
Intracerebral Administration of a Ligand-ASO Conjugate Selectively Reduces α-Synuclein Accumulation in Monoamine Neurons of Double Mutant Human A30P*A53T*α-Synuclein Transgenic Mice

 , ,
, , 
Abstract
1. Introduction
2. Results
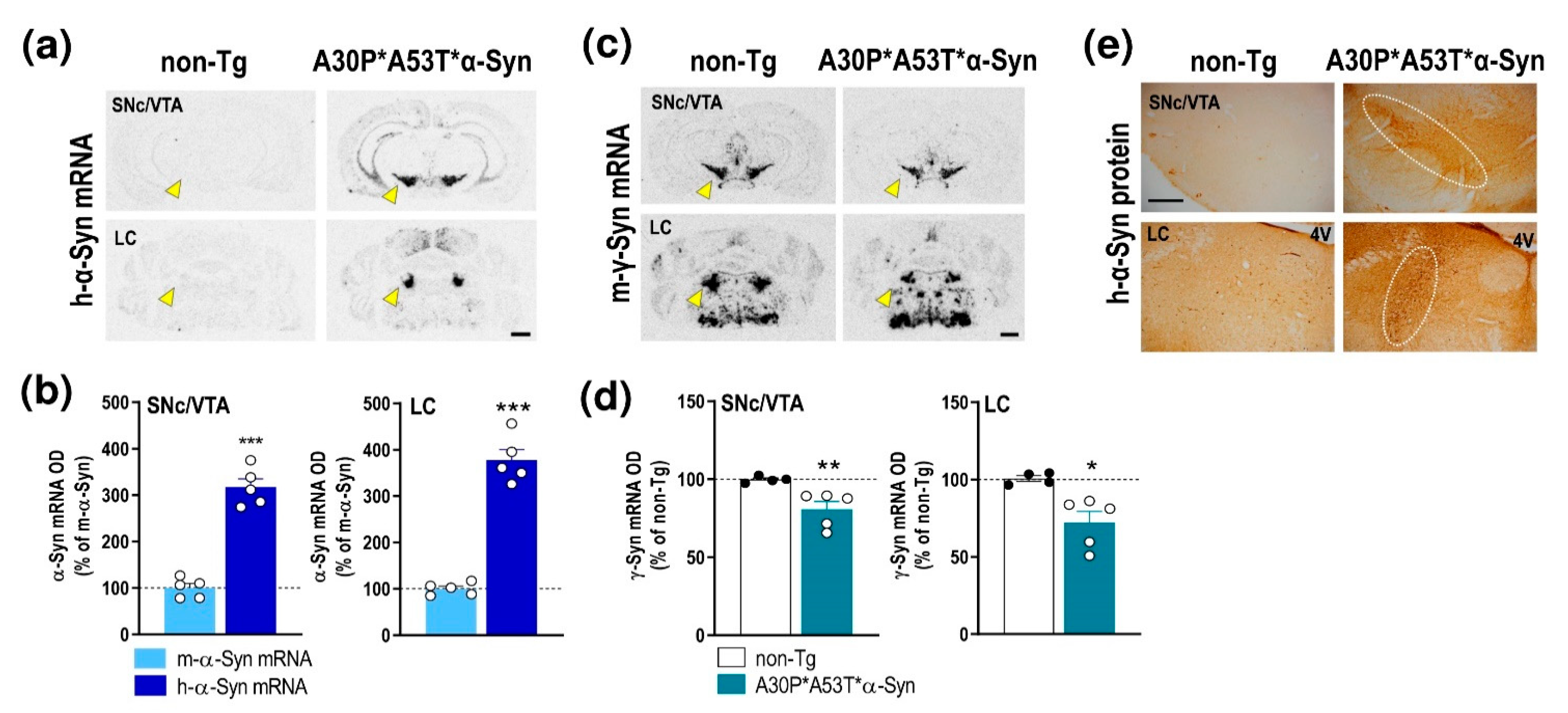
2.1. α-Syn Expression Profile in Brain Areas of A30P*A53T*α-Syn Transgenic Mice
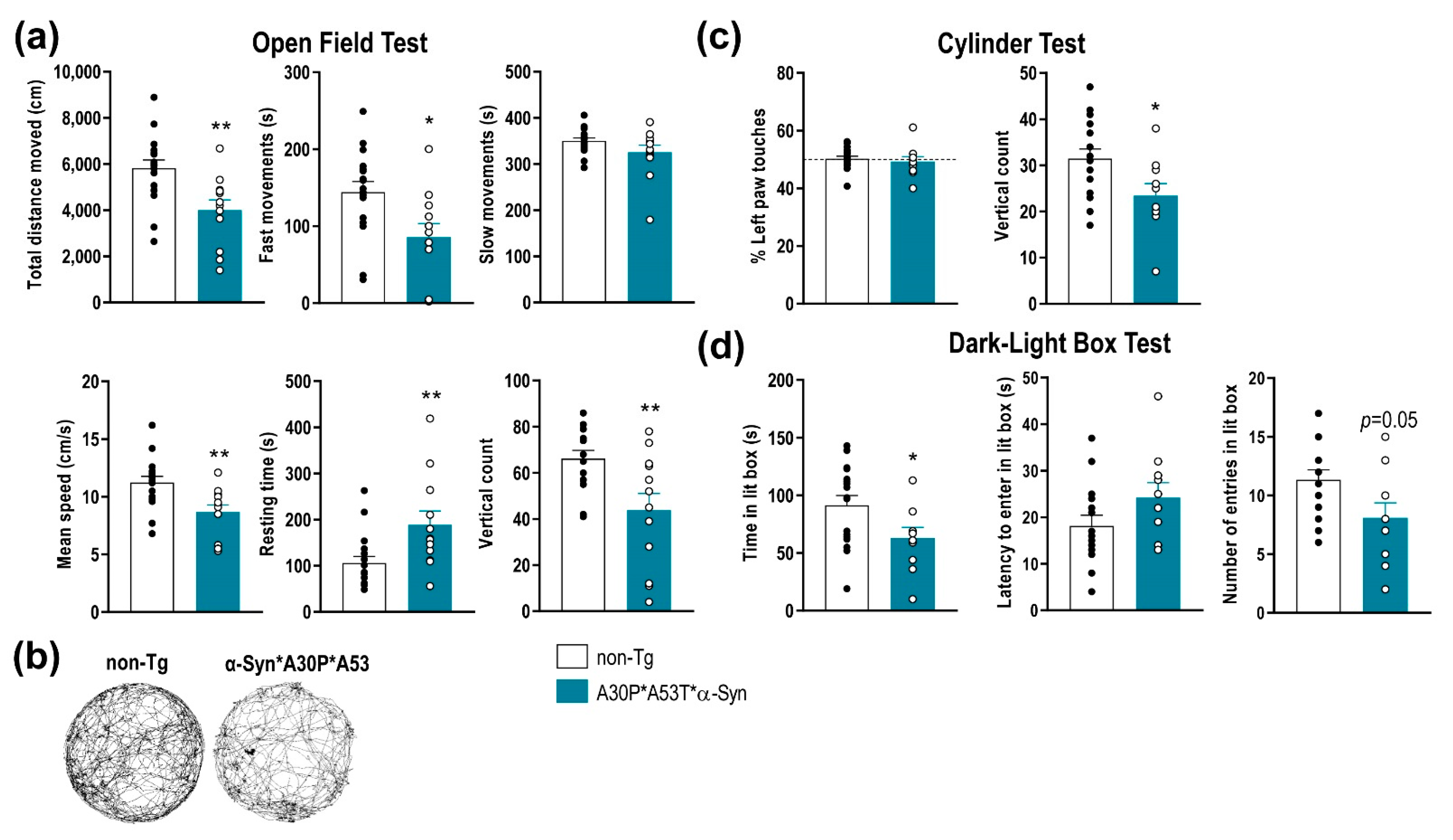
2.2. A30P*A53T*α-Syn Transgenic Mice Show Motor Deficits and an Anxiety-Like Phenotype
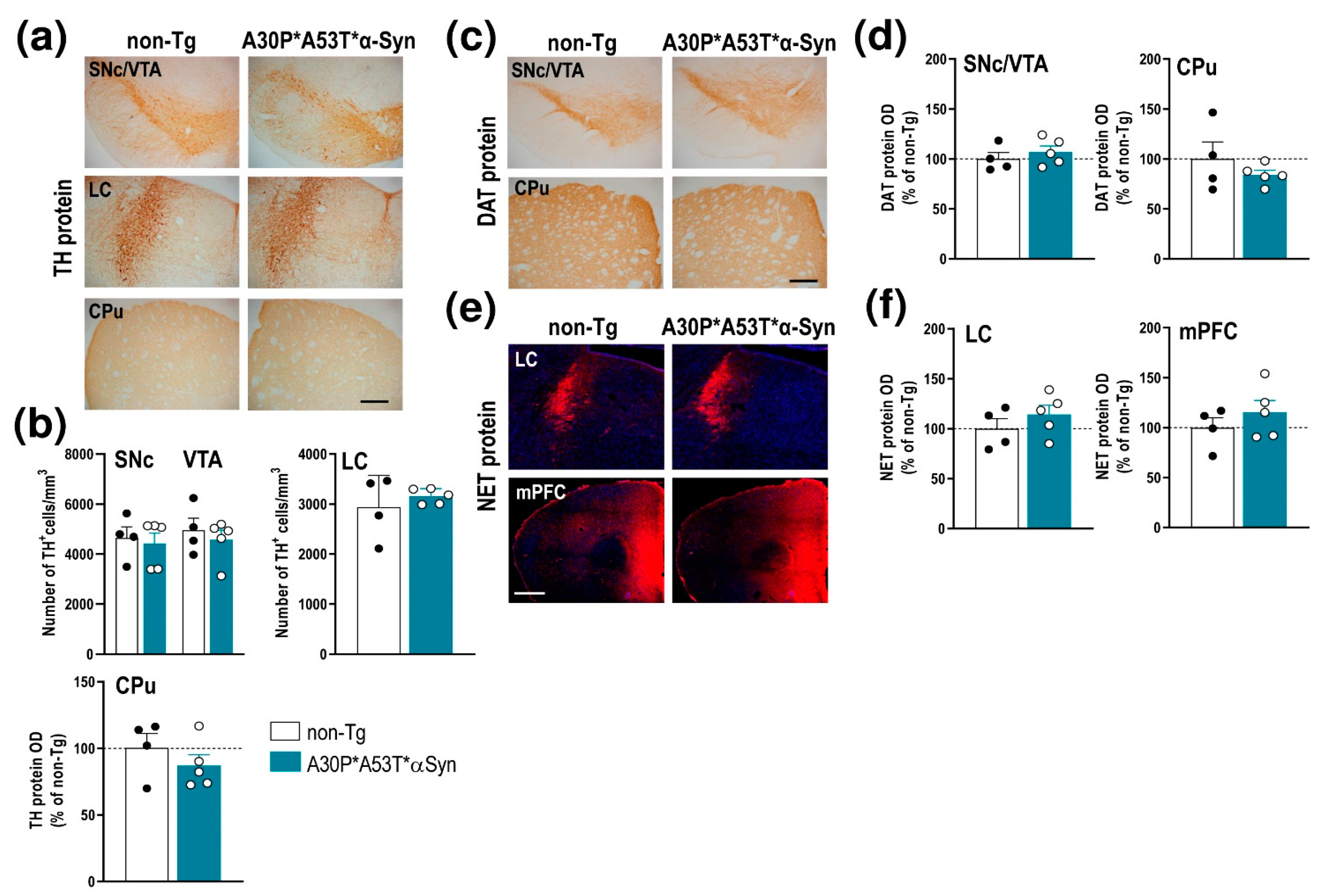
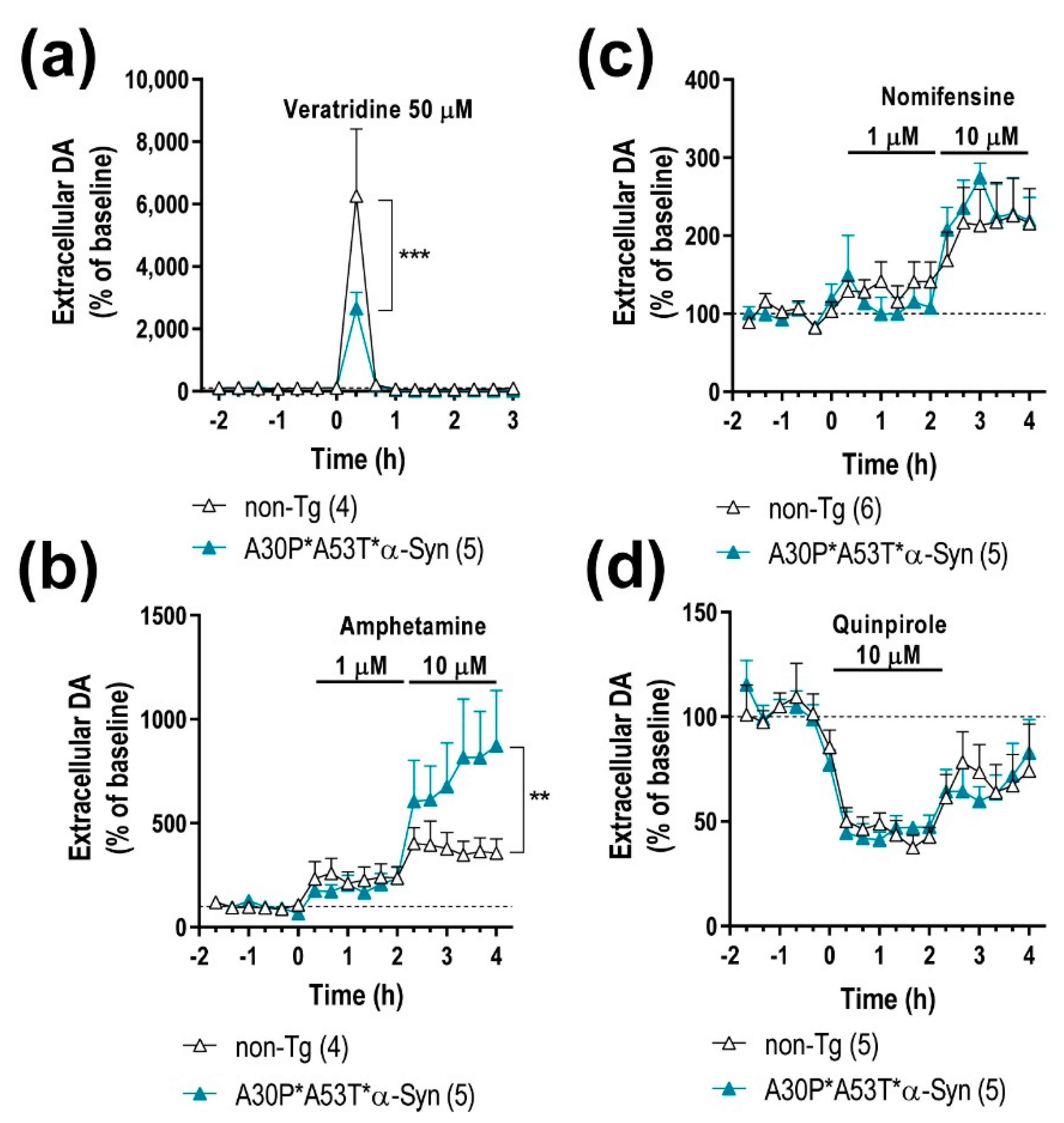
2.3. A30P*A53T*α-Syn Overexpression in TH+ Neurons Impairs DA Neurotransmission in the Nigrostriatal Pathway
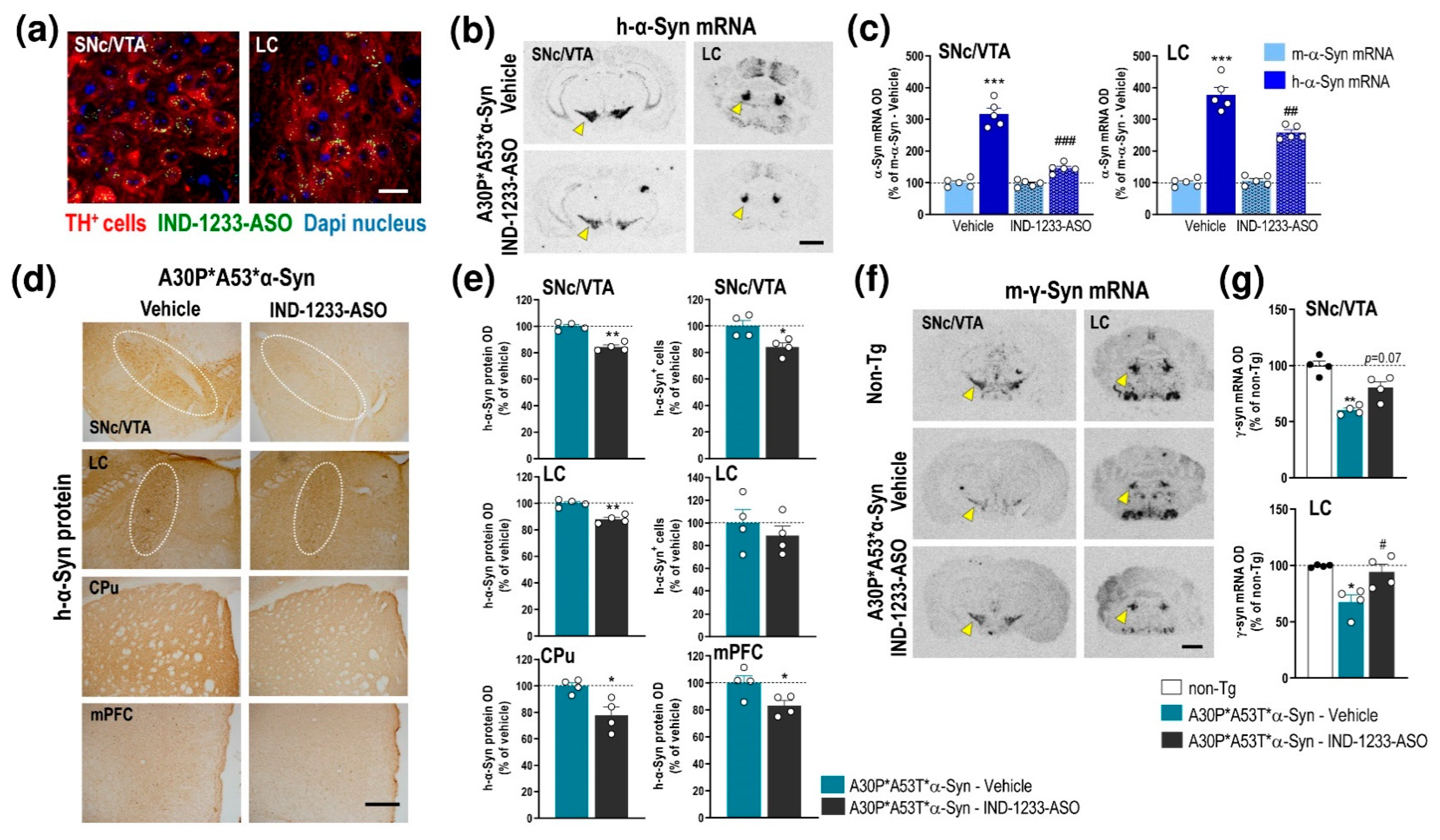
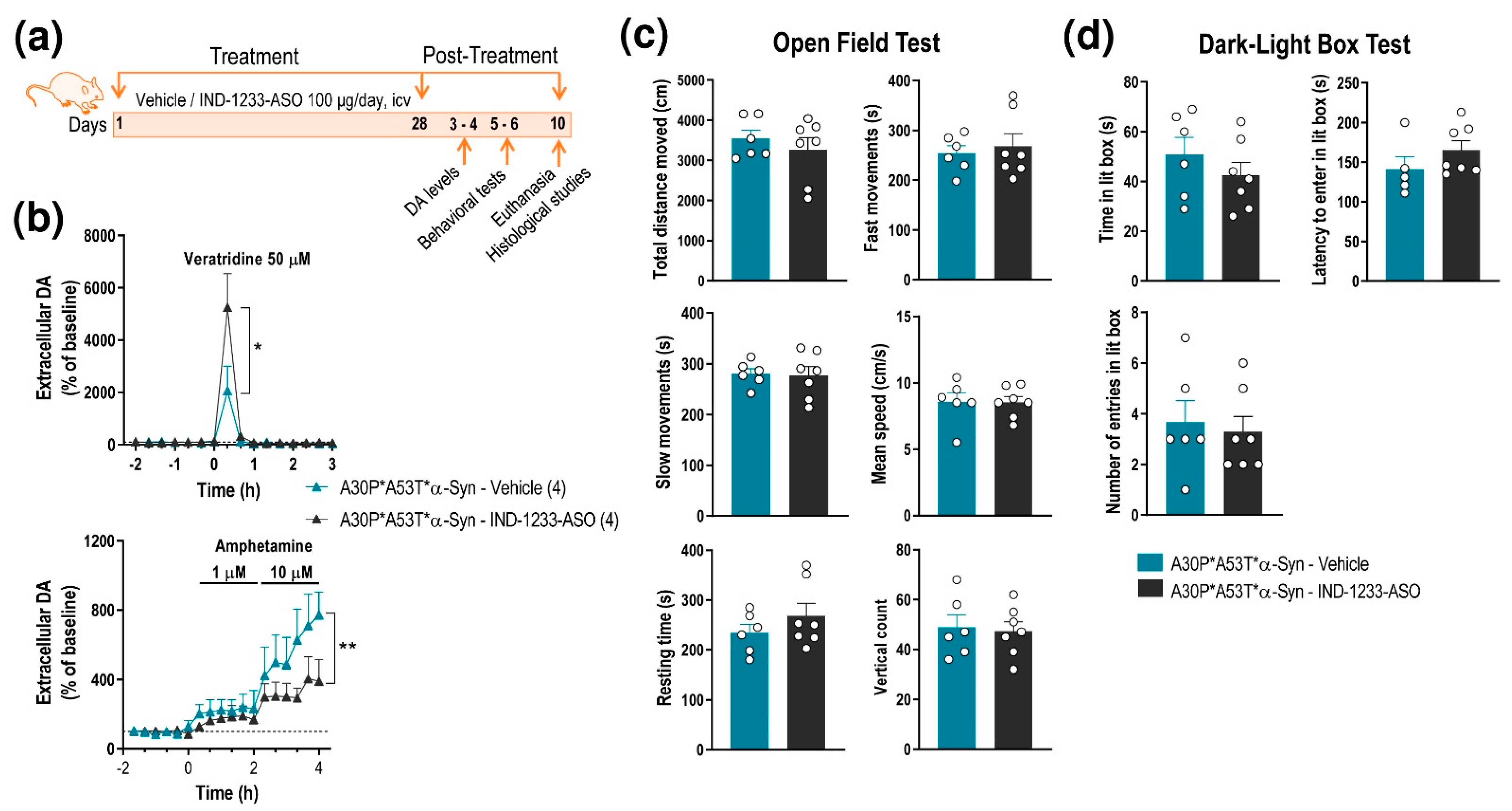
2.4. ASO Therapy Reduces the Accumulation of Human α-Syn and Normalizes DA Neurotransmission Deficits but Not the Behavioral Phenotype in A30P*A53T*α-Syn Transgenic Mice
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Conjugated Antisense Oligonucleotide
4.3. Mouse Treatments
4.4. In Situ Hybridization
4.5. Immunohistochemistry and Immunofluorescence
4.6. Confocal Fluorescence Microscopy
4.7. In Vivo Microdialysis
4.8. Behavioral Testing
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Braak, H.; Del Tredici, K.; Bohl, J.; Bratzke, H.; Braak, E. Pathological changes in the parahippocampal region in select non-Alzheimer’s dementias. Ann. N. Y. Acad. Sci. 2000, 911, 221–239. [Google Scholar] [CrossRef]
- Reeve, A.; Simcox, E.; Turnbull, D. Ageing and Parkinson’s disease: Why is advancing age the biggest risk factor? Ageing Res. Rev. 2014, 14, 19–30. [Google Scholar] [CrossRef]
- Braak, H.; Gai, W.P.; Del Tredici, K. Idiopathic Parkinson’s disease: Possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J. Neural Transm. 2003, 110, 517–536. [Google Scholar] [CrossRef]
- Li, J.Y.; Englund, E.; Holton, J.L.; Soulet, D.; Hagell, P.; Lees, A.J.; Lashley, T.; Quinn, N.P.; Rehncrona, S.; Björklund, A.; et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat. Med. 2008, 14, 501–503. [Google Scholar] [CrossRef]
- Desplats, P.; Lee, H.J.; Bae, E.-J.; Patrick, C.; Rockenstein, E.; Crews, L.; Spencer, B.; Masliah, E.; Lee, S.-J. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of -synuclein. Proc. Natl. Acad. Sci. USA 2009, 106, 13010–13015. [Google Scholar] [CrossRef]
- Kordower, J.H.; Brundin, P. Propagation of host disease to grafted neurons: Accumulating evidence. Exp. Neurol. 2009, 220, 224–225. [Google Scholar] [CrossRef] [PubMed]
- Kish, S.J.; Tong, J.; Hornykiewicz, O.; Rajput, A.; Chang, L.J.; Guttman, M.; Furukawa, Y. Preferential loss of serotonin markers in caudate versus putamen in Parkinson’s disease. Brain 2007, 131, 120–131. [Google Scholar] [CrossRef]
- Wilson, H.; Dervenoulas, G.; Pagano, G.; Koros, C.; Yousaf, T.; Picillo, M.; Polychronis, S.; Simitsi, A.; Giordano, B.; Chappell, Z.; et al. Serotonergic pathology and disease burden in the premotor and motor phase of A53T α-synuclein parkinsonism: A cross-sectional study. Lancet Neurol. 2019, 18, 748–759. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef]
- Krüger, R.; Kuhn, W.; Müller, T.; Woitalla, D.; Graeber, M.; Kösel, S.; Przuntek, H.; Epplen, J.T.; Schöls, L.; Riess, O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet. 1998, 18, 106–108. [Google Scholar] [CrossRef] [PubMed]
- George, J.M.; Clayton, D.F. Songbirds, synelfin, and neurodegenerative disease. Neurosci. News 1998, 1, 12–17. [Google Scholar]
- Murphy, D.D.; Rueter, S.M.; Trojanowski, J.Q.; Lee, V.M.Y. Synucleins Are Developmentally Expressed, and α-Synuclein Regulates the Size of the Presynaptic Vesicular Pool in Primary Hippocampal Neurons. J. Neurosci. 2000, 20, 3214–3220. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.; Roy, S. α-Synuclein inhibits intersynaptic vesicle mobility and maintains recycling-pool homeostasis. J. Neurosci. 2012, 32, 10129–10135. [Google Scholar] [CrossRef] [PubMed]
- Gispert, S.; Del Turco, D.; Garrett, L.; Chen, A.; Bernard, D.J.; Hamm-Clement, J.; Korf, H.W.; Deller, T.; Braak, H.; Auburger, G.; et al. Transgenic mice expressing mutant A53T human alpha-synuclein show neuronal dysfunction in the absence of ag-gregate formation. Mol. Cell Neurosci. 2003, 24, 419–429. [Google Scholar] [CrossRef]
- Giasson, B.I.; Duda, J.E.; Quinn, S.M.; Zhang, B.; Trojanowski, J.Q.; Lee, V.M.-Y. Neuronal α-Synucleinopathy with Severe Movement Disorder in Mice Expressing A53T Human α-Synuclein. Neuron 2002, 34, 521–533. [Google Scholar] [CrossRef]
- Emmer, K.L.; Waxman, E.A.; Covy, J.P.; Giasson, B.I. E46K Human α-Synuclein Transgenic Mice Develop Lewy-like and Tau Pathology Associated with Age-dependent, Detrimental Motor Impairment. J. Biol. Chem. 2011, 286, 35104–35118. [Google Scholar] [CrossRef]
- Ikeda, M.; Kawarabayashi, T.; Harigaya, Y.; Sasaki, A.; Yamada, S.; Matsubara, E.; Murakami, T.; Tanaka, Y.; Kurata, T.; Wuhua, X.; et al. Motor impairment and aberrant production of neurochemicals in human α-synuclein A30P+A53T transgenic mice with α-synuclein pathology. Brain Res. 2009, 1250, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.K.; Stirling, W.; Xu, Y.; Xu, X.; Qui, D.; Mandir, A.S.; Dawson, T.M.; Copeland, N.G.; Jenkins, N.A.; Price, D.L. Human alpha-synuclein-harboring familial Parkinson’s disease-linked Ala-53 --> Thr mutation causes neurodegenerative disease with alpha-synuclein aggregation in transgenic mice. Proc. Natl. Acad. Sci. USA 2002, 25, 8968–8973. [Google Scholar] [CrossRef]
- Oaks, A.W.; Frankfurt, M.; Finkelstein, D.I.; Sidhu, A. Age-dependent effects of A53T alpha-synuclein on behavior and do-paminergic function. PLoS ONE 2013, 8, e60378. [Google Scholar] [CrossRef]
- Richfield, E.K.; Thiruchelvam, M.J.; Cory-Slechta, D.A.; Wuertzerc, C.; Gainetdinov, R.R.; Caron, M.G.; Di Monte, D.A.; Federoff, H.J. Behavioral and Neurochemical Effects of Wild-Type and Mutated Human α-Synuclein in Transgenic Mice. Exp. Neurol. 2002, 175, 35–48. [Google Scholar] [CrossRef]
- Thiruchelvam, M.J.; Powers, J.M.; Cory-Slechta, D.A.; Richfield, E.K. Risk factors for dopaminergic neuron loss in human α-synuclein transgenic mice. Eur. J. Neurosci. 2004, 19, 845–854. [Google Scholar] [CrossRef]
- Kilpeläinen, T.; Julku, U.H.; Svarcbahs, R.; Myöhänen, T.T. Behavioural and dopaminergic changes in double mutated human A30P*A53T alpha-synuclein transgenic mouse model of Parkinson’s disease. Sci. Rep. 2019, 9, 17382. [Google Scholar] [CrossRef]
- Sardi, S.P.; Cedarbaum, J.M.; Brundin, P. Targeted Therapies for Parkinson’s Disease: From Genetics to the Clinic. Mov. Disord. 2018, 33, 684–696. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.; Melrose, H.; Bumcrot, D.; Hope, A.; Zehr, C.; Lincoln, S.; Braithwaite, A.; Heather, M.; Ogholikhan, S.; Hinkle, K.; et al. In vivo silencing of alpha-synuclein using naked siRNA. Mol. Neurodegener. 2008, 3, 19. [Google Scholar] [CrossRef]
- Junn, E.; Lee, K.W.; Jeong, B.S.; Chan, T.W.; Im, J.Y.; Mouradian, M.M. Repression of -synuclein expression and toxicity by microRNA-7. Proc. Natl. Acad. Sci. USA 2009, 106, 13052–13057. [Google Scholar] [CrossRef] [PubMed]
- McCormack, A.L.; Mak, S.K.; Henderson, J.M.; Bumcrot, D.; Farrer, M.J.; Di Monte, D.A. Alpha-synuclein suppression by targeted small interfering RNA in the primate substantia nigra. PLoS ONE 2010, 5, e12122. [Google Scholar] [CrossRef] [PubMed]
- Zharikov, A.; Bai, Q.; De Miranda, B.R.; Van Laar, A.; Greenamyre, J.T.; Burton, E.A. Long-term RNAi knockdown of α-synuclein in the adult rat substantia nigra without neurodegeneration. Neurobiol. Dis. 2019, 125, 146–153. [Google Scholar] [CrossRef]
- Cole, T.; Paumier, K.; Zhao, H.; Weinhofen, A.; Kordasiewicz, H.; Swyze, E. Snca targeted antisense oligonucleotides mediate progression of pathological deposition in alpha synuclein rodent transmission models of Parkinson’s disease. Neurology 2016, 86, P6.239. [Google Scholar]
- Helmschrodt, C.; Höbel, S.; Schöniger, S.; Bauer, A.; Bonicelli, J.; Gringmuth, M.; Fietz, S.A.; Aigner, A.; Richter, A.; Richter, F. Polyethylenimine Nanoparticle-Mediated siRNA Delivery to Reduce α-Synuclein Expression in a Model of Parkinson’s Disease. Mol. Ther. Nucleic Acids 2017, 9, 57–68. [Google Scholar] [CrossRef]
- Nakamori, M.; Junn, E.; Mochizuki, H.; Mouradian, M.M. Nucleic acid-based therapeutics for Parkinson’s disease. Neutherapeutics 2019, 16, 287. [Google Scholar] [CrossRef]
- Spencer, B.; Trinh, I.; Rockenstein, E.; Mante, M.; Florio, J.; Adame, A.; El-Agnaf, O.M.A.; Kim, C.; Masliah, E.; Rissman, R.A. Systemic peptide mediated delivery of a siRNA targeting a-synuclein in the CNS ameliorates the neurodegenerative process in a transgenic model of Lewy body disease. Neurobiol. Dis. 2019, 127, 163–177. [Google Scholar] [CrossRef] [PubMed]
- Bortolozzi, A.; Castañé, A.; Semakova, J.; Santana, N.; Alvarado, G.; Cortés, R.; Ferrés-Coy, A.; Fernández, G.; Carmona, M.C.; Toth, M.; et al. Selective siRNA-mediated suppression of 5-HT1A autoreceptors evokes strong anti-depressant-like effects. Mol. Psychiatry 2012, 17, 612–623. [Google Scholar] [CrossRef]
- Ferrés-Coy, A.; Galofré, M.; Pilar-Cuéllar, F.; Vidal, R.; Paz, V.; Ruiz-Bronchal, E.; Campa, L.; Pazos, A.; Caso, J.R.; Leza, J.C.; et al. Therapeutic antidepressant potential of a conjugated siRNA silencing the serotonin transporter after intranasal administration. Mol. Psychiatry 2016, 21, 328–338. [Google Scholar] [CrossRef]
- Alarcón-Arís, D.; Recasens, A.; Galofré, M.; Carballo-Carbajal, I.; Zacchi, N.; Ruiz-Bronchal, E.; Pavia-Collado, R.; Chica, R.; Ferrés-Coy, A.; Santos, M.; et al. Selective α-synuclein knockdown in monoamine neurons by intranasal oligonucleotide de-livery: Potential therapy for Parkinson’s disease. Mol. Ther. 2018, 26, 550–567. [Google Scholar] [CrossRef] [PubMed]
- Fullana, M.N.; Ferrés-Coy, A.; Ortega, J.E.; Ruiz-Bronchal, E.; Paz, V.; Meana, J.J.; Artigas, F.; Bortolozzi, A. Selective Knockdown of TASK3 Potassium Channel in Monoamine Neurons: A New Therapeutic Approach for Depression. Mol. Neurobiol. 2018, 56, 3038–3052. [Google Scholar] [CrossRef]
- Alarcón-Arís, D.; Pavia-Collado, R.; Miquel-Rio, L.; Coppola-Segovia, V.; Ferrés-Coy, A.; Ruiz-Bronchal, E.; Galofré, M.; Paz, V.; Campa, L.; Revilla, R.; et al. Anti-α-synuclein ASO delivered to monoamine neurons prevents α-synuclein accumulation in a Parkinson’s disease-like mouse model and in monkeys. EBioMedicine 2020, 59, 102944. [Google Scholar] [CrossRef] [PubMed]
- Fernagut, P.-O.; Chesselet, M.-F. Alpha-synuclein and transgenic mouse models. Neurobiol. Dis. 2004, 17, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Lengyel, K.; Pieschl, R.; Strong, T.; Molski, T.; Mattson, G.; Lodge, N.J.; Li, Y.-W. Ex vivo assessment of binding site occupancy of monoamine reuptake inhibitors: Methodology and biological significance. Neuropharmacology 2008, 55, 63–70. [Google Scholar] [CrossRef]
- Javitch, J.; Strittmatter, S.; Snyder, S. Differential visualization of dopamine and norepinephrine uptake sites in rat brain using [3H]mazindol autoradiography. J. Neurosci. 1985, 5, 1513–1521. [Google Scholar] [CrossRef] [PubMed]
- Oaks, A.W.; Sidhu, A. Synuclein modulation of monoamine transporters. FEBS Lett. 2011, 585, 1001–1006. [Google Scholar] [CrossRef]
- Nishina, K.; Piao, W.; Yoshida-Tanaka, K.; Sujino, Y.; Nishina, T.; Yamamoto, T.; Nitta, K.; Yoshioka, K.; Kuwahara, H.; Yasuhara, H.; et al. DNA/RNA heteroduplex oligonucleotide for highly efficient gene silencing. Nat. Commun. 2015, 6, 7969. [Google Scholar] [CrossRef] [PubMed]
- Gorbatyuk, O.S.; Li, S.; Nash, K.; Gorbatyuk, M.; Lewin, A.S.; Sullivan, L.F.; Mandel, R.J.; Chen, W.; Meyers, C.; Manfredsson, F.P.; et al. In Vivo RNAi-Mediated α-Synuclein Silencing Induces Nigrostriatal Degeneration. Mol. Ther. 2010, 18, 1450–1457. [Google Scholar] [CrossRef]
- Khodr, C.E.; Sapru, M.K.; Pedapati, J.; Han, Y.; West, N.C.; Kells, A.P.; Bankiewicz, K.S.; Bohn, M.C. An α-synuclein AAV gene silencing vector ameliorates a behavioral deficit in a rat model of Parkinson’s disease, but displays toxicity in dopamine neurons. Brain Res. 2011, 1395, 94–107. [Google Scholar] [CrossRef] [PubMed]
- Collier, T.J.; Redmond, D.E., Jr.; Steece-Collier, K.; Lipton, J.W.; Manfredsson, F.P. Is alpha-synuclein loss-of-function a con-tributor to parkinsonian pathology? Evidence from non-human primates. Front. Neurosci. 2016, 10, 12. [Google Scholar] [CrossRef] [PubMed]
- Benskey, M.J.; Sellnow, R.C.; Sandoval, I.M.; Sortwell, C.E.; Lipton, J.W.; Manfredsson, F.P. Silencing Alpha Synuclein in Mature Nigral Neurons Results in Rapid Neuroinflammation and Subsequent Toxicity. Front. Mol. Neurosci. 2018, 11, 36. [Google Scholar] [CrossRef]
- Hardee, C.L.; Arévalo-Soliz, L.M.; Hornstein, B.D.; Zechiedrich, L. Advances in non-viral DNA vectors for gene therapy. Genes 2017, 8, 65. [Google Scholar] [CrossRef]
- Imbert, M.; Dias-Florencio, G.; Goyenvalle, A. Viral Vector-Mediated Antisense Therapy for Genetic Diseases. Genes 2017, 8, 51. [Google Scholar] [CrossRef] [PubMed]
- Nyamay’Antu, A.; Dumont, M.; Kedinger, V.; Erbacher, P. Non-Viral Vector Mediated Gene Delivery: The Outsider to Watch Out For in Gene Therapy. Cell Gene Ther. Insights 2019, 5, 51–57. [Google Scholar] [CrossRef]
- Smith, C.E.; Zain, R. Therapeutic Oligonucleotides: State of the Art. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 605–630. [Google Scholar] [CrossRef]
- Juliano, R.L. The delivery of therapeutic oligonucleotides. Nucleic Acids Res. 2016, 44, 6518–6548. [Google Scholar] [CrossRef]
- Doxakis, E. Therapeutic antisense oligonucleotides for movement disorders. Med. Res. Rev. 2020, 1–33. [Google Scholar] [CrossRef]
- Nakagawa, O.; Ming, X.; Huang, L.; Juliano, R.L. Targeted Intracellular Delivery of Antisense Oligonucleotides via Conjugation with Small-Molecule Ligands. J. Am. Chem. Soc. 2010, 132, 8848–8849. [Google Scholar] [CrossRef]
- Willibald, J.; Harder, J.; Sparrer, K.; Conzelmann, K.-K.; Carell, T. Click-Modified Anandamide siRNA Enables Delivery and Gene Silencing in Neuronal and Immune Cells. J. Am. Chem. Soc. 2012, 134, 12330–12333. [Google Scholar] [CrossRef] [PubMed]
- Kurz, A.; Double, K.L.; Lastres-Becker, I.; Tozzi, A.; Tantucci, M.; Bockhart, V.; Bonin, M.; García-Arencibia, M.; Nuber, S.; Schlaudraff, F.; et al. A53T-Alpha-Synuclein Overexpression Impairs Dopamine Signaling and Striatal Synaptic Plasticity in Old Mice. PLoS ONE 2010, 5, e11464. [Google Scholar] [CrossRef] [PubMed]
- Abeliovich, A.; Schmitz, Y.; Fariñas, I.; Choi-Lundberg, D.; Ho, W.H.; Castillo, P.E.; Shinsky, N.; Verdugo, J.M.G.; Armanini, M.; Ryan, A.; et al. Mice Lacking α-Synuclein Display Functional Deficits in the Nigrostriatal Dopamine System. Neuron 2000, 25, 239–252. [Google Scholar] [CrossRef]
- Sidhu, A.; Wersinger, C.; Vernier, P. Does α-synuclein modulate dopaminergic synaptic content and tone at the synapse? FASEB J. 2004, 18, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Wersinger, C.; Prou, D.; Vernier, P.; Niznik, H.B.; Sidhu, A. Mutations in the lipid-binding domain of α-synuclein confer overlapping, yet distinct, functional properties in the regulation of dopamine transporter activity. Mol. Cell. Neurosci. 2003, 24, 91–105. [Google Scholar] [CrossRef]
- Lotharius, J.; Brundin, P. Pathogenesis of Parkinson’s disease: Dopamine, vesicles and alpha-synuclein. Nat. Rev. Neurosci. 2002, 3, 932–942. [Google Scholar] [CrossRef]
- Lotharius, J.; Barg, S.; Wiekop, P.; Lundberg, C.; Raymon, H.K.; Brundin, P. Effect of Mutant α-Synuclein on Dopamine Homeostasis in a New Human Mesencephalic Cell Line. J. Biol. Chem. 2002, 277, 38884–38894. [Google Scholar] [CrossRef]
- Franklin, K.B.J.; Paxinos, G. The Mouse Brain in Stereotaxic Coordinates, 3rd ed.; Academic Press: San Diego, CA, USA, 2008; pp. 1–256. [Google Scholar]
- Morton, D.B.; Griffiths, P.H. Guidelines on the recognition of pain, distress and discomfort in experimental animals and an hypothesis for assessment. Vet. Rec. 1985, 116, 431–436. [Google Scholar] [CrossRef]
- Li, Y.; Lium, W.; Oo, T.F.; Wang, L.; Tang, Y.; Jackson-Lewis, V.; Zhou, C.; Geghman, K.; Bogdanov, M.; Przedborski, S.; et al. Mutant LRRK2(R1441G) BAC transgenic mice recapitulate cardinal features of Parkinson’s disease. Nat. Neurosci. 2009, 12, 826–828. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mice | Experimental Conditions | Baseline DA | Baseline DOPAC |
|---|---|---|---|
| non-Tg | aCSF | 10.3 ± 1.6 (15) | 0.8 ± 0.1 (15) |
| aCSF + DMSO 1% | 7.2 ± 2.2 (4) | 1.6 ± 0.4 (4) | |
| A30P*A53T*α-Syn | aCSF | 8.5 ± 1.2 (15) | 1.1 ± 0.1 (15) |
| aCSF + DMSO 1% | 6.6 ± 1.7 (5) | 1.8 ± 0.5 (5) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pavia-Collado, R.; Cóppola-Segovia, V.; Miquel-Rio, L.; Alarcón-Aris, D.; Rodríguez-Aller, R.; Torres-López, M.; Paz, V.; Ruiz-Bronchal, E.; Campa, L.; Artigas, F.; et al. Intracerebral Administration of a Ligand-ASO Conjugate Selectively Reduces α-Synuclein Accumulation in Monoamine Neurons of Double Mutant Human A30P*A53T*α-Synuclein Transgenic Mice. Int. J. Mol. Sci. 2021, 22, 2939. https://doi.org/10.3390/ijms22062939
Pavia-Collado R, Cóppola-Segovia V, Miquel-Rio L, Alarcón-Aris D, Rodríguez-Aller R, Torres-López M, Paz V, Ruiz-Bronchal E, Campa L, Artigas F, et al. Intracerebral Administration of a Ligand-ASO Conjugate Selectively Reduces α-Synuclein Accumulation in Monoamine Neurons of Double Mutant Human A30P*A53T*α-Synuclein Transgenic Mice. International Journal of Molecular Sciences. 2021; 22(6):2939. https://doi.org/10.3390/ijms22062939
Chicago/Turabian StylePavia-Collado, Rubén, Valentín Cóppola-Segovia, Lluís Miquel-Rio, Diana Alarcón-Aris, Raquel Rodríguez-Aller, María Torres-López, Verónica Paz, Esther Ruiz-Bronchal, Leticia Campa, Francesc Artigas, and et al. 2021. "Intracerebral Administration of a Ligand-ASO Conjugate Selectively Reduces α-Synuclein Accumulation in Monoamine Neurons of Double Mutant Human A30P*A53T*α-Synuclein Transgenic Mice" International Journal of Molecular Sciences 22, no. 6: 2939. https://doi.org/10.3390/ijms22062939
APA StylePavia-Collado, R., Cóppola-Segovia, V., Miquel-Rio, L., Alarcón-Aris, D., Rodríguez-Aller, R., Torres-López, M., Paz, V., Ruiz-Bronchal, E., Campa, L., Artigas, F., Montefeltro, A., Revilla, R., & Bortolozzi, A. (2021). Intracerebral Administration of a Ligand-ASO Conjugate Selectively Reduces α-Synuclein Accumulation in Monoamine Neurons of Double Mutant Human A30P*A53T*α-Synuclein Transgenic Mice. International Journal of Molecular Sciences, 22(6), 2939. https://doi.org/10.3390/ijms22062939