter, J. Neurodegenerative disease: Models, mechanisms, and a new hope. Dis. Model Mech. 2017, 10, 499–502. [Google Scholar] [CrossRef] [PubMed]
- Wyss-Coray, T. Ageing, neurodegeneration and brain rejuvenation. Nature 2016, 539, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef]
- Canter, R.G.; Penney, J.; Tsai, L.H. The road to restoring neural circuits for the treatment of Alzheimer’s disease. Nature 2016, 539, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Thanan, R.; Oikawa, S.; Hiraku, Y.; Ohnishi, S.; Ma, N.; Pinlaor, S.; Yongvanit, P.; Kawanishi, S.; Murata, M. Oxidative stress and its significant roles in neurodegenerative diseases and cancer. Int. J. Mol. Sci. 2014, 16, 193–217. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.X.; Wang, Y.; Qin, Z.H. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 2009, 30, 379–387. [Google Scholar] [CrossRef]
- Ridler, C. Neurodegenerative disease: Proteome points to synaptic dysfunction in dementia. Nat. Rev. Neurol. 2018, 14, 128. [Google Scholar] [CrossRef]
- Gilhus, N.E.; Deuschl, G. Neuroinflammation—A common thread in neurological disorders. Nat. Rev. Neurol. 2019, 15, 429–430. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A.; Kwon, Y.T. Degradation of misfolded proteins in neurodegenerative diseases: Therapeutic targets and strategies. Exp. Mol. Med. 2015, 47, e147. [Google Scholar] [CrossRef]
- Hetz, C.; Saxena, S. ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol. 2017, 13, 477–491. [Google Scholar] [CrossRef] [PubMed]
- Norat, P.; Soldozy, S.; Sokolowski, J.D.; Gorick, C.M.; Kumar, J.S.; Chae, Y.; Yagmurlu, K.; Prada, F.; Walker, M.; Levitt, M.R.; et al. Mitochondrial dysfunction in neurological disorders: Exploring mitochondrial transplantation. NPJ Regen. Med. 2020, 5, 22. [Google Scholar] [CrossRef]
- Yu, C.; Li, C.H.; Chen, S.; Yoo, H.; Qin, X.; Park, H. Decreased BDNF Release in Cortical Neurons of a Knock-in Mouse Model of Huntington’s Disease. Sci. Rep. 2018, 8, 16976. [Google Scholar] [CrossRef] [PubMed]
- Zuccato, C.; Marullo, M.; Vitali, B.; Tarditi, A.; Mariotti, C.; Valenza, M.; Lahiri, N.; Wild, E.J.; Sassone, J.; Ciammola, A.; et al. Brain-derived neurotrophic factor in patients with Huntington’s disease. PLoS ONE 2011, 6, e22966. [Google Scholar] [CrossRef]
- Hock, C.; Heese, K.; Hulette, C.; Rosenberg, C.; Otten, U. Region-specific neurotrophin imbalances in Alzheimer disease: Decreased levels of brain-derived neurotrophic factor and increased levels of nerve growth factor in hippocampus and cortical areas. Arch. Neurol. 2000, 57, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, I.; Marin, C.; Rey, M.J.; Ribalta, T.; Goutan, E.; Blanco, R.; Tolosa, E.; Marti, E. BDNF and full-length and truncated TrkB expression in Alzheimer disease. Implications in therapeutic strategies. J. Neuropathol. Exp. Neurol. 1999, 58, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Wuu, J.; Mufson, E.J.; Fahnestock, M. Precursor form of brain-derived neurotrophic factor and mature brain-derived neurotrophic factor are decreased in the pre-clinical stages of Alzheimer’s disease. J. Neurochem. 2005, 93, 1412–1421. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, N.B.; Siegel, G.J.; Lee, J.M. Depletion of glial cell line-derived neurotrophic factor in substantia nigra neurons of Parkinson’s disease brain. J. Chem. Neuroanat. 2001, 21, 277–288. [Google Scholar] [CrossRef]
- Nagahara, A.H.; Merrill, D.A.; Coppola, G.; Tsukada, S.; Schroeder, B.E.; Shaked, G.M.; Wang, L.; Blesch, A.; Kim, A.; Conner, J.M.; et al. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat. Med. 2009, 15, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Garcia, P.; Youssef, I.; Utvik, J.K.; Florent-Bechard, S.; Barthelemy, V.; Malaplate-Armand, C.; Kriem, B.; Stenger, C.; Koziel, V.; Olivier, J.L.; et al. Ciliary neurotrophic factor cell-based delivery prevents synaptic impairment and improves memory in mouse models of Alzheimer’s disease. J. Neurosci. 2010, 30, 7516–7527. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Li, S.; Xu, X.; Li, Y.; Guan, K.; Arnold, E.; Ding, J. Structural basis for the unique biological function of small GTPase RHEB. J. Biol. Chem. 2005, 280, 17093–17100. [Google Scholar] [CrossRef]
- Aspuria, P.J.; Tamanoi, F. The Rheb family of GTP-binding proteins. Cell Signal. 2004, 16, 1105–1112. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Araki, Y.; Kontani, K.; Nishina, H.; Katada, T. Novel role of the small GTPase Rheb: Its implication in endocytic pathway independent of the activation of mammalian target of rapamycin. J. Biochem. 2005, 137, 423–430. [Google Scholar] [CrossRef]
- Yamagata, K.; Sanders, L.K.; Kaufmann, W.E.; Yee, W.; Barnes, C.A.; Nathans, D.; Worley, P.F. rheb, a growth factor- and synaptic activity-regulated gene, encodes a novel Ras-related protein. J. Biol. Chem. 1994, 269, 16333–16339. [Google Scholar] [CrossRef]
- Karassek, S.; Berghaus, C.; Schwarten, M.; Goemans, C.G.; Ohse, N.; Kock, G.; Jockers, K.; Neumann, S.; Gottfried, S.; Herrmann, C.; et al. Ras homolog enriched in brain (Rheb) enhances apoptotic signaling. J. Biol. Chem. 2010, 285, 33979–33991. [Google Scholar] [CrossRef]
- Wu, D.; Klaw, M.C.; Connors, T.; Kholodilov, N.; Burke, R.E.; Tom, V.J. Expressing Constitutively Active Rheb in Adult Neurons after a Complete Spinal Cord Injury Enhances Axonal Regeneration beyond a Chondroitinase-Treated Glial Scar. J. Neurosci. 2015, 35, 11068–11080. [Google Scholar] [CrossRef]
- Avruch, J.; Hara, K.; Lin, Y.; Liu, M.; Long, X.; Ortiz-Vega, S.; Yonezawa, K. Insulin and amino-acid regulation of mTOR signaling and kinase activity through the Rheb GTPase. Oncogene 2006, 25, 6361–6372. [Google Scholar] [CrossRef]
- Patel, P.H.; Thapar, N.; Guo, L.; Martinez, M.; Maris, J.; Gau, C.L.; Lengyel, J.A.; Tamanoi, F. Drosophila Rheb GTPase is required for cell cycle progression and cell growth. J. Cell Sci. 2003, 116, 3601–3610. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Rubinsztein, D.C. Small molecule enhancers of autophagy for neurodegenerative diseases. Mol. Biosyst. 2008, 4, 895–901. [Google Scholar] [CrossRef]
- Swiech, L.; Perycz, M.; Malik, A.; Jaworski, J. Role of mTOR in physiology and pathology of the nervous system. Biochim. Biophys. Acta 2008, 1784, 116–132. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 2006, 443, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Trojanowski, J.Q.; Lee, V.M. Protein transmission in neurodegenerative disease. Nat. Rev. Neurol. 2020, 16, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Meade, R.M.; Fairlie, D.P.; Mason, J.M. Alpha-synuclein structure and Parkinson’s disease—Lessons and emerging principles. Mol. Neurodegener. 2019, 14, 29. [Google Scholar] [CrossRef]
- Sowa, A.S.; Martin, E.; Martins, I.M.; Schmidt, J.; Depping, R.; Weber, J.J.; Rother, F.; Hartmann, E.; Bader, M.; Riess, O.; et al. Karyopherin alpha-3 is a key protein in the pathogenesis of spinocerebellar ataxia type 3 controlling the nuclear localization of ataxin-3. Proc. Natl. Acad. Sci. USA 2018, 115, E2624–E2633. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Tecedor, L.; Chen, Y.H.; Monteys, A.M.; Sowada, M.J.; Thompson, L.M.; Davidson, B.L. Reinstating aberrant mTORC1 activity in Huntington’s disease mice improves disease phenotypes. Neuron 2015, 85, 303–315. [Google Scholar] [CrossRef] [PubMed]
- Shahani, N.; Pryor, W.; Swarnkar, S.; Kholodilov, N.; Thinakaran, G.; Burke, R.E.; Subramaniam, S. Rheb GTPase regulates beta-secretase levels and amyloid beta generation. J. Biol. Chem. 2014, 289, 5799–5808. [Google Scholar] [CrossRef] [PubMed]
- Jeon, M.T.; Moon, G.J.; Kim, S.; Choi, M.; Oh, Y.S.; Kim, D.W.; Kim, H.J.; Lee, K.J.; Choe, Y.; Ha, C.M.; et al. Neurotrophic interactions between neurons and astrocytes following AAV1-Rheb(S16H) transduction in the hippocampus in vivo. Br. J. Pharmacol. 2020, 177, 668–686. [Google Scholar] [CrossRef]
- Jeon, M.T.; Nam, J.H.; Shin, W.H.; Leem, E.; Jeong, K.H.; Jung, U.J.; Bae, Y.S.; Jin, Y.H.; Kholodilov, N.; Burke, R.E.; et al. In vivo AAV1 transduction with hRheb(S16H) protects hippocampal neurons by BDNF production. Mol Ther. 2015, 23, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Moon, G.J.; Oh, Y.S.; Park, J.; Shin, W.H.; Jeong, J.Y.; Choi, K.S.; Jin, B.K.; Kholodilov, N.; Burke, R.E.; et al. Protection of nigral dopaminergic neurons by AAV1 transduction with Rheb(S16H) against neurotoxic inflammation in vivo. Exp. Mol. Med. 2018, 50, e440. [Google Scholar] [CrossRef] [PubMed]
- Moon, G.J.; Kim, S.; Jeon, M.T.; Lee, K.J.; Jang, I.S.; Nakamura, M.; Kim, S.R. Therapeutic Potential of AAV1-Rheb(S16H) Transduction Against Alzheimer’s Disease. J. Clin. Med. 2019, 8, 2053. [Google Scholar] [CrossRef]
- Chan-Palay, V. Alterations in the locus coeruleus in dementias of Alzheimer’s and Parkinson’s disease. Prog. Brain Res. 1991, 88, 625–630. [Google Scholar] [CrossRef]
- Burke, W.J.; Park, D.H.; Chung, H.D.; Marshall, G.L.; Haring, J.H.; Joh, T.H. Evidence for decreased transport of tryptophan hydroxylase in Alzheimer’s disease. Brain Res. 1990, 537, 83–87. [Google Scholar] [CrossRef]
- Burke, W.J.; Chung, H.D.; Marshall, G.L.; Gillespie, K.N.; Joh, T.H. Evidence for decreased transport of PNMT protein in advanced Alzheimer’s disease. J. Am. Geriatr. Soc. 1990, 38, 1275–1282. [Google Scholar] [CrossRef] [PubMed]
- Tabaton, M.; Schenone, A.; Romagnoli, P.; Mancardi, G.L. A quantitative and ultrastructural study of substantia nigra and nucleus centralis superior in Alzheimer’s disease. Acta Neuropathol. 1985, 68, 218–223. [Google Scholar] [CrossRef]
- Whitehouse, P.J.; Price, D.L.; Struble, R.G.; Clark, A.W.; Coyle, J.T.; Delon, M.R. Alzheimer’s disease and senile dementia: Loss of neurons in the basal forebrain. Science 1982, 215, 1237–1239. [Google Scholar] [CrossRef] [PubMed]
- Cheshire, P.; Ayton, S.; Bertram, K.L.; Ling, H.; Li, A.; McLean, C.; Halliday, G.M.; O’Sullivan, S.S.; Revesz, T.; Finkelstein, D.I.; et al. Serotonergic markers in Parkinson’s disease and levodopa-induced dyskinesias. Mov. Disord. 2015, 30, 796–804. [Google Scholar] [CrossRef]
- Hall, H.; Reyes, S.; Landeck, N.; Bye, C.; Leanza, G.; Double, K.; Thompson, L.; Halliday, G.; Kirik, D. Hippocampal Lewy pathology and cholinergic dysfunction are associated with dementia in Parkinson’s disease. Brain 2014, 137, 2493–2508. [Google Scholar] [CrossRef] [PubMed]
- Damier, P.; Hirsch, E.C.; Agid, Y.; Graybiel, A.M. The substantia nigra of the human brain. II. Patterns of loss of dopamine-containing neurons in Parkinson’s disease. Brain 1999, 122, 1437–1448. [Google Scholar] [CrossRef]
- Halliday, G.M.; Li, Y.W.; Blumbergs, P.C.; Joh, T.H.; Cotton, R.G.; Howe, P.R.; Blessing, W.W.; Geffen, L.B. Neuropathology of immunohistochemically identified brainstem neurons in Parkinson’s disease. Ann. Neurol. 1990, 27, 373–385. [Google Scholar] [CrossRef]
- Mann, D.M.; Yates, P.O. Pathological basis for neurotransmitter changes in Parkinson’s disease. Neuropathol. Appl. Neurobiol. 1983, 9, 3–19. [Google Scholar] [CrossRef]
- Deng, Y.P.; Reiner, A. Cholinergic interneurons in the Q140 knockin mouse model of Huntington’s disease: Reductions in dendritic branching and thalamostriatal input. J. Comp. Neurol. 2016, 524, 3518–3529. [Google Scholar] [CrossRef]
- Reinius, B.; Blunder, M.; Brett, F.M.; Eriksson, A.; Patra, K.; Jonsson, J.; Jazin, E.; Kullander, K. Conditional targeting of medium spiny neurons in the striatal matrix. Front. Behav. Neurosci. 2015, 9, 71. [Google Scholar] [CrossRef]
- Rikani, A.A.; Choudhry, Z.; Choudhry, A.M.; Rizvi, N.; Ikram, H.; Mobassarah, N.J.; Tulli, S. The mechanism of degeneration of striatal neuronal subtypes in Huntington disease. Ann. Neurosci. 2014, 21, 112–114. [Google Scholar] [CrossRef]
- Huot, P.; Levesque, M.; Parent, A. The fate of striatal dopaminergic neurons in Parkinson’s disease and Huntington’s chorea. Brain 2007, 130, 222–232. [Google Scholar] [CrossRef]
- Behrens, P.F.; Franz, P.; Woodman, B.; Lindenberg, K.S.; Landwehrmeyer, G.B. Impaired glutamate transport and glutamate-glutamine cycling: Downstream effects of the Huntington mutation. Brain 2002, 125, 1908–1922. [Google Scholar] [CrossRef]
- Jimcy Platholi, F.S.L. Neurotrophic Factors. In Handbook of Developmental Neurotoxicology; Academic Press: Cambridge, MA, USA, 2018; pp. 55–64. [Google Scholar]
- Huang, E.J.; Reichardt, L.F. Neurotrophins: Roles in neuronal development and function. Annu. Rev. Neurosci. 2001, 24, 677–736. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.S.; Shen, L.L.; Zhu, C.; Bu, X.L.; Liu, Y.H.; Liu, C.H.; Yao, X.Q.; Zhang, L.L.; Zhou, H.D.; Walker, D.G.; et al. Brain-derived neurotrophic factor protects against tau-related neurodegeneration of Alzheimer’s disease. Transl. Psychiatry 2016, 6, e907. [Google Scholar] [CrossRef] [PubMed]
- Konishi, Y.; Yang, L.B.; He, P.; Lindholm, K.; Lu, B.; Li, R.; Shen, Y. Deficiency of GDNF Receptor GFRalpha1 in Alzheimer’s Neurons Results in Neuronal Death. J. Neurosci. 2014, 34, 13127–13138. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.S.; Zhang, Z.; Liu, X.; Manfredsson, F.P.; Benskey, M.J.; Cao, X.; Xu, J.; Sun, Y.E.; Ye, K. TrkB neurotrophic activities are blocked by alpha-synuclein, triggering dopaminergic cell death in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, 10773–10778. [Google Scholar] [CrossRef] [PubMed]
- Littrell, O.M.; Granholm, A.C.; Gerhardt, G.A.; Boger, H.A. Glial cell-line derived neurotrophic factor (GDNF) replacement attenuates motor impairments and nigrostriatal dopamine deficits in 12-month-old mice with a partial deletion of GDNF. Pharmacol. Biochem. Behav. 2013, 104, 10–19. [Google Scholar] [CrossRef]
- Howells, D.W.; Porritt, M.J.; Wong, J.Y.; Batchelor, P.E.; Kalnins, R.; Hughes, A.J.; Donnan, G.A. Reduced BDNF mRNA expression in the Parkinson’s disease substantia nigra. Exp. Neurol. 2000, 166, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Canals, J.M.; Pineda, J.R.; Torres-Peraza, J.F.; Bosch, M.; Martin-Ibanez, R.; Munoz, M.T.; Mengod, G.; Ernfors, P.; Alberch, J. Brain-derived neurotrophic factor regulates the onset and severity of motor dysfunction associated with enkephalinergic neuronal degeneration in Huntington’s disease. J. Neurosci. 2004, 24, 7727–7739. [Google Scholar] [CrossRef] [PubMed]
- Zuccato, C.; Ciammola, A.; Rigamonti, D.; Leavitt, B.R.; Goffredo, D.; Conti, L.; MacDonald, M.E.; Friedlander, R.M.; Silani, V.; Hayden, M.R.; et al. Loss of huntingtin-mediated BDNF gene transcription in Huntington’s disease. Science 2001, 293, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Schiaffino, L.; Bonafede, R.; Scambi, I.; Parrella, E.; Pizzi, M.; Mariotti, R. Acetylation state of RelA modulated by epigenetic drugs prolongs survival and induces a neuroprotective effect on ALS murine model. Sci. Rep. 2018, 8, 12875. [Google Scholar] [CrossRef] [PubMed]
- Shruthi, S.; Sumitha, R.; Varghese, A.M.; Ashok, S.; Chandrasekhar Sagar, B.K.; Sathyaprabha, T.N.; Nalini, A.; Kramer, B.W.; Raju, T.R.; Vijayalakshmi, K.; et al. Brain-Derived Neurotrophic Factor Facilitates Functional Recovery from ALS-Cerebral Spinal Fluid-Induced Neurodegenerative Changes in the NSC-34 Motor Neuron Cell Line. Neurodegener. Dis. 2017, 17, 44–58. [Google Scholar] [CrossRef]
- Yun, D.; Jeon, M.T.; Kim, H.J.; Moon, G.J.; Lee, S.; Ha, C.M.; Shin, M.; Kim, S.R. Induction of GDNF and GFRalpha-1 Following AAV1-Rheb(S16H) Administration in the Hippocampus in vivo. Exp. Neurobiol. 2020, 29, 164–175. [Google Scholar] [CrossRef]
- Fan, C.H.; Ting, C.Y.; Lin, C.Y.; Chan, H.L.; Chang, Y.C.; Chen, Y.Y.; Liu, H.L.; Yeh, C.K. Noninvasive, Targeted, and Non-Viral Ultrasound-Mediated GDNF-Plasmid Delivery for Treatment of Parkinson’s Disease. Sci. Rep. 2016, 6, 19579. [Google Scholar] [CrossRef]
- Decressac, M.; Ulusoy, A.; Mattsson, B.; Georgievska, B.; Romero-Ramos, M.; Kirik, D.; Bjorklund, A. GDNF fails to exert neuroprotection in a rat alpha-synuclein model of Parkinson’s disease. Brain 2011, 134, 2302–2311. [Google Scholar] [CrossRef] [PubMed]
- Lo Bianco, C.; Deglon, N.; Pralong, W.; Aebischer, P. Lentiviral nigral delivery of GDNF does not prevent neurodegeneration in a genetic rat model of Parkinson’s disease. Neurobiol. Dis. 2004, 17, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Jeong, K.H.; Nam, J.H.; Jin, B.K.; Kim, S.R. Activation of CNTF/CNTFRalpha signaling pathway by hRheb(S16H) transduction of dopaminergic neurons in vivo. PLoS ONE 2015, 10, e0121803. [Google Scholar] [CrossRef]
- Nam, J.H.; Leem, E.; Jeon, M.T.; Jeong, K.H.; Park, J.W.; Jung, U.J.; Kholodilov, N.; Burke, R.E.; Jin, B.K.; Kim, S.R. Induction of GDNF and BDNF by hRheb(S16H) transduction of SNpc neurons: Neuroprotective mechanisms of hRheb(S16H) in a model of Parkinson’s disease. Mol. Neurobiol. 2015, 51, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, L.; Guidotti, G.; Martorana, F.; Iyer, A.M.; Aronica, E.; Valori, C.F.; Rossi, D. Disruption of the astrocytic TNFR1-GDNF axis accelerates motor neuron degeneration and disease progression in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2016, 25, 3080–3095. [Google Scholar] [CrossRef][Green Version]
- Wang, L.J.; Lu, Y.Y.; Muramatsu, S.; Ikeguchi, K.; Fujimoto, K.; Okada, T.; Mizukami, H.; Matsushita, T.; Hanazono, Y.; Kume, A.; et al. Neuroprotective effects of glial cell line-derived neurotrophic factor mediated by an adeno-associated virus vector in a transgenic animal model of amyotrophic lateral sclerosis. J. Neurosci. 2002, 22, 6920–6928. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, G.M.; Alkaslasi, M.; Vit, J.P.; Lawless, G.; Godoy, M.; Gowing, G.; Shelest, O.; Svendsen, C.N. Systemic injection of AAV9-GDNF provides modest functional improvements in the SOD1(G93A) ALS rat but has adverse side effects. Gene Ther. 2017, 24, 245–252. [Google Scholar] [CrossRef]
- Korkmaz, O.T.; Aytan, N.; Carreras, I.; Choi, J.K.; Kowall, N.W.; Jenkins, B.G.; Dedeoglu, A. 7,8-Dihydroxyflavone improves motor performance and enhances lower motor neuronal survival in a mouse model of amyotrophic lateral sclerosis. Neurosci. Lett. 2014, 566, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef]
- Cummings, J.L. Alzheimer’s disease. N. Engl. J. Med. 2004, 351, 56–67. [Google Scholar] [CrossRef]
- Kawas, C.H. Clinical practice. Early Alzheimer’s disease. N. Engl. J. Med. 2003, 349, 1056–1063. [Google Scholar] [CrossRef]
- Congdon, E.E.; Sigurdsson, E.M. Tau-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 399–415. [Google Scholar] [CrossRef]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Mesulam, M.M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 2018, 141, 1917–1933. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, T.W.; Nathan Spreng, R.; The Alzheimer’s Disease Neuroimaging Initiative. Basal forebrain degeneration precedes and predicts the cortical spread of Alzheimer’s pathology. Nat. Commun. 2016, 7, 13249. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Fukumoto, H.; Orne, J.; Klucken, J.; Raju, S.; Vanderburg, C.R.; Irizarry, M.C.; Hyman, B.T.; Ingelsson, M. Decreased levels of BDNF protein in Alzheimer temporal cortex are independent of BDNF polymorphisms. Exp. Neurol. 2005, 194, 91–96. [Google Scholar] [CrossRef]
- Wang, Z.H.; Xiang, J.; Liu, X.; Yu, S.P.; Manfredsson, F.P.; Sandoval, I.M.; Wu, S.; Wang, J.Z.; Ye, K. Deficiency in BDNF/TrkB Neurotrophic Activity Stimulates delta-Secretase by Upregulating C/EBPbeta in Alzheimer’s Disease. Cell Rep. 2019, 28, 655–669.e5. [Google Scholar] [CrossRef] [PubMed]
- Revilla, S.; Sunol, C.; Garcia-Mesa, Y.; Gimenez-Llort, L.; Sanfeliu, C.; Cristofol, R. Physical exercise improves synaptic dysfunction and recovers the loss of survival factors in 3xTg-AD mouse brain. Neuropharmacology 2014, 81, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Straten, G.; Eschweiler, G.W.; Maetzler, W.; Laske, C.; Leyhe, T. Glial cell-line derived neurotrophic factor (GDNF) concentrations in cerebrospinal fluid and serum of patients with early Alzheimer’s disease and normal controls. J. Alzheimers Dis. 2009, 18, 331–337. [Google Scholar] [CrossRef]
- Li, W.; Wei, D.; Lin, J.; Liang, J.; Xie, X.; Song, K.; Huang, L. Dl-3-n-Butylphthalide Reduces Cognitive Impairment Induced by Chronic Cerebral Hypoperfusion Through GDNF/GFRalpha1/Ret Signaling Preventing Hippocampal Neuron Apoptosis. Front. Cell Neurosci. 2019, 13, 351. [Google Scholar] [CrossRef]
- Scholz, D.; Chernyshova, Y.; Leist, M. Control of Abeta release from human neurons by differentiation status and RET signaling. Neurobiol. Aging 2013, 34, 184–199. [Google Scholar] [CrossRef] [PubMed]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef]
- Tysnes, O.B.; Storstein, A. Epidemiology of Parkinson’s disease. J. Neural Transm. 2017, 124, 901–905. [Google Scholar] [CrossRef] [PubMed]
- Buddhala, C.; Loftin, S.K.; Kuley, B.M.; Cairns, N.J.; Campbell, M.C.; Perlmutter, J.S.; Kotzbauer, P.T. Dopaminergic, serotonergic, and noradrenergic deficits in Parkinson disease. Ann. Clin. Transl. Neurol. 2015, 2, 949–959. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.L.; Bohnen, N.I. Cholinergic dysfunction in Parkinson’s disease. Curr. Neurol. Neurosci. Rep. 2013, 13, 377. [Google Scholar] [CrossRef] [PubMed]
- Tan, E.K.; Chao, Y.X.; West, A.; Chan, L.L.; Poewe, W.; Jankovic, J. Parkinson disease and the immune system—Associations, mechanisms and therapeutics. Nat. Rev. Neurol. 2020, 16, 303–318. [Google Scholar] [CrossRef] [PubMed]
- Sampaio, T.B.; Savall, A.S.; Gutierrez, M.E.Z.; Pinton, S. Neurotrophic factors in Alzheimer’s and Parkinson’s diseases: Implications for pathogenesis and therapy. Neural Regen. Res. 2017, 12, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Tome, D.; Fonseca, C.P.; Campos, F.L.; Baltazar, G. Role of Neurotrophic Factors in Parkinson’s Disease. Curr. Pharm. Des. 2017, 23, 809–838. [Google Scholar] [CrossRef]
- Jiang, L.; Zhang, H.; Wang, C.; Ming, F.; Shi, X.; Yang, M. Serum level of brain-derived neurotrophic factor in Parkinson’s disease: A meta-analysis. Prog. Neuropsychopharmacol. Biol. Psychiatry 2019, 88, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Scalzo, P.; Kummer, A.; Bretas, T.L.; Cardoso, F.; Teixeira, A.L. Serum levels of brain-derived neurotrophic factor correlate with motor impairment in Parkinson’s disease. J. Neurol. 2010, 257, 540–545. [Google Scholar] [CrossRef]
- Rahmani, F.; Saghazadeh, A.; Rahmani, M.; Teixeira, A.L.; Rezaei, N.; Aghamollaii, V.; Ardebili, H.E. Plasma levels of brain-derived neurotrophic factor in patients with Parkinson disease: A systematic review and meta-analysis. Brain Res. 2019, 1704, 127–136. [Google Scholar] [CrossRef]
- Enterria-Morales, D.; Lopez-Lopez, I.; Lopez-Barneo, J.; d’Anglemont de Tassigny, X. Role of Glial Cell Line-Derived Neurotrophic Factor in the Maintenance of Adult Mesencephalic Catecholaminergic Neurons. Mov. Disord. 2020, 35, 565–576. [Google Scholar] [CrossRef]
- Pascual, A.; Hidalgo-Figueroa, M.; Piruat, J.I.; Pintado, C.O.; Gomez-Diaz, R.; Lopez-Barneo, J. Absolute requirement of GDNF for adult catecholaminergic neuron survival. Nat. Neurosci. 2008, 11, 755–761. [Google Scholar] [CrossRef]
- Grandoso, L.; Ponce, S.; Manuel, I.; Arrue, A.; Ruiz-Ortega, J.A.; Ulibarri, I.; Orive, G.; Hernandez, R.M.; Rodriguez, A.; Rodriguez-Puertas, R.; et al. Long-term survival of encapsulated GDNF secreting cells implanted within the striatum of parkinsonized rats. Int. J. Pharm. 2007, 343, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Kopra, J.; Vilenius, C.; Grealish, S.; Harma, M.A.; Varendi, K.; Lindholm, J.; Castren, E.; Voikar, V.; Bjorklund, A.; Piepponen, T.P.; et al. GDNF is not required for catecholaminergic neuron survival in vivo. Nat. Neurosci. 2015, 18, 319–322. [Google Scholar] [CrossRef] [PubMed]
- Decressac, M.; Kadkhodaei, B.; Mattsson, B.; Laguna, A.; Perlmann, T.; Bjorklund, A. α-Synuclein-induced down-regulation of Nurr1 disrupts GDNF signaling in nigral dopamine neurons. Sci. Transl. Med. 2012, 4, 163ra156. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Fischer, D.L.; Li, X.; Bankiewicz, K.; Sortwell, C.E.; Federoff, H.J. Alpha-Synuclein mRNA Is Not Increased in Sporadic PD and Alpha-Synuclein Accumulation Does Not Block GDNF Signaling in Parkinson’s Disease and Disease Models. Mol. Ther. 2017, 25, 2231–2235. [Google Scholar] [CrossRef]
- Chmielarz, P.; Er, S.; Konovalova, J.; Bandres, L.; Hlushchuk, I.; Albert, K.; Panhelainen, A.; Luk, K.; Airavaara, M.; Domanskyi, A. GDNF/RET Signaling Pathway Activation Eliminates Lewy Body Pathology in Midbrain Dopamine Neurons. Mov. Disord. 2020, 35, 2279–2289. [Google Scholar] [CrossRef]
- Nutt, J.G.; Burchiel, K.J.; Comella, C.L.; Jankovic, J.; Lang, A.E.; Laws, E.R., Jr.; Lozano, A.M.; Penn, R.D.; Simpson, R.K., Jr.; Stacy, M.; et al. Randomized, double-blind trial of glial cell line-derived neurotrophic factor (GDNF) in PD. Neurology 2003, 60, 69–73. [Google Scholar] [CrossRef]
- Gill, S.S.; Patel, N.K.; Hotton, G.R.; O’Sullivan, K.; McCarter, R.; Bunnage, M.; Brooks, D.J.; Svendsen, C.N.; Heywood, P. Direct brain infusion of glial cell line-derived neurotrophic factor in Parkinson disease. Nat. Med. 2003, 9, 589–595. [Google Scholar] [CrossRef]
- Love, S.; Plaha, P.; Patel, N.K.; Hotton, G.R.; Brooks, D.J.; Gill, S.S. Glial cell line-derived neurotrophic factor induces neuronal sprouting in human brain. Nat. Med. 2005, 11, 703–704. [Google Scholar] [CrossRef]
- Slevin, J.T.; Gash, D.M.; Smith, C.D.; Gerhardt, G.A.; Kryscio, R.; Chebrolu, H.; Walton, A.; Wagner, R.; Young, A.B. Unilateral intraputamenal glial cell line-derived neurotrophic factor in patients with Parkinson disease: Response to 1 year of treatment and 1 year of withdrawal. J. Neurosurg. 2007, 106, 614–620. [Google Scholar] [CrossRef]
- Slevin, J.T.; Gerhardt, G.A.; Smith, C.D.; Gash, D.M.; Kryscio, R.; Young, B. Improvement of bilateral motor functions in patients with Parkinson disease through the unilateral intraputaminal infusion of glial cell line-derived neurotrophic factor. J. Neurosurg. 2005, 102, 216–222. [Google Scholar] [CrossRef]
- Lang, A.E.; Gill, S.; Patel, N.K.; Lozano, A.; Nutt, J.G.; Penn, R.; Brooks, D.J.; Hotton, G.; Moro, E.; Heywood, P.; et al. Randomized controlled trial of intraputamenal glial cell line-derived neurotrophic factor infusion in Parkinson disease. Ann. Neurol. 2006, 59, 459–466. [Google Scholar] [CrossRef]
- Patel, N.K.; Pavese, N.; Javed, S.; Hotton, G.R.; Brooks, D.J.; Gill, S.S. Benefits of putaminal GDNF infusion in Parkinson disease are maintained after GDNF cessation. Neurology 2013, 81, 1176–1178. [Google Scholar] [CrossRef] [PubMed]
- Whone, A.L.; Boca, M.; Luz, M.; Woolley, M.; Mooney, L.; Dharia, S.; Broadfoot, J.; Cronin, D.; Schroers, C.; Barua, N.U.; et al. Extended Treatment with Glial Cell Line-Derived Neurotrophic Factor in Parkinson’s Disease. J. Parkinsons Dis. 2019, 9, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Whone, A.; Luz, M.; Boca, M.; Woolley, M.; Mooney, L.; Dharia, S.; Broadfoot, J.; Cronin, D.; Schroers, C.; Barua, N.U.; et al. Randomized trial of intermittent intraputamenal glial cell line-derived neurotrophic factor in Parkinson’s disease. Brain 2019, 142, 512–525. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.R.; Hardiman, O.; Benatar, M.; Brooks, B.R.; Chio, A.; de Carvalho, M.; Ince, P.G.; Lin, C.; Miller, R.G.; Mitsumoto, H.; et al. Controversies and priorities in amyotrophic lateral sclerosis. Lancet Neurol. 2013, 12, 310–322. [Google Scholar] [CrossRef]
- Cleveland, D.W.; Rothstein, J.D. From Charcot to Lou Gehrig: Deciphering selective motor neuron death in ALS. Nat. Rev. Neurosci. 2001, 2, 806–819. [Google Scholar] [CrossRef]
- Koliatsos, V.E.; Clatterbuck, R.E.; Winslow, J.W.; Cayouette, M.H.; Price, D.L. Evidence that brain-derived neurotrophic factor is a trophic factor for motor neurons in vivo. Neuron 1993, 10, 359–367. [Google Scholar] [CrossRef]
- Oppenheim, R.W.; Yin, Q.W.; Prevette, D.; Yan, Q. Brain-derived neurotrophic factor rescues developing avian motoneurons from cell death. Nature 1992, 360, 755–757. [Google Scholar] [CrossRef] [PubMed]
- Almeida, R.D.; Manadas, B.J.; Melo, C.V.; Gomes, J.R.; Mendes, C.S.; Graos, M.M.; Carvalho, R.F.; Carvalho, A.P.; Duarte, C.B. Neuroprotection by BDNF against glutamate-induced apoptotic cell death is mediated by ERK and PI3-kinase pathways. Cell Death Differ. 2005, 12, 1329–1343. [Google Scholar] [CrossRef]
- Guerzoni, L.P.; Nicolas, V.; Angelova, A. In Vitro Modulation of TrkB Receptor Signaling upon Sequential Delivery of Curcumin-DHA Loaded Carriers Towards Promoting Neuronal Survival. Pharm. Res. 2017, 34, 492–505. [Google Scholar] [CrossRef]
- Mojsilovic-Petrovic, J.; Jeong, G.B.; Crocker, A.; Arneja, A.; David, S.; Russell, D.S.; Kalb, R.G. Protecting motor neurons from toxic insult by antagonism of adenosine A2a and Trk receptors. J. Neurosci. 2006, 26, 9250–9263. [Google Scholar] [CrossRef]
- Hu, P.; Kalb, R.G. BDNF heightens the sensitivity of motor neurons to excitotoxic insults through activation of TrkB. J. Neurochem. 2003, 84, 1421–1430. [Google Scholar] [CrossRef]
- Kafitz, K.W.; Rose, C.R.; Thoenen, H.; Konnerth, A. Neurotrophin-evoked rapid excitation through TrkB receptors. Nature 1999, 401, 918–921. [Google Scholar] [CrossRef] [PubMed]
- Mojsilovic-Petrovic, J.; Arneja, A.; Kalb, R.G. Enprofylline protects motor neurons from in vitro excitotoxic challenge. Neurodegener. Dis. 2005, 2, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Fryer, H.J.; Wolf, D.H.; Knox, R.J.; Strittmatter, S.M.; Pennica, D.; O’Leary, R.M.; Russell, D.S.; Kalb, R.G. Brain-derived neurotrophic factor induces excitotoxic sensitivity in cultured embryonic rat spinal motor neurons through activation of the phosphatidylinositol 3-kinase pathway. J. Neurochem. 2000, 74, 582–595. [Google Scholar] [CrossRef]
- Kust, B.M.; Copray, J.C.; Brouwer, N.; Troost, D.; Boddeke, H.W. Elevated levels of neurotrophins in human biceps brachii tissue of amyotrophic lateral sclerosis. Exp. Neurol. 2002, 177, 419–427. [Google Scholar] [CrossRef]
- Grundstrom, E.; Lindholm, D.; Johansson, A.; Blennow, K.; Askmark, H. GDNF but not BDNF is increased in cerebrospinal fluid in amyotrophic lateral sclerosis. Neuroreport 2000, 11, 1781–1783. [Google Scholar] [CrossRef] [PubMed]
- Saba, J.; Lopez Couselo, F.; Turati, J.; Carniglia, L.; Durand, D.; de Laurentiis, A.; Lasaga, M.; Caruso, C. Astrocytes from cortex and striatum show differential responses to mitochondrial toxin and BDNF: Implications for protection of striatal neurons expressing mutant huntingtin. J. Neuroinflamm. 2020, 17, 290. [Google Scholar] [CrossRef]
- Kowianski, P.; Lietzau, G.; Czuba, E.; Waskow, M.; Steliga, A.; Morys, J. BDNF: A Key Factor with Multipotent Impact on Brain Signaling and Synaptic Plasticity. Cell Mol. Neurobiol. 2018, 38, 579–593. [Google Scholar] [CrossRef]
- Causing, C.G.; Gloster, A.; Aloyz, R.; Bamji, S.X.; Chang, E.; Fawcett, J.; Kuchel, G.; Miller, F.D. Synaptic innervation density is regulated by neuron-derived BDNF. Neuron 1997, 18, 257–267. [Google Scholar] [CrossRef]
- Talbott, J.F.; Cao, Q.; Bertram, J.; Nkansah, M.; Benton, R.L.; Lavik, E.; Whittemore, S.R. CNTF promotes the survival and differentiation of adult spinal cord-derived oligodendrocyte precursor cells in vitro but fails to promote remyelination in vivo. Exp. Neurol. 2007, 204, 485–489. [Google Scholar] [CrossRef] [PubMed]
- Duarte Azevedo, M.; Sander, S.; Tenenbaum, L. GDNF, A Neuron-Derived Factor Upregulated in Glial Cells during Disease. J. Clin. Med. 2020, 9, 456. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Schwartz, J.P. Gene expression profiles of reactive astrocytes in dopamine-depleted striatum. Brain Pathol. 2004, 14, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.P.; Fang, K.M.; Lee, T.I.; Tzeng, S.F. Regulation of microglial activities by glial cell line derived neurotrophic factor. J. Cell Biochem. 2006, 97, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Longo, F.M.; Yang, T.; Knowles, J.K.; Xie, Y.; Moore, L.A.; Massa, S.M. Small molecule neurotrophin receptor ligands: Novel strategies for targeting Alzheimer’s disease mechanisms. Curr. Alzheimer Res. 2007, 4, 503–506. [Google Scholar] [CrossRef] [PubMed]
- Poduslo, J.F.; Curran, G.L. Permeability at the blood-brain and blood-nerve barriers of the neurotrophic factors: NGF, CNTF, NT-3, BDNF. Brain Res. Mol. Brain Res. 1996, 36, 280–286. [Google Scholar] [CrossRef]
- Abramson, J.H. The four basic types of evaluation: Clinical reviews, clinical trials, program reviews, and program trials. Public Health Rep. 1979, 94, 210–215. [Google Scholar] [PubMed]
- Thoenen, H.; Sendtner, M. Neurotrophins: From enthusiastic expectations through sobering experiences to rational therapeutic approaches. Nat. Neurosci. 2002, 5, 1046–1050. [Google Scholar] [CrossRef] [PubMed]
- Henriques, A.; Pitzer, C.; Schneider, A. Neurotrophic growth factors for the treatment of amyotrophic lateral sclerosis: Where do we stand? Front. Neurosci. 2010, 4, 32. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.; Wang, W.; Cao, Q.; Gu, J.; Mi, X.; Wang, K.; Chen, G.; Wang, X. Insulin growth factor-1 (IGF-1) enhances hippocampal excitatory and seizure activity through IGF-1 receptor-mediated mechanisms in the epileptic brain. Clin. Sci. 2015, 129, 1047–1060. [Google Scholar] [CrossRef]
- Binder, D.K.; Croll, S.D.; Gall, C.M.; Scharfman, H.E. BDNF and epilepsy: Too much of a good thing? Trends Neurosci. 2001, 24, 47–53. [Google Scholar] [CrossRef]
- Dibble, C.C.; Elis, W.; Menon, S.; Qin, W.; Klekota, J.; Asara, J.M.; Finan, P.M.; Kwiatkowski, D.J.; Murphy, L.O.; Manning, B.D. TBC1D7 is a third subunit of the TSC1-TSC2 complex upstream of mTORC1. Mol. Cell 2012, 47, 535–546. [Google Scholar] [CrossRef]
- Dibble, C.C.; Cantley, L.C. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 2015, 25, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Dey, C.S. PTEN, a widely known negative regulator of insulin/PI3K signaling, positively regulates neuronal insulin resistance. Mol. Biol. Cell 2012, 23, 3882–3898. [Google Scholar] [CrossRef] [PubMed]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol 2011, 12, 21–35. [Google Scholar] [CrossRef]
- Kim, S.R.; Kareva, T.; Yarygina, O.; Kholodilov, N.; Burke, R.E. AAV transduction of dopamine neurons with constitutively active Rheb protects from neurodegeneration and mediates axon regrowth. Mol. Ther. 2012, 20, 275–286. [Google Scholar] [CrossRef]
- Kim, S.R.; Chen, X.; Oo, T.F.; Kareva, T.; Yarygina, O.; Wang, C.; During, M.; Kholodilov, N.; Burke, R.E. Dopaminergic pathway reconstruction by Akt/Rheb-induced axon regeneration. Ann. Neurol. 2011, 70, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Moon, G.J.; Shin, M.; Kim, S.R. Upregulation of Neuronal Rheb(S16H) for Hippocampal Protection in the Adult Brain. Int. J. Mol. Sci. 2020, 21, 2023. [Google Scholar] [CrossRef] [PubMed]
- Park, K.K.; Liu, K.; Hu, Y.; Smith, P.D.; Wang, C.; Cai, B.; Xu, B.; Connolly, L.; Kramvis, I.; Sahin, M.; et al. Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science 2008, 322, 963–966. [Google Scholar] [CrossRef]
- Li, Y.H.; Werner, H.; Puschel, A.W. Rheb and mTOR regulate neuronal polarity through Rap1B. J. Biol. Chem. 2008, 283, 33784–33792. [Google Scholar] [CrossRef]
- Tavazoie, S.F.; Alvarez, V.A.; Ridenour, D.A.; Kwiatkowski, D.J.; Sabatini, B.L. Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat. Neurosci. 2005, 8, 1727–1734. [Google Scholar] [CrossRef]
- Choi, Y.J.; Di Nardo, A.; Kramvis, I.; Meikle, L.; Kwiatkowski, D.J.; Sahin, M.; He, X. Tuberous sclerosis complex proteins control axon formation. Genes Dev. 2008, 22, 2485–2495. [Google Scholar] [CrossRef]
- Jaworski, J.; Sheng, M. The growing role of mTOR in neuronal development and plasticity. Mol. Neurobiol. 2006, 34, 205–219. [Google Scholar] [CrossRef]
- Banerjee, R.; Beal, M.F.; Thomas, B. Autophagy in neurodegenerative disorders: Pathogenic roles and therapeutic implications. Trends Neurosci. 2010, 33, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Shahani, N.; Huang, W.C.; Varnum, M.; Page, D.T.; Subramaniam, S. Forebrain depletion of Rheb GTPase elicits spatial memory deficits in mice. Neurobiol. Aging 2017, 50, 134–143. [Google Scholar] [CrossRef][Green Version]
- Domanskyi, A.; Geissler, C.; Vinnikov, I.A.; Alter, H.; Schober, A.; Vogt, M.A.; Gass, P.; Parlato, R.; Schutz, G. Pten ablation in adult dopaminergic neurons is neuroprotective in Parkinson’s disease models. FASEB J. 2011, 25, 2898–2910. [Google Scholar] [CrossRef] [PubMed]
- Malagelada, C.; Jin, Z.H.; Greene, L.A. RTP801 is induced in Parkinson’s disease and mediates neuron death by inhibiting Akt phosphorylation/activation. J. Neurosci. 2008, 28, 14363–14371. [Google Scholar] [CrossRef]
- Crews, L.; Spencer, B.; Desplats, P.; Patrick, C.; Paulino, A.; Rockenstein, E.; Hansen, L.; Adame, A.; Galasko, D.; Masliah, E. Selective molecular alterations in the autophagy pathway in patients with Lewy body disease and in models of alpha-synucleinopathy. PLoS ONE 2010, 5, e9313. [Google Scholar] [CrossRef]
- Pan, T.; Rawal, P.; Wu, Y.; Xie, W.; Jankovic, J.; Le, W. Rapamycin protects against rotenone-induced apoptosis through autophagy induction. Neuroscience 2009, 164, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Bockaert, J.; Marin, P. mTOR in Brain Physiology and Pathologies. Physiol. Rev. 2015, 95, 1157–1187. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Liu, C.; Liu, W.; Zhang, H.; Zhang, R.; Liu, J.; Zhang, J.; Xu, C.; Liu, L.; Huang, S.; et al. Rotenone induction of hydrogen peroxide inhibits mTOR-mediated S6K1 and 4E-BP1/eIF4E pathways, leading to neuronal apoptosis. Toxicol. Sci. 2015, 143, 81–96. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Liu, C.; Chen, S.; Ye, Y.; Guo, M.; Ren, Q.; Liu, L.; Zhang, H.; Xu, C.; Zhou, Q.; et al. Activation of AMPK and inactivation of Akt result in suppression of mTOR-mediated S6K1 and 4E-BP1 pathways leading to neuronal cell death in in vitro models of Parkinson’s disease. Cell Signal. 2014, 26, 1680–1689. [Google Scholar] [CrossRef]
- Rodriguez-Blanco, J.; Martin, V.; Garcia-Santos, G.; Herrera, F.; Casado-Zapico, S.; Antolin, I.; Rodriguez, C. Cooperative action of JNK and AKT/mTOR in 1-methyl-4-phenylpyridinium-induced autophagy of neuronal PC12 cells. J. Neurosci. Res. 2012, 90, 1850–1860. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, S.; Sun, Y.; Watt, J.A.; Wang, S.; Lei, S.; Birnbaumer, L.; Singh, B.B. Neurotoxin-induced ER stress in mouse dopaminergic neurons involves downregulation of TRPC1 and inhibition of AKT/mTOR signaling. J. Clin. Invest. 2012, 122, 1354–1367. [Google Scholar] [CrossRef] [PubMed]
- Rieker, C.; Engblom, D.; Kreiner, G.; Domanskyi, A.; Schober, A.; Stotz, S.; Neumann, M.; Yuan, X.; Grummt, I.; Schutz, G.; et al. Nucleolar disruption in dopaminergic neurons leads to oxidative damage and parkinsonism through repression of mammalian target of rapamycin signaling. J. Neurosci. 2011, 31, 453–460. [Google Scholar] [CrossRef]
- Chen, L.; Xu, B.; Liu, L.; Luo, Y.; Yin, J.; Zhou, H.; Chen, W.; Shen, T.; Han, X.; Huang, S. Hydrogen peroxide inhibits mTOR signaling by activation of AMPKalpha leading to apoptosis of neuronal cells. Lab. Invest. 2010, 90, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.C.; Kim, S.H.; Ha, J.Y.; Kim, S.T.; Son, J.H. A novel mTOR activating protein protects dopamine neurons against oxidative stress by repressing autophagy related cell death. J. Neurochem. 2010, 112, 366–376. [Google Scholar] [CrossRef]
- Malagelada, C.; Ryu, E.J.; Biswas, S.C.; Jackson-Lewis, V.; Greene, L.A. RTP801 is elevated in Parkinson brain substantia nigral neurons and mediates death in cellular models of Parkinson’s disease by a mechanism involving mammalian target of rapamycin inactivation. J. Neurosci. 2006, 26, 9996–10005. [Google Scholar] [CrossRef]
- Cheng, H.C.; Kim, S.R.; Oo, T.F.; Kareva, T.; Yarygina, O.; Rzhetskaya, M.; Wang, C.; During, M.; Talloczy, Z.; Tanaka, K.; et al. Akt suppresses retrograde degeneration of dopaminergic axons by inhibition of macroautophagy. J. Neurosci. 2011, 31, 2125–2135. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Krainc, D. alpha-synuclein toxicity in neurodegeneration: Mechanism and therapeutic strategies. Nat. Med. 2017, 23, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Recasens, A.; Dehay, B.; Bove, J.; Carballo-Carbajal, I.; Dovero, S.; Perez-Villalba, A.; Fernagut, P.O.; Blesa, J.; Parent, A.; Perier, C.; et al. Lewy body extracts from Parkinson disease brains trigger alpha-synuclein pathology and neurodegeneration in mice and monkeys. Ann. Neurol. 2014, 75, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Lee, V.M.; Trojanowski, J.Q. Mechanisms of Parkinson’s disease linked to pathological alpha-synuclein: New targets for drug discovery. Neuron 2006, 52, 33–38. [Google Scholar] [CrossRef]
- Ramalingam, M.; Huh, Y.J.; Lee, Y.I. The Impairments of alpha-Synuclein and Mechanistic Target of Rapamycin in Rotenone-Induced SH-SY5Y Cells and Mice Model of Parkinson’s Disease. Front. Neurosci. 2019, 13, 1028. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Duan, C.; Gao, G.; Wang, X.; Yang, H. Alpha-synuclein overexpression negatively regulates insulin receptor substrate 1 by activating mTORC1/S6K1 signaling. Int. J. Biochem. Cell Biol. 2015, 64, 25–33. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Yoon, D.; Jeon, M.T.; Woon Kim, D.; Kim, S.R. Treatment with AAV1-Rheb(S16H) provides neuroprotection in a mouse model of photothrombosis-induced ischemic stroke. Neuroreport 2020, 31, 971–978. [Google Scholar] [CrossRef]
- Jeon, M.T.; Kim, S.R. Roles of Rheb(S16H) in substantia nigra pars compacta dopaminergic neurons in vivo. Biomed. Rep. 2015, 3, 137–140. [Google Scholar] [CrossRef][Green Version]
- Wang, Q.; Liu, Y.; Zhou, J. Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl. Neurodegener. 2015, 4, 19. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Neurodegenerative Diseases | Target System | Target Tissues | References |
|---|---|---|---|
| Alzheimer’s Disease | Cholinergic Neurons | Nucleus basalis (NB) | [43] |
| Noradrenergic Neurons | Locus coeruleus (LC) | [44] | |
| Dopaminergic Neurons | Substantia nigra (SN) | [45] | |
| Serotonergic Neurons | Dorsal raphe nucleus (DRN) | [46] | |
| Adrenergic Neurons | Rostral ventral lateral medulla C-1 neurons | [47] | |
| Parkinson’s Disease | Cholinergic Neurons | Nucleus basalis (NB) | [50] |
| Noradrenergic Neurons | Locus coeruleus (LC) | [52] | |
| Dopaminergic Neurons | Substantia nigra (SN) | [49] | |
| Serotonergic Neurons | Dorsal raphe nucleus (DRN) | [48] | |
| Adrenergic Neurons | Rostral ventral lateral medulla C-1 neurons | [51] | |
| Huntington’s Chorea | Dopaminergic Neurons | Striatum | [56] |
| Cholinergic Neurons | Thalamostriatal axodendritic terminals | [53] | |
| GABAergic Neurons | striatum | [54,55] | |
| Glutamate Neurons | Striatum | [57] |
| Diseases | Animal Model | Neurotrophic Factor | Effect | Reference |
|---|---|---|---|---|
| Alzheimer’s Disease | J20 (human APP mutant) | BDNF | Improve | [21] |
| Tg2576 | CNTF | Improve | [22] | |
| Thrombin | BDNF CNTF | Improve | [39,40] | |
| 5XFAD | BDNF CNTF | Improve | [42] | |
| P301L | BDNF | Improve | [60] | |
| Thrombin | GDNF | Improve | [69] | |
| Parkinson’s Disease | Inflammation (pKr-2) | GDNF BDNF | Improve | [41] |
| Gdnf(+/−) | GDNF | Improve | [63] | |
| 6-OHDA | GDNF | Improve | [70] | |
| α-synuclein | GDNF | Worsen | [71,72] | |
| MPP+ | CNTF | Improve | [73] | |
| MPP+ | GDNF BDNF | Improve | [74] | |
| Huntington’s Disease | bdnf(+/−) | BDNF | Improve | [65] |
| Amyotrophic lateral sclerosis | SOD1(G93A) | GDNF | Improve | [75,76,77] |
| BDNF | Improve | [78] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nam, Y.; Moon, G.J.; Kim, S.R. Therapeutic Potential of AAV1-Rheb(S16H) Transduction against Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 3064. https://doi.org/10.3390/ijms22063064
Nam Y, Moon GJ, Kim SR. Therapeutic Potential of AAV1-Rheb(S16H) Transduction against Neurodegenerative Diseases. International Journal of Molecular Sciences. 2021; 22(6):3064. https://doi.org/10.3390/ijms22063064
Chicago/Turabian StyleNam, Youngpyo, Gyeong Joon Moon, and Sang Ryong Kim. 2021. "Therapeutic Potential of AAV1-Rheb(S16H) Transduction against Neurodegenerative Diseases" International Journal of Molecular Sciences 22, no. 6: 3064. https://doi.org/10.3390/ijms22063064
APA StyleNam, Y., Moon, G. J., & Kim, S. R. (2021). Therapeutic Potential of AAV1-Rheb(S16H) Transduction against Neurodegenerative Diseases. International Journal of Molecular Sciences, 22(6), 3064. https://doi.org/10.3390/ijms22063064