
IRE1α Is a Therapeutic Target for Cystic Fibrosis Airway Inflammation



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
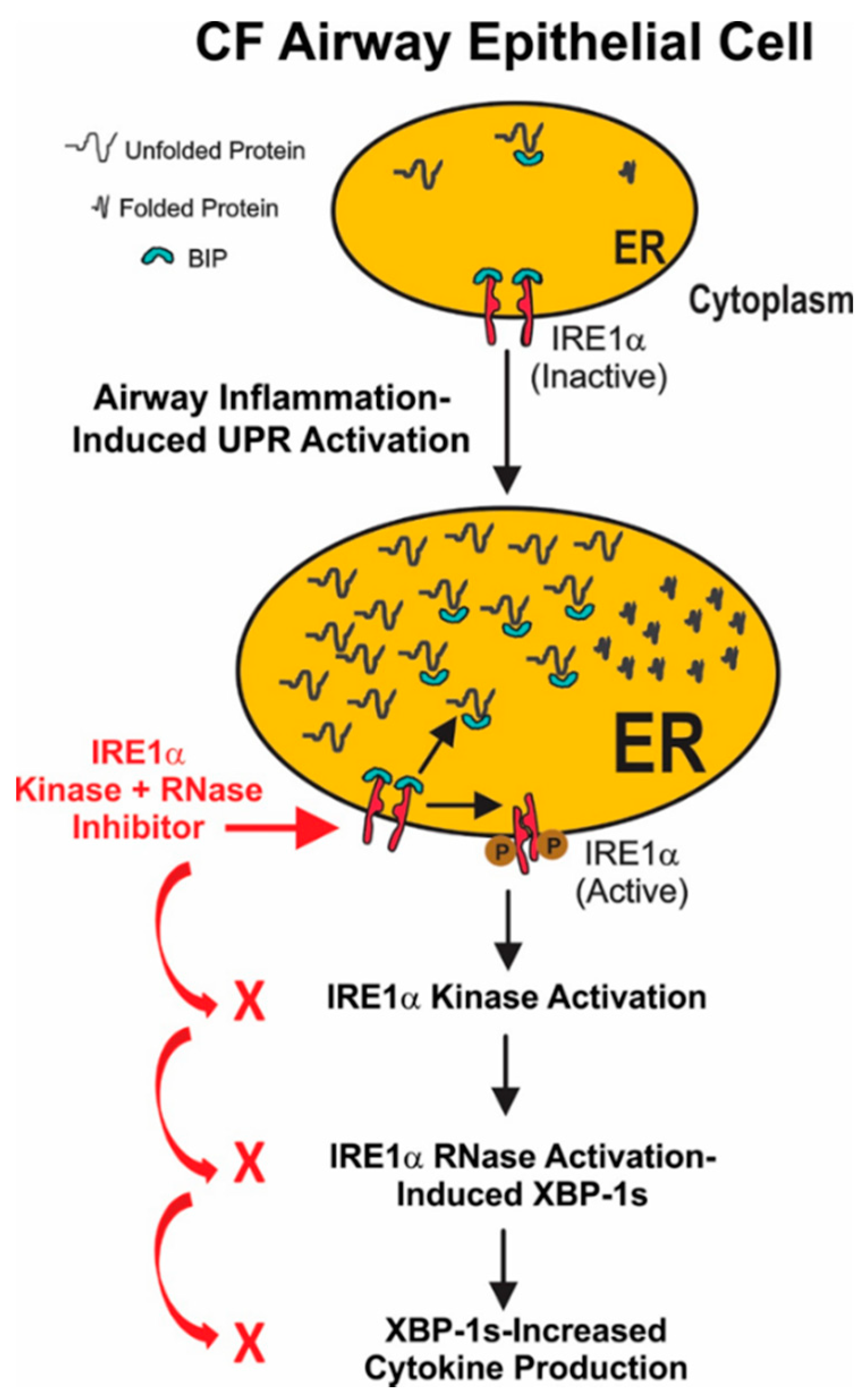
1. Introduction
2. Results
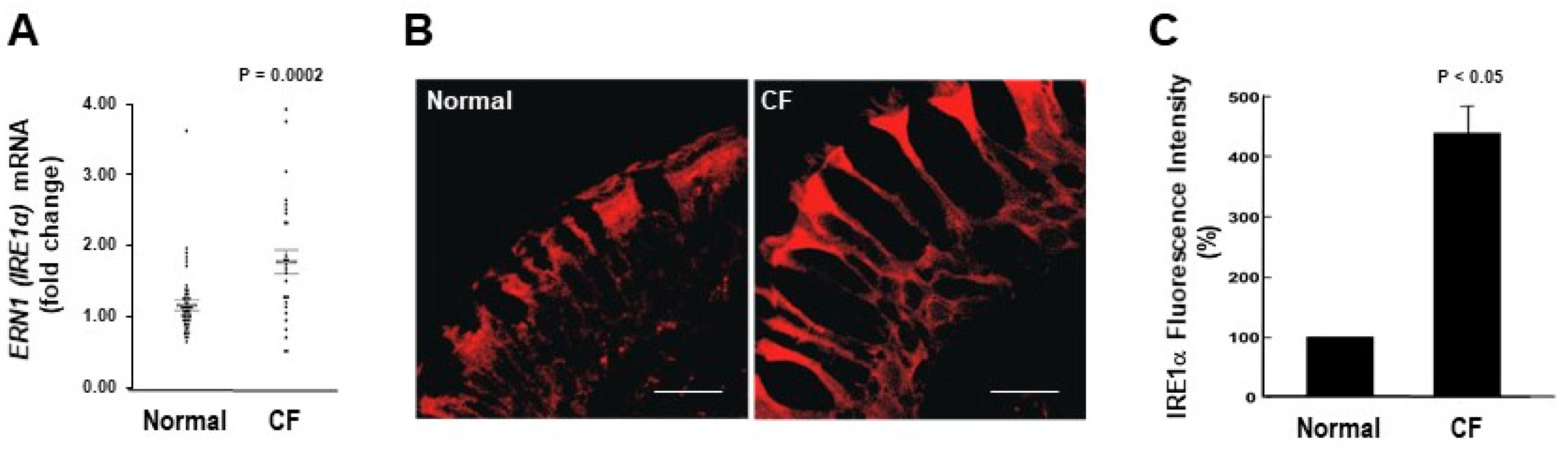
2.1. IRE1α mRNA and Protein Expression Levels Are Up-Regulated in CF Human Airway Epithelia
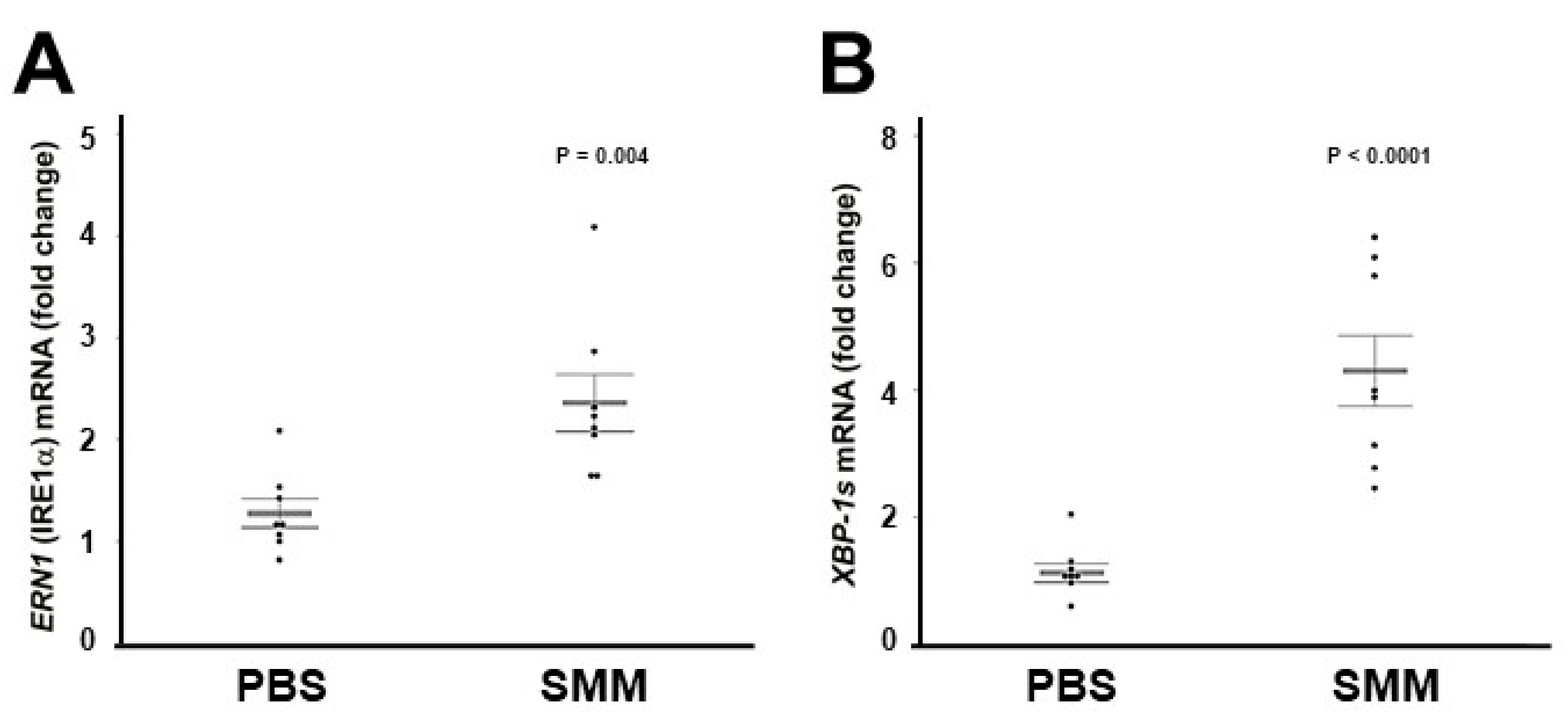
2.2. The Up-Regulation of IRE1α and XBP-1s Found in Freshly Isolated CF HBE Is Reproduced in Primary Normal HBE Cultures Exposed to SMM
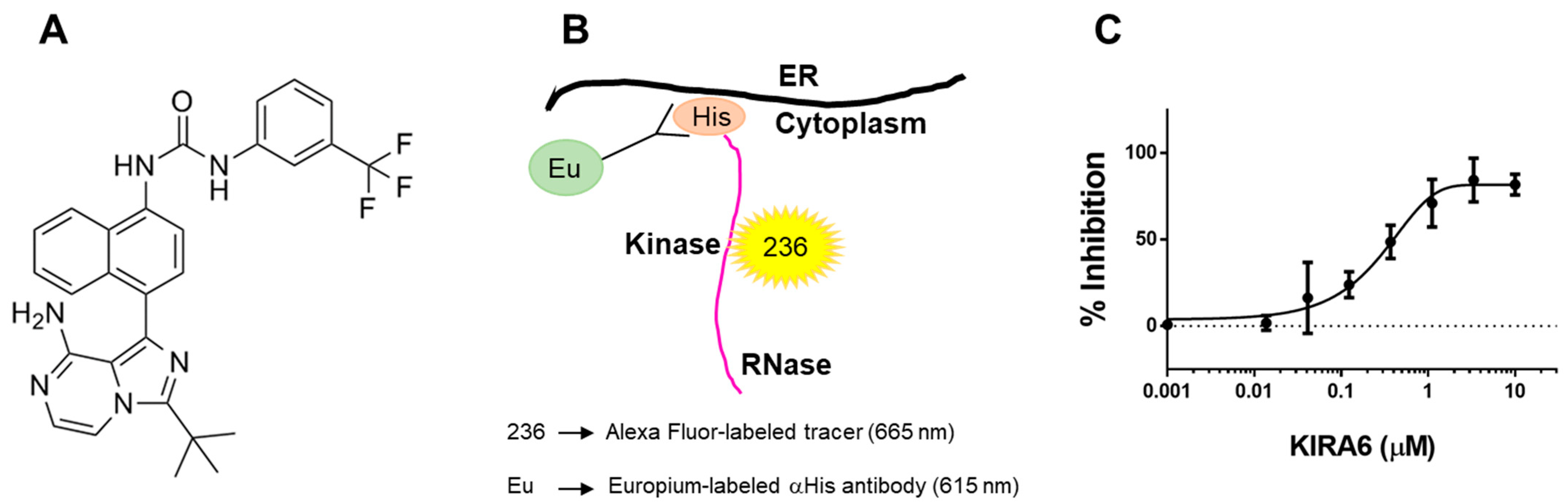
2.3. The Small Molecule IRE1α Kinase and RNase Inhibitor KIRA6 (Kinase Inhibiting RNase Attenuating 6) Binds Directly to IRE1α Kinase
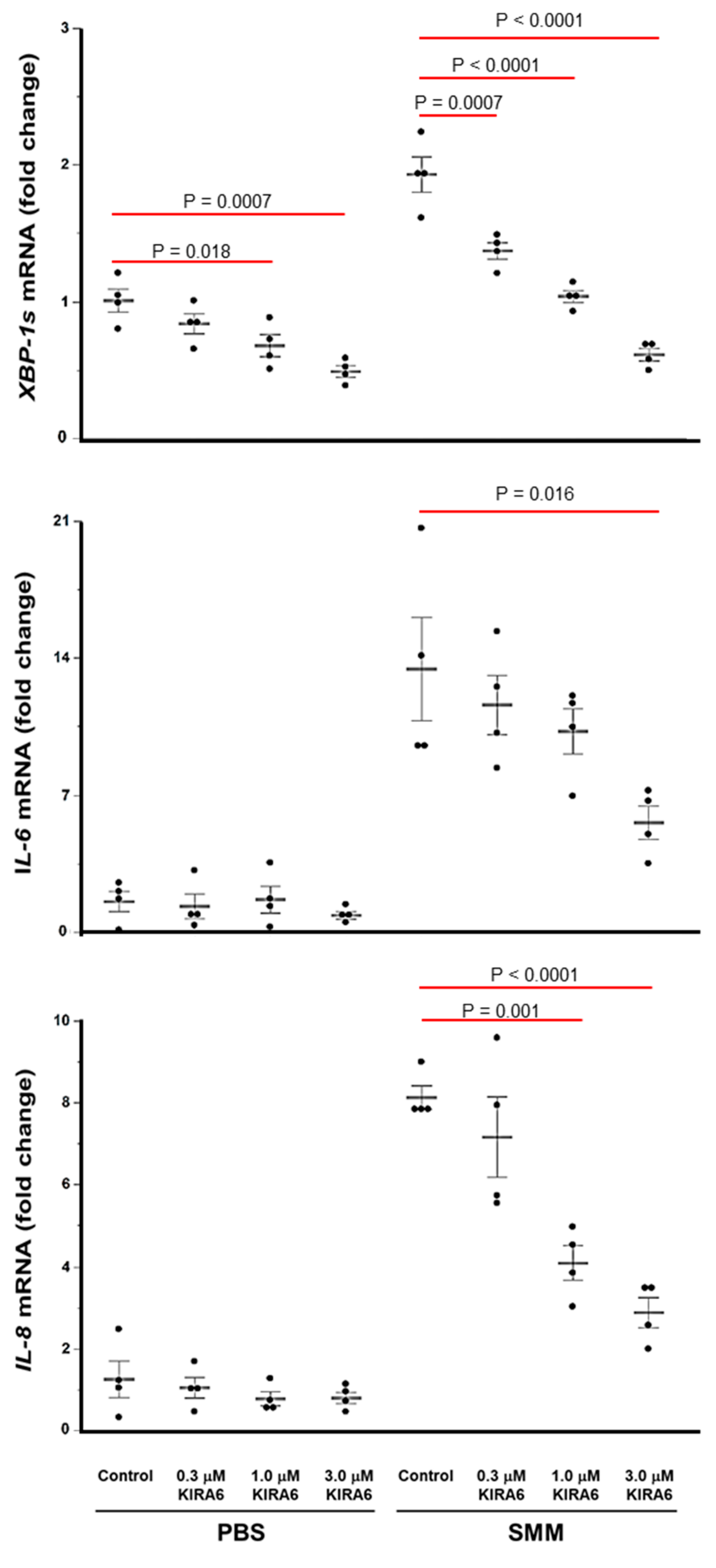
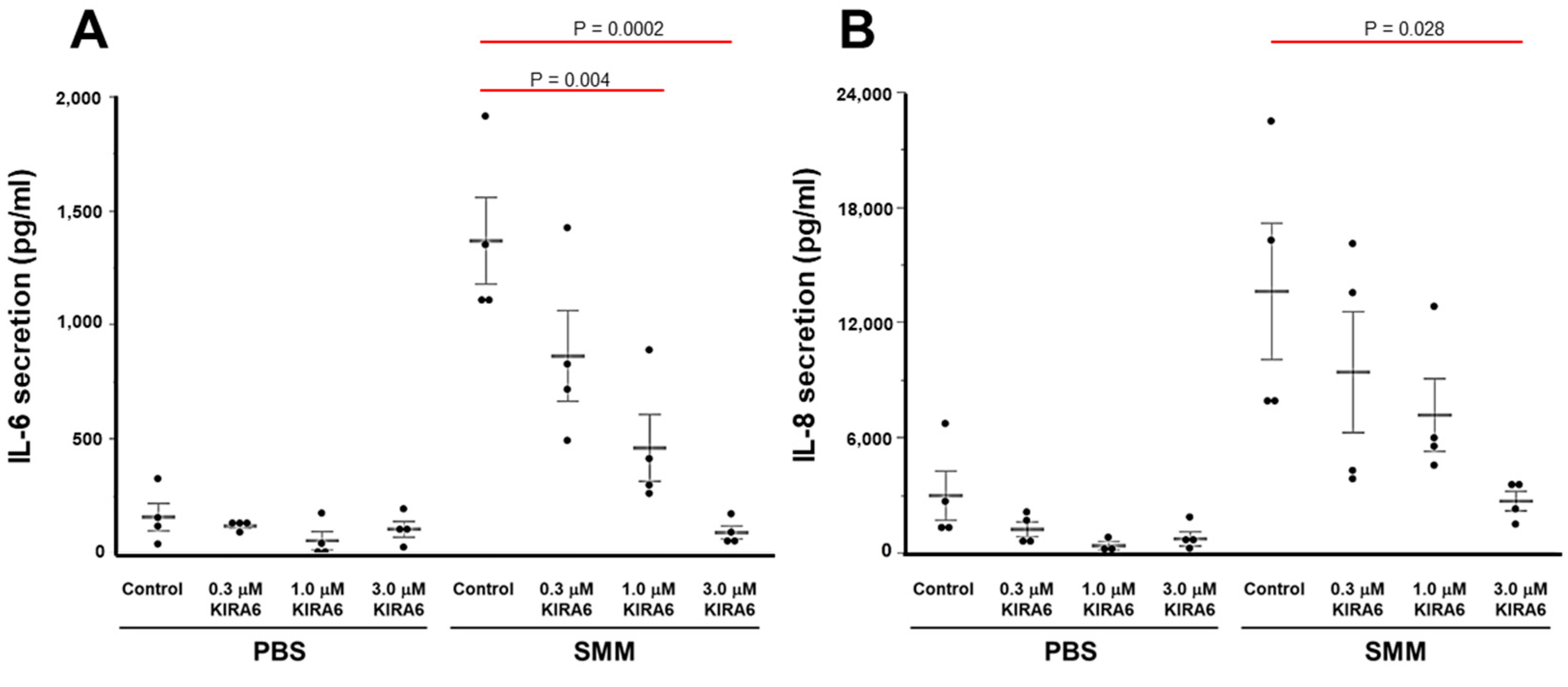
2.4. Proof-of-Concept That Pharmacological Inhibition of IRE1α Kinase and RNase Blocks XBP-1s Expression Coupled to Cytokine Production in CF HBE
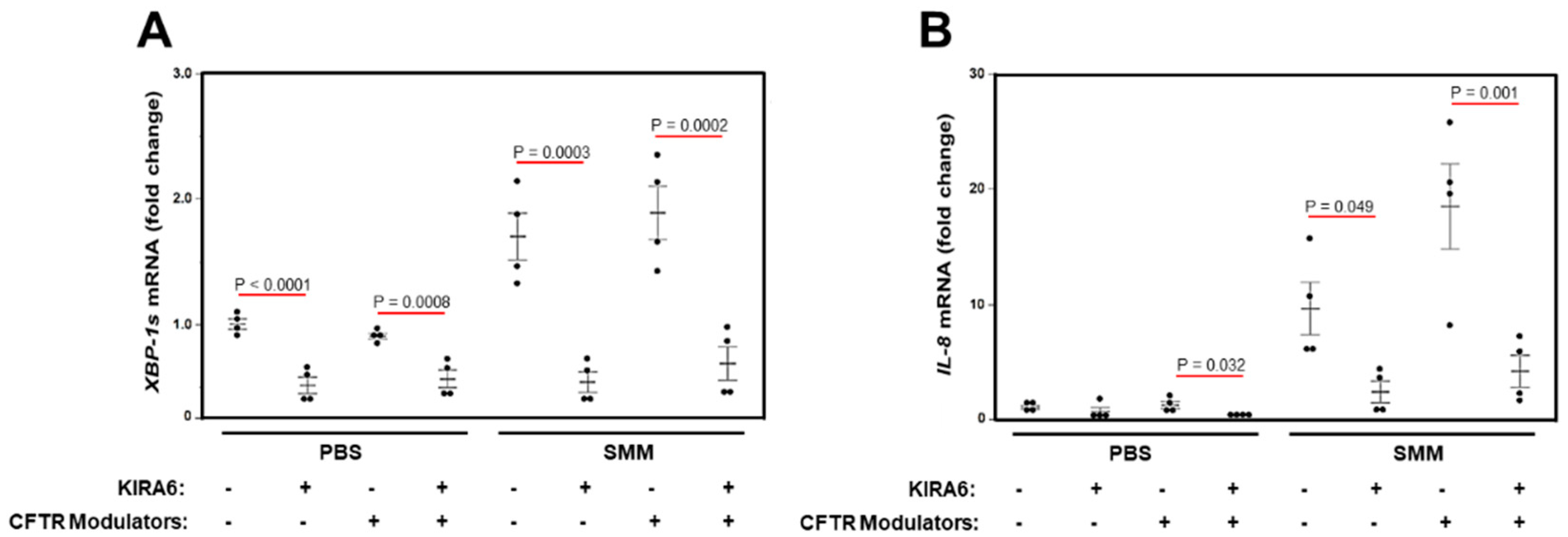
2.5. A Triple Combination of CFTR Modulators Does Not Blunt SMM-Increased XBP-1s and Cytokine Production in CF HBE
3. Discussion
4. Materials and Methods
4.1. Tissue Harvesting
4.2. Supernatant of Mucopurulent Material (SMM) from CF Airways
4.3. Cell Culture and Treatments
4.4. Immunofluorescence and Quantification of IRE1α Expression
4.5. Real Time Polymerase Chain Reaction (RT-PCR)
4.6. Expression of the Cytoplasmic Domain of IRE1α
4.7. Purification of Cytoplasmic IRE1α for Kinase Assays
4.8. Time Resolved—Fluorescence Resonance Energy Transfer (TR-FRET) IRE1α Kinase Assay
4.9. Cytokine Secretion
4.10. Statistical Analyses
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boucher, R.C.; Cotton, C.U.; Gatzy, J.T.; Knowles, M.R.; Yankaskas, J.R. Evidence for reduced Cl- and increased Na+ permeability in cystic fibrosis human primary cell cultures. J. Physiol. 1988, 405, 77–103. [Google Scholar] [CrossRef] [PubMed]
- Keiser, N.W.; Engelhardt, J.F. New animal models of cystic fibrosis: What are they teaching us? Curr. Opin. Pulm. Med. 2011, 17, 478–483. [Google Scholar] [CrossRef]
- Matsui, H.; Grubb, B.R.; Tarran, R.; Randell, S.H.; Gatzy, J.T.; Davis, C.W.; Boucher, R.C. Evidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis of cystic fibrosis airways disease. Cell 1998, 95, 1005–1015. [Google Scholar] [CrossRef]
- Boucher, R.C. Evidence for airway surface dehydration as the initiating event in CF airway disease. J. Intern. Med. 2007, 261, 5–16. [Google Scholar] [CrossRef]
- Hobbs, C.A.; Da Tan, C.; Tarran, R. Does epithelial sodium channel hyperactivity contribute to cystic fibrosis lung disease? J. Physiol. 2013, 591, 4377–4387. [Google Scholar] [CrossRef] [PubMed]
- Mall, M.A.; Hartl, D. CFTR: Cystic fibrosis and beyond. Eur. Respir. J. 2014, 44, 1042–1054. [Google Scholar] [CrossRef]
- Konstan, M.W.; Hilliard, K.A.; Norvell, T.M.; Berger, M. Bronchoalveolar lavage findings in cystic fibrosis patients with stable, clinically mild lung disease suggest ongoing infection and inflammation. Am. J. Respir. Crit. Care Med. 1994, 150, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.Z.; Wagener, J.S.; Bost, T.; Martinez, J.; Accurso, F.J.; Riches, D.W. Early pulmonary inflammation in infants with cystic fibrosis. Am. J. Respir. Crit. Care Med. 1995, 151, 1075–1082. [Google Scholar] [CrossRef] [PubMed]
- Koller, D.Y.; Nething, I.; Otto, J.; Urbanek, R.; Eichler, I. Cytokine concentrations in sputum from patients with cystic fibrosis and their relation to eosinophil activity. Am. J. Respir. Crit. Care Med. 1997, 155, 1050–1054. [Google Scholar] [CrossRef]
- Muhlebach, M.S.; Stewart, P.W.; Leigh, M.W.; Noah, T.L. Quantitation of inflammatory responses to bacteria in young cystic fibrosis and control patients. Am. J. Respir. Crit. Care Med. 1999, 160, 186–191. [Google Scholar] [CrossRef]
- Taggart, C.; Coakley, R.J.; Greally, P.; Canny, G.; O’Neill, S.J.; McElvaney, N.G. Increased elastase release by CF neutrophils is mediated by tumor necrosis factor-alpha and interleukin-8. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 278, L33–L41. [Google Scholar] [CrossRef]
- Muhlebach, M.S.; Noah, T.L. Endotoxin activity and inflammatory markers in the airways of young patients with cystic fibrosis. Am. J. Respir. Crit. Care Med. 2002, 165, 911–915. [Google Scholar] [CrossRef]
- Elizur, A.; Cannon, C.L.; Ferkol, T.W. Airway inflammation in cystic fibrosis. Chest 2008, 133, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, S.C.; Parsons, F.; Gangell, C.; Brennan, S.; Stick, S.M.; Sly, P.D. Evolution of pulmonary inflammation and nutritional status in infants and young children with cystic fibrosis. Thorax 2011, 66, 408–413. [Google Scholar] [CrossRef] [PubMed]
- Sagel, S.D.; Wagner, B.D.; Anthony, M.M.; Emmett, P.; Zemanick, E.T. Sputum biomarkers of inflammation and lung function decline in children with cystic fibrosis. Am. J. Respir. Crit. Care Med. 2012, 186, 857–865. [Google Scholar] [CrossRef] [PubMed]
- Elborn, J.S. Cystic fibrosis. Lancet 2016, 388, 2519–2531. [Google Scholar] [CrossRef]
- Roesch, E.A.; Nichols, D.P.; Chmiel, J.F. Inflammation in cystic fibrosis: An update. Pediatr. Pulmonol. 2018, 53, S30–S50. [Google Scholar] [CrossRef]
- Bergeron, C.; Cantin, A.M. Cystic fibrosis: Pathophysiology of lung disease. Semin. Respir. Crit. Care Med. 2019, 40, 715–726. [Google Scholar] [CrossRef]
- Cabrini, G.; Rimessi, A.; Borgatti, M.; Lampronti, I.; Finotti, A.; Pinton, P.; Gambari, R. Role of cystic fibrosis bronchial epithelium in neutrophil chemotaxis. Front. Immunol. 2020, 11, 1438. [Google Scholar] [CrossRef] [PubMed]
- Prandini, P.; De Logu, F.; Fusi, C.; Provezza, L.; Nassini, R.; Montagner, G.; Materazzi, S.; Munari, S.; Gilioli, E.; Bezzerri, V.; et al. Transient receptor potential ankyrin 1 channels modulate inflammatory response in respiratory cells from patients with cystic fibrosis. Am. J. Respir. Cell Mol. Biol. 2016, 55, 645–656. [Google Scholar] [CrossRef]
- Rimessi, A.; Bezzerri, V.; Salvatori, F.; Tamanini, A.; Nigro, F.; Dechecchi, M.C.; Santangelo, A.; Prandini, P.; Munari, S.; Provezza, L.; et al. PLCB3 loss-of-function reduces P. aeruginosa-dependent IL-8 release in cystic fibrosis. Am. J. Respir. Cell Mol. Biol. 2018, 59, 428–436. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, C.M.; Paradiso, A.M.; Schwab, U.; Perez-Vilar, J.; Jones, L.; O’Neal, W.; Boucher, R.C. Chronic airway infection/inflammation induces a Ca2+i-dependent hyperinflammatory response in human cystic fibrosis airway epithelia. J. Biol. Chem. 2005, 280, 17798–17806. [Google Scholar] [CrossRef] [PubMed]
- Tabary, O.; Escotte, S.; Couetil, J.P.; Hubert, D.; Dusser, D.; Puchelle, E.; Jacquot, J. High susceptibility for cystic fibrosis human airway gland cells to produce IL-8 through the I kappa B kinase alpha pathway in response to extracellular NaCl content. J. Immunol. 2000, 164, 3377–3384. [Google Scholar] [CrossRef]
- Tabary, O.; Zahm, J.M.; Hinnrasky, J.; Couetil, J.P.; Cornillet, P.; Guenounou, M.; Gaillard, D.; Puchelle, E.; Jacquot, J. Selective up-regulation of chemokine IL-8 expression in cystic fibrosis bronchial gland cells in vivo and in vitro. Am. J. Pathol. 1998, 153, 921–930. [Google Scholar] [CrossRef]
- Bonfield, T.L.; Konstan, M.W.; Berger, M. Altered respiratory epithelial cell cytokine production in cystic fibrosis. J. Allergy Clin. Immunol. 1999, 104, 72–78. [Google Scholar] [CrossRef]
- Ribeiro, C.M.; Paradiso, A.M.; Carew, M.A.; Shears, S.B.; Boucher, R.C. Cystic fibrosis airway epithelial Ca2+ i signaling: The mechanism for the larger agonist-mediated Ca2+ i signals in human cystic fibrosis airway epithelia. J. Biol. Chem. 2005, 280, 10202–10209. [Google Scholar] [CrossRef]
- Ribeiro, C.M.; Lubamba, B.A. Role of IRE1alpha/XBP-1 in cystic fibrosis airway inflammation. Int. J. Mol. Sci. 2017, 18, 118. [Google Scholar] [CrossRef]
- Martino, M.E.; Olsen, J.C.; Fulcher, N.B.; Wolfgang, M.C.; O’Neal, W.K.; Ribeiro, C.M. Airway epithelial inflammation-induced endoplasmic reticulum Ca2+ store expansion is mediated by X-box binding protein-1. J. Biol. Chem. 2009, 284, 14904–14913. [Google Scholar] [CrossRef]
- Ribeiro, C.M.; Boucher, R.C. Role of endoplasmic reticulum stress in cystic fibrosis-related airway inflammatory responses. Proc. Am. Thorac. Soc. 2010, 7, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Lubamba, B.A.; Jones, L.C.; O’Neal, W.K.; Boucher, R.C.; Ribeiro, C.M. X-box-binding protein 1 and innate Immune responses of human cystic fibrosis alveolar macrophages. Am. J. Respir. Crit. Care Med. 2015, 192, 1449–1461. [Google Scholar] [CrossRef] [PubMed]
- Iwawaki, T.; Hosoda, A.; Okuda, T.; Kamigori, Y.; Nomura-Furuwatari, C.; Kimata, Y.; Tsuru, A.; Kohno, K. Translational control by the ER transmembrane kinase/ribonuclease IRE1 under ER stress. Nat. Cell Biol. 2001, 3, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Martino, M.B.; Jones, L.; Brighton, B.; Ehre, C.; Abdulah, L.; Davis, C.W.; Ron, D.; O’Neal, W.K.; Ribeiro, C.M. The ER stress transducer IRE1beta is required for airway epithelial mucin production. Mucosal Immunol. 2013, 6, 639–654. [Google Scholar] [CrossRef]
- Hetz, C.; Chevet, E.; Harding, H.P. Targeting the unfolded protein response in disease. Nat. Rev. Drug Discov. 2013, 12, 703–719. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002, 415, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Niwa, M.; Koong, A.C. Targeting the IRE1α-XBP1 branch of the unfolded protein response in human diseases. Semin. Cancer Biol. 2015, 33, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Logue, S.E.; McGrath, E.P.; Cleary, P.; Greene, S.; Mnich, K.; Almanza, A.; Chevet, E.; Dwyer, R.M.; Oommen, A.; Legembre, P.; et al. Inhibition of IRE1 RNase activity modulates the tumor cell secretome and enhances response to chemotherapy. Nat. Commun. 2018, 9, 3267. [Google Scholar] [CrossRef]
- Lu, Y.; Liang, F.X.; Wang, X. A synthetic biology approach identifies the mammalian UPR RNA ligase RtcB. Mol. Cell 2014, 55, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, L.H.; Coakley, R.; Webster, M.J.; Zhu, Y.; Tarran, R.; Radicioni, G.; Kesimer, M.; Boucher, R.C.; Davis, C.W.; Ribeiro, C.M.P. Mucin production and hydration responses to mucopurulent materials in normal versus cystic fibrosis airway epithelia. Am. J. Respir. Crit. Care Med. 2018, 197, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, C.M.; Hurd, H.; Wu, Y.; Martino, M.E.; Jones, L.; Brighton, B.; Boucher, R.C.; O’Neal, W.K. Azithromycin treatment alters gene expression in inflammatory, lipid metabolism, and cell cycle pathways in well-differentiated human airway epithelia. PLoS ONE 2009, 4, e5806. [Google Scholar] [CrossRef]
- Ghosh, R.; Wang, L.; Wang, E.S.; Perera, B.G.; Igbaria, A.; Morita, S.; Prado, K.; Thamsen, M.; Caswell, D.; Macias, H.; et al. Allosteric inhibition of the IRE1alpha RNase preserves cell viability and function during endoplasmic reticulum stress. Cell 2014, 158, 534–548. [Google Scholar] [CrossRef]
- Wang, L.; Perera, B.G.; Hari, S.B.; Bhhatarai, B.; Backes, B.J.; Seeliger, M.A.; Schurer, S.C.; Oakes, S.A.; Papa, F.R.; Maly, D.J. Divergent allosteric control of the IRE1alpha endoribonuclease using kinase inhibitors. Nat. Chem. Biol. 2012, 8, 982–989. [Google Scholar] [CrossRef]
- Bhullar, K.S.; Lagarón, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef] [PubMed]
- Mahameed, M.; Wilhelm, T.; Darawshi, O.; Obiedat, A.; Tommy, W.S.; Chintha, C.; Schubert, T.; Samali, A.; Chevet, E.; Eriksson, L.A.; et al. The unfolded protein response modulators GSK2606414 and KIRA6 are potent KIT inhibitors. Cell Death Dis. 2019, 10, 300. [Google Scholar] [CrossRef] [PubMed]
- PubChem. PubChem Compound Summary for CID 73425700. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/kira6 (accessed on 21 January 2021).
- McElvaney, O.J.; Wade, P.; Murphy, M.; Reeves, E.P.; McElvaney, N.G. Targeting airway inflammation in cystic fibrosis. Expert Rev. Respir. Med. 2019, 13, 1041–1055. [Google Scholar] [CrossRef]
- Flume, P.A.; O’Sullivan, B.P.; Robinson, K.A.; Goss, C.H.; Mogayzel, P.J., Jr.; Willey-Courand, D.B.; Bujan, J.; Finder, J.; Lester, M.; Quittell, L.; et al. Cystic fibrosis pulmonary guidelines: Chronic medications for maintenance of lung health. Am. J. Respir. Crit. Care Med. 2007, 176, 957–969. [Google Scholar] [CrossRef] [PubMed]
- Mogayzel, P.J., Jr.; Naureckas, E.T.; Robinson, K.A.; Mueller, G.; Hadjiliadis, D.; Hoag, J.B.; Lubsch, L.; Hazle, L.; Sabadosa, K.; Marshall, B. Cystic fibrosis pulmonary guidelines. Chronic medications for maintenance of lung health. Am. J. Respir. Crit. Care Med. 2013, 187, 680–689. [Google Scholar] [CrossRef] [PubMed]
- Konstan, M.W.; Schluchter, M.D.; Xue, W.; Davis, P.B. Clinical use of Ibuprofen is associated with slower FEV1 decline in children with cystic fibrosis. Am. J. Respir. Crit. Care Med. 2007, 176, 1084–1089. [Google Scholar] [CrossRef] [PubMed]
- Konstan, M.W.; VanDevanter, D.R.; Sawicki, G.S.; Pasta, D.J.; Foreman, A.J.; Neiman, E.A.; Morgan, W.J. Association of high-dose ibuprofen use, lung function decline, and long-term survival in children with cystic fibrosis. Ann. Am. Thorac. Soc. 2018, 15, 485–493. [Google Scholar] [CrossRef]
- Marjanovic, N.; Bosnar, M.; Michielin, F.; Wille, D.R.; Anic-Milic, T.; Culic, O.; Popovic-Grle, S.; Bogdan, M.; Parnham, M.J.; Erakovic Haber, V. Macrolide antibiotics broadly and distinctively inhibit cytokine and chemokine production by COPD sputum cells in vitro. Pharmacol. Res. 2011, 63, 389–397. [Google Scholar] [CrossRef]
- Southern, K.W.; Barker, P.M.; Solis-Moya, A.; Patel, L. Macrolide antibiotics for cystic fibrosis. Cochrane Database Syst. Rev. 2012, 11, CD002203. [Google Scholar] [CrossRef] [PubMed]
- Saiman, L.; Marshall, B.C.; Mayer-Hamblett, N.; Burns, J.L.; Quittner, A.L.; Cibene, D.A.; Coquillette, S.; Fieberg, A.Y.; Accurso, F.J.; Campbell, P.W., 3rd. Azithromycin in patients with cystic fibrosis chronically infected with Pseudomonas aeruginosa: A randomized controlled trial. JAMA 2003, 290, 1749–1756. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, C.M.; O’Neal, W.K. Endoplasmic reticulum stress in chronic obstructive lung diseases. Curr. Mol. Med. 2012, 12, 872–882. [Google Scholar] [CrossRef]
- Chen, G.; Sun, L.; Kato, T.; Okuda, K.; Martino, M.B.; Abzhanova, A.; Lin, J.M.; Gilmore, R.C.; Batson, B.D.; O’Neal, Y.K.; et al. IL-1beta dominates the promucin secretory cytokine profile in cystic fibrosis. J. Clin. Investig. 2019, 129, 4433–4450. [Google Scholar] [CrossRef]
- Korennykh, A.V.; Korostelev, A.A.; Egea, P.F.; Finer-Moore, J.; Stroud, R.M.; Zhang, C.; Shokat, K.M.; Walter, P. Structural and functional basis for RNA cleavage by Ire1. BMC Biol. 2011, 9, 47. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, I.; Denko, N.C.; Olson, M.; Van Melckebeke, H.; Lust, S.; Tam, A.; Solow-Cordero, D.E.; Bouley, D.M.; Offner, F.; Niwa, M.; et al. Identification of an Ire1alpha endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood 2011, 117, 1311–1314. [Google Scholar] [CrossRef] [PubMed]
- Cross, B.C.; Bond, P.J.; Sadowski, P.G.; Jha, B.K.; Zak, J.; Goodman, J.M.; Silverman, R.H.; Neubert, T.A.; Baxendale, I.R.; Ron, D.; et al. The molecular basis for selective inhibition of unconventional mRNA splicing by an IRE1-binding small molecule. Proc. Natl. Acad. Sci. USA 2012, 109, E869–E878. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Holmes, I.P.; Hochgräfe, F.; Walker, S.R.; Ali, N.A.; Humphrey, E.S.; Wu, J.; de Silva, M.; Kersten, W.J.; Connor, T.; et al. Characterization of the novel broad-spectrum kinase inhibitor CTx-0294885 as an affinity reagent for mass spectrometry-based kinome profiling. J. Proteome Res. 2013, 12, 3104–3116. [Google Scholar] [CrossRef]
- Feldman, H.C.; Tong, M.; Wang, L.; Meza-Acevedo, R.; Gobillot, T.A.; Lebedev, I.; Gliedt, M.J.; Hari, S.B.; Mitra, A.K.; Backes, B.J.; et al. Structural and functional analysis of the allosteric inhibition of IRE1alpha with ATP-competitive ligands. ACS Chem. Biol. 2016, 11, 2195–2205. [Google Scholar] [CrossRef] [PubMed]
- Esther, C.R., Jr.; Alexis, N.E.; Clas, M.L.; Lazarowski, E.R.; Donaldson, S.H.; Ribeiro, C.M.; Moore, C.G.; Davis, S.D.; Boucher, R.C. Extracellular purines are biomarkers of neutrophilic airway inflammation. Eur. Respir. J. 2008, 31, 949–956. [Google Scholar] [CrossRef]
- Cantin, A.M.; Hartl, D.; Konstan, M.W.; Chmiel, J.F. Inflammation in cystic fibrosis lung disease: Pathogenesis and therapy. J. Cyst. Fibros. 2015, 14, 419–430. [Google Scholar] [CrossRef]
- Gentzsch, M.; Cholon, D.M.; Quinney, N.L.; Martino, M.E.B.; Minges, J.T.; Boyles, S.E.; Guhr Lee, T.N.; Esther, C.R.; Ribeiro, C.M.P. Airway epithelial inflammation in vitro augments the rescue of mutant CFTR by current CFTR modulator therapies. Front. Pharmacol. 2021, 12, 628722. [Google Scholar] [CrossRef]
- Harris, J.K.; Wagner, B.D.; Zemanick, E.T.; Robertson, C.E.; Stevens, M.J.; Heltshe, S.L.; Rowe, S.M.; Sagel, S.D. Changes in airway microbiome and inflammation with ivacaftor treatment in patients with cystic fibrosis and the G551D mutation. Ann. Am. Thorac. Soc. 2020, 17, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Rowe, S.M.; Heltshe, S.L.; Gonska, T.; Donaldson, S.H.; Borowitz, D.; Gelfond, D.; Sagel, S.D.; Khan, U.; Mayer-Hamblett, N.; Van Dalfsen, J.M.; et al. Clinical mechanism of the cystic fibrosis transmembrane conductance regulator potentiator ivacaftor in G551D-mediated cystic fibrosis. Am. J. Respir. Crit. Care Med. 2014, 190, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Hisert, K.B.; Heltshe, S.L.; Pope, C.; Jorth, P.; Wu, X.; Edwards, R.M.; Radey, M.; Accurso, F.J.; Wolter, D.J.; Cooke, G.; et al. Restoring cystic fibrosis transmembrane conductance regulator function reduces airway bacteria and inflammation in people with cystic fibrosis and chronic lung infections. Am. J. Respir. Crit. Care Med. 2017, 195, 1617–1628. [Google Scholar] [CrossRef]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef]
- Nishitoh, H.; Matsuzawa, A.; Tobiume, K.; Saegusa, K.; Takeda, K.; Inoue, K.; Hori, S.; Kakizuka, A.; Ichijo, H. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002, 16, 1345–1355. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, M.; Niinuma, Y.; Nomura, Y. Activation signal of nuclear factor-kappa B in response to endoplasmic reticulum stress is transduced via IRE1 and tumor necrosis factor receptor-associated factor 2. Biol. Pharm. Bull. 2003, 26, 931–935. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Han, Z.; Couvillon, A.D.; Kaufman, R.J.; Exton, J.H. Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1alpha-mediated NF-kappaB activation and down-regulation of TRAF2 expression. Mol. Cell. Biol. 2006, 26, 3071–3084. [Google Scholar] [CrossRef] [PubMed]
- Oh-Hashi, K.; Kohno, H.; Kandeel, M.; Hirata, Y. Characterization of IRE1α in Neuro2a cells by pharmacological and CRISPR/Cas9 approaches. Mol. Cell. Biochem. 2020, 465, 53–64. [Google Scholar] [CrossRef]
- Ranatunga, S.; Tang, C.H.; Kang, C.W.; Kriss, C.L.; Kloppenburg, B.J.; Hu, C.C.; Del Valle, J.R. Synthesis of novel tricyclic chromenone-based inhibitors of IRE-1 RNase activity. J. Med. Chem. 2014, 57, 4289–4301. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hull-Ryde, E.A.; Minges, J.T.; Martino, M.E.B.; Kato, T.; Norris-Drouin, J.L.; Ribeiro, C.M.P. IRE1α Is a Therapeutic Target for Cystic Fibrosis Airway Inflammation. Int. J. Mol. Sci. 2021, 22, 3063. https://doi.org/10.3390/ijms22063063
Hull-Ryde EA, Minges JT, Martino MEB, Kato T, Norris-Drouin JL, Ribeiro CMP. IRE1α Is a Therapeutic Target for Cystic Fibrosis Airway Inflammation. International Journal of Molecular Sciences. 2021; 22(6):3063. https://doi.org/10.3390/ijms22063063
Chicago/Turabian StyleHull-Ryde, Emily A., John T. Minges, Mary E. B. Martino, Takafumi Kato, Jacqueline L. Norris-Drouin, and Carla M. P. Ribeiro. 2021. "IRE1α Is a Therapeutic Target for Cystic Fibrosis Airway Inflammation" International Journal of Molecular Sciences 22, no. 6: 3063. https://doi.org/10.3390/ijms22063063
APA StyleHull-Ryde, E. A., Minges, J. T., Martino, M. E. B., Kato, T., Norris-Drouin, J. L., & Ribeiro, C. M. P. (2021). IRE1α Is a Therapeutic Target for Cystic Fibrosis Airway Inflammation. International Journal of Molecular Sciences, 22(6), 3063. https://doi.org/10.3390/ijms22063063