
Incorporating Machine Learning into Established Bioinformatics Frameworks



Abstract
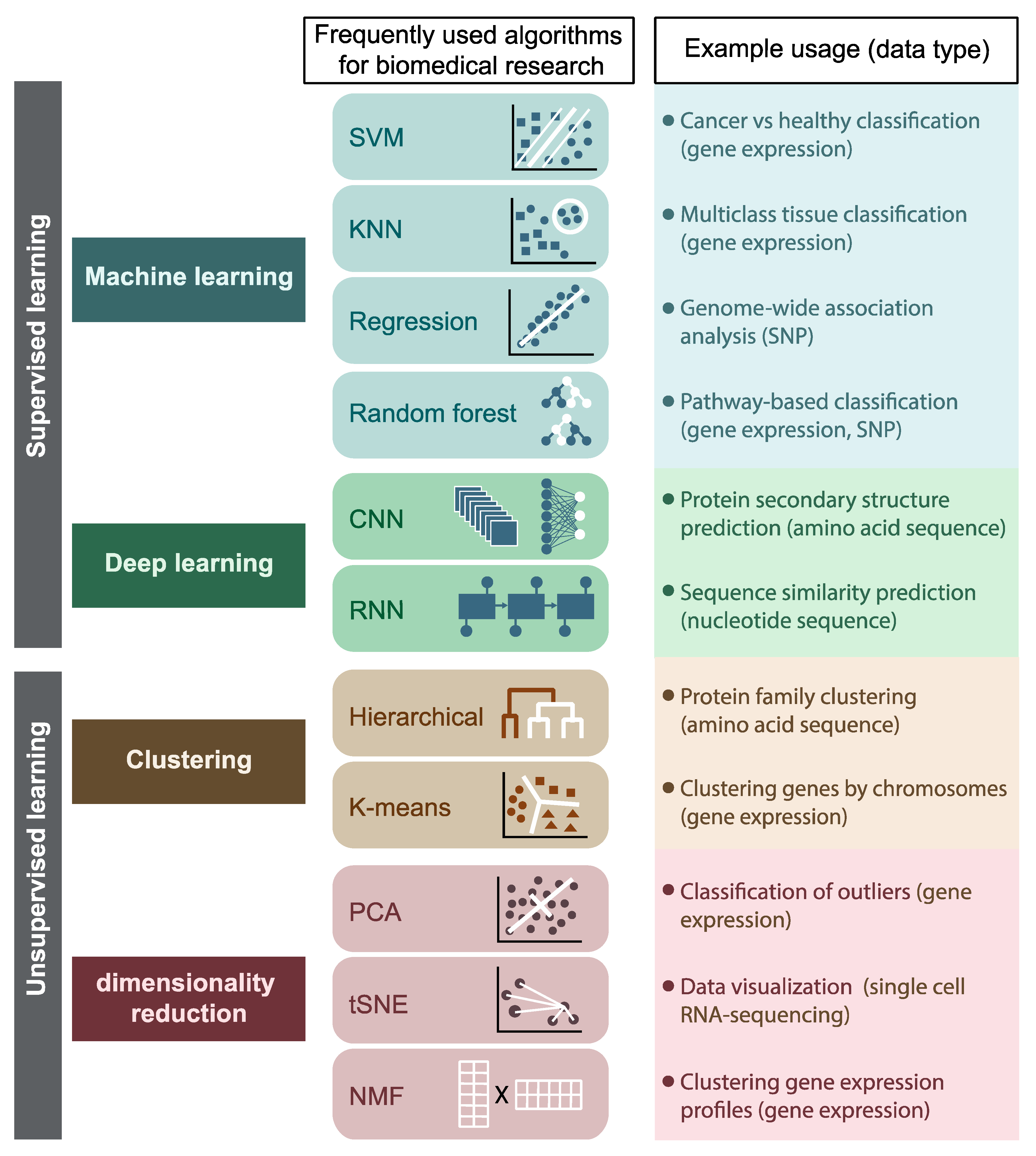
1. Introduction
1.1. Integrating Machine-Learning into Molecular Evolution Research
1.2. Integrating Machine-Learning with Protein Structure Analysis
1.3. Integrating ML into Systems Biology
1.4. Integrating ML with Genomics and Biomarker Analysis for Disease Research
1.5. Key Challenges and Future Directions

{kind=link}
{kind=link}
| Problem | Bottleneck | Example Solutions | Potential Integrated ML/DL and Bioinformatics Solutions |
|---|---|---|---|
| Small and dependent datasets | Data availability | Restricting the number of parameters [27,190] | Neural network architectures for small and sparse datasets |
| Separating training and test sets by phylogenetic similarity [27] | Methods to evaluate data dependency by protein and sequence similarities | ||
| Biological sequence representation | Methodological | NLP with neural networks-based modeling [191,192,193,194] | Incorporating amino acid substitution and codon usage matrices to representation frameworks |
| Incorporating conserved domain databases to the training framework | |||
| Incorporation of different data types | Methodological | Integration of multi-omics datasets through existing network topologies | |
| Reproducibility | Acceptance | Documentation and deposition of the processed data [195] | - |
| Benchmarking of the processing pipeline and optimized parameters [196] | - | ||
| Interpretability | Acceptance | Incorporation of established bioinformatic methods and databases with ML and DL frameworks [128,196] | |
| Generation of interpretable DL models [197,198,199] | |||
2. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pevsner, J. Funtional Genomics. In Bioinformatics and Functional Genomics; John Wiley & Sons: Hoboken, NJ, USA, 2015; ISBN 9781118581780. [Google Scholar]
- Ayyildiz, D.; Piazza, S. Introduction to Bioinformatics. In Methods in Molecular Biology; Oxford University Press: Oxford, UK, 2019. [Google Scholar]
- Wodarz, D.; Komarova, N. Computational Biology of Cancer; World Scientific: Singapore, 2005. [Google Scholar]
- Lecun, Y.; Bengio, Y.; Hinton, G. Deep Learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef]
- Butler, K.T.; Davies, D.W.; Cartwright, H.; Isayev, O.; Walsh, A. Machine learning for molecular and materials science. Nat. Cell Biol. 2018, 559, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Schuster, M.; Chen, Z.; Le, Q.V.; Norouzi, M.; Macherey, W.; Krikun, M.; Cao, Y.; Gao, Q.; Macherey, K.; et al. Google’s NMT. arXiv 2016, arXiv:1609.08144v2. [Google Scholar]
- Bhattacharjee, A.; Bayzid, M.S. Machine Learning Based Imputation Techniques for Estimating Phylogenetic Trees from Incomplete Distance Matrices. BMC Genom. 2020, 21, 497. [Google Scholar] [CrossRef]
- Abadi, S.; Avram, O.; Rosset, S.; Pupko, T.; Mayrose, I. ModelTeller: Model Selection for Optimal Phylogenetic Reconstruction Using Machine Learning. Mol. Biol. Evol. 2020, 37, 3338–3352. [Google Scholar] [CrossRef]
- Suvorov, A.; Hochuli, J.; Schrider, D.R. Accurate Inference of Tree Topologies from Multiple Sequence Alignments Using Deep Learning. Syst. Biol. 2019, 69, 221–233. [Google Scholar] [CrossRef]
- Azer, E.S.; Ebrahimabadi, M.H.; Malikić, S.; Khardon, R.; Sahinalp, S.C. Tumor Phylogeny Topology Inference via Deep Learning. iScience 2020, 23, 101655. [Google Scholar] [CrossRef]
- Jafari, R.; Javidi, M.M.; Kuchaki Rafsanjani, M. Using Deep Reinforcement Learning Approach for Solving the Multiple Sequence Alignment Problem. SN Appl. Sci. 2019, 1, 592. [Google Scholar] [CrossRef]
- Yu, X. Introduction to Evolutionary Algorithms; Springer: Berlin/Heidelberg, Germany, 2010. [Google Scholar]
- Fortin, F.A.; De Rainville, F.M.; Gardner, M.A.; Parizeau, M.; Gagńe, C. DEAP: Evolutionary Algorithms Made Easy. J. Mach. Learn. Res. 2012, 13, 2171–2175. [Google Scholar]
- Whitley, D. A genetic algorithm tutorial. Stat. Comput. 1994, 4, 65–85. [Google Scholar] [CrossRef]
- Pal, S.K.; Bandyopadhyay, S.; Ray, S.S. Evolutionary Computation in Bioinformatics: A Review. IEEE Trans. Syst. Man Cybern. Part C Appl. Rev. 2006, 36, 601–615. [Google Scholar] [CrossRef]
- Sivanandam, S.N.; Deepa, S.N. Introduction to Genetic Algorithms; Springer: Berlin/Heidelberg, Germany, 2008; ISBN 9783540731894. [Google Scholar]
- Audet, C.; Hare, W. Genetic Algorithms. In Springer Series in Operations Research and Financial Engineering; Springer: Berlin/Heidelberg, Germany, 2017. [Google Scholar]
- Strodthoff, N.; Wagner, P.; Wenzel, M.; Samek, W. UDSMProt: Universal deep sequence models for protein classification. Bioinformatics 2020, 36, 2401–2409. [Google Scholar] [CrossRef] [PubMed]
- Liu, B. BioSeq-Analysis: A platform for DNA, RNA and protein sequence analysis based on machine learning approaches. Briefings Bioinform. 2017, 20, 1280–1294. [Google Scholar] [CrossRef]
- Gussow, A.B.; Auslander, N.; Faure, G.; Wolf, Y.I.; Zhang, F.; Koonin, E.V. Genomic Determinants of Pathogenicity in SARS-CoV-2 and Other Human Coronaviruses. Proc. Natl. Acad. Sci. USA 2020, 117, 15193–15199. [Google Scholar] [CrossRef]
- Auslander, N.; Wolf, Y.I.; Shabalina, S.A.; Koonin, E.V. A unique insert in the genomes of high-risk human papillomaviruses with a predicted dual role in conferring oncogenic risk. F1000Research 2019, 8, 1000. [Google Scholar] [CrossRef]
- Gussow, A.B.; Auslander, N.; Wolf, Y.I.; Koonin, E.V. Prediction of the incubation period for COVID-19 and future virus disease outbreaks. BMC Biol. 2020, 18, 186. [Google Scholar] [CrossRef] [PubMed]
- Abadi, S.; Yan, W.X.; Amar, D.; Mayrose, I. A machine learning approach for predicting CRISPR-Cas9 cleavage efficiencies and patterns underlying its mechanism of action. PLoS Comput. Biol. 2017, 13, e1005807. [Google Scholar] [CrossRef]
- Gussow, A.B.; Park, A.E.; Borges, A.L.; Shmakov, S.A.; Makarova, K.S.; Wolf, Y.I.; Bondy-Denomy, J.; Koonin, E.V. Machine-Learning Approach Expands the Repertoire of Anti-CRISPR Protein Families. Nat. Commun. 2020, 11, 3784. [Google Scholar] [CrossRef] [PubMed]
- Eitzinger, S.; Asif, A.; Watters, K.E.; Iavarone, A.T.; Knott, G.J.; Doudna, J.A.; Minhas, F.; Ul, A.A. Machine Learning Predicts New Anti-CRISPR Proteins. Nucleic Acids Res. 2020, 48, 4698–4708. [Google Scholar] [CrossRef]
- Solis-Reyes, S.; Avino, M.; Poon, A.; Kari, L. An Open-Source k-Mer Based Machine Learning Tool for Fast and Accurate Subtyping of HIV-1 Genomes. PLoS ONE 2018, 13, e0206409. [Google Scholar] [CrossRef]
- Auslander, N.; Gussow, A.B.; Benler, S.; Wolf, Y.I.; Koonin, E.V. Seeker: Alignment-Free Identification of Bacteriophage Genomes by Deep Learning. Nucleic Acids Res. 2020, 48, e121. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Tan, J.; Wu, S.; Li, M.; Xu, C.; Xie, Z.; Zhu, H. PPR-Meta: A tool for identifying phages and plasmids from metagenomic fragments using deep learning. GigaScience 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Song, K.; Deng, C.; Ahlgren, N.A.; Fuhrman, J.A.; Li, Y.; Xie, X.; Poplin, R.; Sun, F. Identifying viruses from metagenomic data using deep learning. Quant. Biol. 2020, 8, 64–77. [Google Scholar] [CrossRef]
- Seo, S.; Oh, M.; Park, Y.; Kim, S. DeepFam: Deep Learning Based Alignment-Free Method for Protein Family Modeling and Prediction. Bioinformatics 2018, 34, i254–i262. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Thakur, V.; Raghava, G.P.S. COPid: Composition Based Protein Identification. In Silico Biol. 2008, 8, 121–128. [Google Scholar]
- Liu, X.L. Deep Recurrent Neural Network for Protein Function Prediction from Sequence. arXiv 2017, arXiv:1701.08318. [Google Scholar]
- Hamid, M.-N.; Friedberg, I. Identifying antimicrobial peptides using word embedding with deep recurrent neural networks. Bioinformmatics 2019, 35, 2009–2016. [Google Scholar] [CrossRef]
- Zacharaki, E.I. Prediction of protein function using a deep convolutional neural network ensemble. PeerJ Comput. Sci. 2017, 3. [Google Scholar] [CrossRef]
- Nguyen, A.; Dosovitskiy, A.; Yosinski, J.; Brox, T.; Clune, J. Synthesizing the Preferred Inputs for Neurons in Neural Networks via Deep Generator Networks. arXiv 2016, arXiv:1605.09304. [Google Scholar]
- Le, N.Q.K.; Yapp, E.K.Y.; Nagasundaram, N.; Yeh, H.-Y. Classifying Promoters by Interpreting the Hidden Information of DNA Sequences via Deep Learning and Combination of Continuous FastText N-Grams. Front. Bioeng. Biotechnol. 2019, 7, 305. [Google Scholar] [CrossRef] [PubMed]
- Umarov, R.K.; Solovyev, V.V. Recognition of Prokaryotic and Eukaryotic Promoters Using Convolutional Deep Learning Neural Networks. PLoS ONE 2017, 12, e0171410. [Google Scholar] [CrossRef]
- Le, N.Q.K.; Ho, Q.-T.; Nguyen, T.-T.-D.; Ou, Y.-Y. A transformer architecture based on BERT and 2D convolutional neural network to identify DNA enhancers from sequence information. Briefings Bioinform. 2021. [Google Scholar] [CrossRef] [PubMed]
- Min, X.; Zeng, W.; Chen, S.; Chen, N.; Chen, T.; Jiang, R. Predicting Enhancers with Deep Convolutional Neural Networks. BMC Bioinform. 2017, 18, 478. [Google Scholar] [CrossRef]
- Xu, Y.; Zhao, X.; Liu, S.; Zhang, W. Predicting Long Non-Coding RNAs through Feature Ensemble Learning. BMC Genom. 2020, 21, 865. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Liu, H.; Zhang, L.; Meng, J. LncRScan-SVM: A Tool for Predicting Long Non-Coding RNAs Using Support Vector Machine. PLoS ONE 2015, 10, e0139654. [Google Scholar] [CrossRef] [PubMed]
- Schneider, H.W.; Raiol, T.; Brigido, M.M.; Walter, M.E.M.T.; Stadler, P.F. A Support Vector Machine Based Method to Distinguish Long Non-Coding RNAs from Protein Coding Transcripts. BMC Genom. 2017, 18, 804. [Google Scholar] [CrossRef]
- Hu, L.; Xu, Z.; Hu, B.; Lu, Z.J. COME: A Robust Coding Potential Calculation Tool for LncRNA Identification and Characterization Based on Multiple Features. Nucleic Acids Res. 2017, 45, e2. [Google Scholar] [CrossRef]
- Zhao, J.; Song, X.; Wang, K. lncScore: Alignment-free identification of long noncoding RNA from assembled novel transcripts. Sci. Rep. 2016, 6, 34838. [Google Scholar] [CrossRef]
- Wen, M.; Cong, P.; Zhang, Z.; Lu, H.; Li, T. DeepMirTar: A Deep-Learning Approach for Predicting Human MiRNA Targets. Bioinformatics 2018, 34, 3781–3787. [Google Scholar] [CrossRef]
- Zheng, X.; Chen, L.; Li, X.; Zhang, Y.; Xu, S.; Huang, X. Prediction of MiRNA Targets by Learning from Interaction Sequences. PLoS ONE 2020, 15, e0232578. [Google Scholar] [CrossRef] [PubMed]
- Mitrofanov, A.; Alkhnbashi, O.S.; Shmakov, S.A.; Makarova, K.S.; Koonin, E.V.; Backofen, R. CRISPRidentify: Identification of CRISPR arrays using machine learning approach. Nucleic Acids Res. 2021, 49, e20. [Google Scholar] [CrossRef]
- Blom, N.; Sicheritz-Pontén, T.; Gupta, R.; Gammeltoft, S.; Brunak, S. Prediction of Post-Translational Glycosylation and Phosphorylation of Proteins from the Amino Acid Sequence. Proteomics 2004, 4, 1633–1649. [Google Scholar] [CrossRef]
- Huang, G.; Li, J. Feature Extractions for Computationally Predicting Protein Post- Translational Modifications. Curr. Bioinform. 2018, 13, 387–395. [Google Scholar] [CrossRef]
- Wang, D.; Liu, D.; Yuchi, J.; He, F.; Jiang, Y.; Cai, S.; Li, J.; Xu, D. MusiteDeep: A deep-learning based webserver for protein post-translational modification site prediction and visualization. Nucleic Acids Res. 2020, 48, W140–W146. [Google Scholar] [CrossRef] [PubMed]
- Duan, G.; Walther, D. The Roles of Post-translational Modifications in the Context of Protein Interaction Networks. PLoS Comput. Biol. 2015, 11, e1004049. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.; Zuo, Y.; Zou, Q. O-GlcNAcPRED-II: An integrated classification algorithm for identifying O-GlcNAcylation sites based on fuzzy undersampling and a K-means PCA oversampling technique. Bioinformatics 2018, 34, 2029–2036. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Thelen, J.J.; Dunker, A.K.; Xu, D. Musite, a Tool for Global Prediction of General and Kinase-specific Phosphorylation Sites. Mol. Cell. Proteom. 2010, 9, 2586–2600. [Google Scholar] [CrossRef] [PubMed]
- Caragea, C.; Sinapov, J.; Silvescu, A.; Dobbs, D.; Honavar, V. Glycosylation site prediction using ensembles of Support Vector Machine classifiers. BMC Bioinform. 2007, 8, 438. [Google Scholar] [CrossRef]
- Luo, F.; Wang, M.; Liu, Y.; Zhao, X.-M.; Li, A. DeepPhos: Prediction of protein phosphorylation sites with deep learning. Bioinformatics 2019, 35, 2766–2773. [Google Scholar] [CrossRef]
- Kotidis, P.; Kontoravdi, C. Harnessing the potential of artificial neural networks for predicting protein glycosylation. Metab. Eng. Commun. 2020, 10, e00131. [Google Scholar] [CrossRef]
- Hameduh, T.; Haddad, Y.; Adam, V.; Heger, Z. Homology Modeling in the Time of Collective and Artificial Intelligence. Comput. Struct. Biotechnol. J. 2020, 18, 3494–3506. [Google Scholar] [CrossRef] [PubMed]
- Torrisi, M.; Pollastri, G.; Le, Q. Deep Learning Methods in Protein Structure Prediction. Comput. Struct. Biotechnol. J. 2020, 18, 1301–1310. [Google Scholar] [CrossRef] [PubMed]
- Shakhnovich, B.E. Protein Structure and Evolutionary History Determine Sequence Space Topology. Genome Res. 2005, 15, 385–392. [Google Scholar] [CrossRef]
- Muhammed, M.T.; Aki-Yalcin, E. Homology Modeling in Drug Discovery: Overview, Current Applications, and Future Perspectives. Chem. Biol. Drug Des. 2019, 93, 12–20. [Google Scholar] [CrossRef]
- Lazaridis, T.; Karplus, M. Effective Energy Functions for Protein Structure Prediction. Curr. Opin. Struct. Biol. 2000, 10, 139–145. [Google Scholar] [CrossRef]
- Snow, C.D.; Sorin, E.J.; Rhee, Y.M.; Pande, V.S. How Well Can Simulation Predict Protein Folding Kinetics and Thermodynamics? Annu. Rev. Biophys. Biomol. Struct. 2005, 34, 43–69. [Google Scholar] [CrossRef]
- Spassov, V.Z.; Flook, P.K.; Yan, L. LOOPER: A molecular mechanics-based algorithm for protein loop prediction. Protein Eng. Des. Sel. 2008, 21, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Kryshtafovych, A.; Schwede, T.; Topf, M.; Fidelis, K.; Moult, J. Critical Assessment of Methods of Protein Structure Prediction (CASP)—Round XIII. Proteins Struct. Funct. Bioinform. 2019, 87, 1011–1020. [Google Scholar] [CrossRef]
- Xu, J.; Wang, S. Analysis of Distance-based Protein Structure Prediction by Deep Learning in CASP13. Proteins Struct. Funct. Bioinform. 2019, 87, 1069–1081. [Google Scholar] [CrossRef]
- Zheng, W.; Li, Y.; Zhang, C.; Pearce, R.; Mortuza, S.M.; Zhang, Y. Deep-learning Contact-map Guided Protein Structure Prediction in CASP13. Proteins Struct. Funct. Bioinform. 2019, 87, 1149–1164. [Google Scholar] [CrossRef]
- Li, Y.; Hu, J.; Zhang, C.; Yu, D.-J.; Zhang, Y. ResPRE: High-Accuracy Protein Contact Prediction by Coupling Precision Matrix with Deep Residual Neural Networks. Bioinformatics 2019, 35, 4647–4655. [Google Scholar] [CrossRef]
- Hou, J.; Wu, T.; Guo, Z.; Quadir, F.; Cheng, J. The MULTICOM Protein Structure Prediction Server Empowered by Deep Learning and Contact Distance Prediction. In Protein Structure Prediction; Humana Press: New York, NY, USA, 2020; pp. 13–26. [Google Scholar]
- Jones, D.T.; Kandathil, S.M. High Precision in Protein Contact Prediction Using Fully Convolutional Neural Networks and Minimal Sequence Features. Bioinformatics 2018, 34, 3308–3315. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, B.; Hou, J.; Cheng, J. DNCON2: Improved Protein Contact Prediction Using Two-Level Deep Convolutional Neural Networks. Bioinformatics 2018, 34, 1466–1472. [Google Scholar] [CrossRef]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Žídek, A.; Nelson, A.W.R.; Bridgland, A.; et al. Protein Structure Prediction Using Multiple Deep Neural Networks in the 13th Critical Assessment of Protein Structure Prediction (CASP13). Proteins Struct. Funct. Bioinform. 2019, 87, 1141–1148. [Google Scholar] [CrossRef]
- Fukuda, H.; Tomii, K. DeepECA: An End-to-End Learning Framework for Protein Contact Prediction from a Multiple Sequence Alignment. BMC Bioinform. 2020, 21, 10. [Google Scholar] [CrossRef] [PubMed]
- Kandathil, S.M.; Greener, J.G.; Jones, D.T. Prediction of Interresidue Contacts with DeepMetaPSICOV in CASP13. Proteins Struct. Funct. Bioinform. 2019, 87, 1092–1099. [Google Scholar] [CrossRef] [PubMed]
- Stahl, K.; Schneider, M.; Brock, O. EPSILON-CP: Using Deep Learning to Combine Information from Multiple Sources for Protein Contact Prediction. BMC Bioinform. 2017, 18, 303. [Google Scholar] [CrossRef]
- Gao, M.; Zhou, H.; Skolnick, J. DESTINI: A Deep-Learning Approach to Contact-Driven Protein Structure Prediction. Sci. Rep. 2019, 9, 3514. [Google Scholar] [CrossRef] [PubMed]
- AlQuraishi, M. AlphaFold at CASP13. Bioinformatics 2019, 35, 4862–4865. [Google Scholar] [CrossRef] [PubMed]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Žídek, A.; Nelson, A.W.R.; Bridgland, A.; et al. Improved Protein Structure Prediction Using Potentials from Deep Learning. Nature 2020, 577, 706–710. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wu, T.; Guo, Z.; Hou, J.; Cheng, J. Improving Protein Tertiary Structure Prediction by Deep Learning and Distance Prediction in CASP14. bioRxiv 2021, 1, 1–10. [Google Scholar] [CrossRef]
- He, K.; Zhang, X.; Ren, S.; Sun, J. Deep Residual Learning for Image Recognition. In Proceedings of the IEEE Computer Society Conference on Computer Vision and Pattern Recognition, Las Vegas, NV, USA, 27–30 June 2016. [Google Scholar]
- Camacho, D.M.; Collins, K.M.; Powers, R.K.; Costello, J.C.; Collins, J.J. Next-Generation Machine Learning for Biological Networks. Cell 2018, 173, 1581–1592. [Google Scholar] [CrossRef]
- Marbach, D.; Costello, J.C.; Küffner, R.; Vega, N.M.; Prill, R.J.; Camacho, D.M.; Allison, K.R.; Kellis, M.; Collins, J.J.; Stolovitzky, G. Wisdom of crowds for robust gene network inference. Nat. Methods 2012, 9, 796–804. [Google Scholar] [CrossRef]
- Mordelet, F.; Vert, J.P. SIRENE: Supervised Inference of Regulatory Networks. Bioinformatics 2008, 24, i76–i82. [Google Scholar] [CrossRef]
- Mignone, P.; Pio, G.; D’Elia, D.; Ceci, M. Exploiting transfer learning for the reconstruction of the human gene regulatory network. Bioinformatics 2019, 36, 1553–1561. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.A.; Castro, D.M.; Saldi, G.-A.; Bonneau, R.; Gresham, D. Gene regulatory network reconstruction using single-cell RNA sequencing of barcoded genotypes in diverse environments. eLife 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Greene, D.; Cagney, G.; Krogan, N.; Cunningham, P. Ensemble non-negative matrix factorization methods for clustering protein–protein interactions. Bioinformatics 2008, 24, 1722–1728. [Google Scholar] [CrossRef]
- Huang, D.-S.; Zhang, L.; Han, K.; Deng, S.; Yang, K.; Zhang, H. Prediction of Protein-Protein Interactions Based on Protein-Protein Correlation Using Least Squares Regression. Curr. Protein Pept. Sci. 2014, 15, 553–560. [Google Scholar] [CrossRef] [PubMed]
- You, Z.-H.; Lei, Y.-K.; Zhu, L.; Xia, J.; Wang, B. Prediction of protein-protein interactions from amino acid sequences with ensemble extreme learning machines and principal component analysis. BMC Bioinform. 2013, 14, S10. [Google Scholar] [CrossRef]
- Zhang, L.; Yu, G.; Xia, D.; Wang, J. Protein–protein interactions prediction based on ensemble deep neural networks. Neurocomputing 2019, 324, 10–19. [Google Scholar] [CrossRef]
- Yang, F.; Fan, K.; Song, D.; Lin, H. Graph-based prediction of Protein-protein interactions with attributed signed graph embedding. BMC Bioinform. 2020, 21, 323. [Google Scholar] [CrossRef]
- Chatterjee, P.; Basu, S.; Kundu, M.; Nasipuri, M.; Plewczynski, D. PPI_SVM: Prediction of protein-protein interactions using machine learning, domain-domain affinities and frequency tables. Cell. Mol. Biol. Lett. 2011, 16, 264–278. [Google Scholar] [CrossRef]
- Chen, C.; Zhang, Q.; Ma, Q.; Yu, B. LightGBM-PPI: Predicting protein-protein interactions through LightGBM with multi-information fusion. Chemom. Intell. Lab. Syst. 2019, 191, 54–64. [Google Scholar] [CrossRef]
- Wang, Y.-B.; You, Z.-H.; Li, X.; Jiang, T.-H.; Chen, X.; Zhou, X.; Wang, L. Predicting protein–protein interactions from protein sequences by a stacked sparse autoencoder deep neural network. Mol. BioSyst. 2017, 13, 1336–1344. [Google Scholar] [CrossRef]
- Du, X.; Sun, S.; Hu, C.; Yao, Y.; Yan, Y.; Zhang, Y. DeepPPI: Boosting Prediction of Protein–Protein Interactions with Deep Neural Networks. J. Chem. Inf. Model. 2017, 57, 1499–1510. [Google Scholar] [CrossRef]
- Lei, H.; Wen, Y.; You, Z.; ElAzab, A.; Tan, E.-L.; Zhao, Y.; Lei, B. Protein–Protein Interactions Prediction via Multimodal Deep Polynomial Network and Regularized Extreme Learning Machine. IEEE J. Biomed. Heal. Inform. 2018, 23, 1290–1303. [Google Scholar] [CrossRef]
- Hashemifar, S.; Neyshabur, B.; Khan, A.A.; Xu, J. Predicting Protein-Protein Interactions through Sequence-Based Deep Learning. Bioinformatics 2018, 34, i802–i810. [Google Scholar] [CrossRef]
- Lu, Y.; Guo, Y.; Korhonen, A. Link prediction in drug-target interactions network using similarity indices. BMC Bioinform. 2017, 18, 39. [Google Scholar] [CrossRef]
- Nascimento, A.C.A.; Prudêncio, R.B.C.; Costa, I.G. A Drug-Target Network-Based Supervised Machine Learning Repurposing Method Allowing the Use of Multiple Heterogeneous Information Sources. In Methods in Molecular Biology; Springer: Berlin/Heidelberg, Germany, 2019. [Google Scholar]
- Aghakhani, S.; Qabaja, A.; Alhajj, R. Integration of k-means clustering algorithm with network analysis for drug-target interactions network prediction. Int. J. Data Min. Bioinform. 2018, 20, 185. [Google Scholar] [CrossRef]
- Madhukar, N.S.; Khade, P.K.; Huang, L.; Gayvert, K.; Galletti, G.; Stogniew, M.; Allen, J.E.; Giannakakou, P.; Elemento, O. A Bayesian machine learning approach for drug target identification using diverse data types. Nat. Commun. 2019, 10, 5221. [Google Scholar] [CrossRef]
- Zeng, X.; Zhu, S.; Hou, Y.; Zhang, P.; Li, L.; Li, J.; Huang, L.F.; Lewis, S.J.; Nussinov, R.; Cheng, F. Network-based prediction of drug–target interactions using an arbitrary-order proximity embedded deep forest. Bioinformatics 2020, 36, 2805–2812. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Li, H.; Li, S.; Leung, K.-S. Improving prediction of phenotypic drug response on cancer cell lines using deep convolutional network. BMC Bioinform. 2019, 20, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Park, H.; Yang, H.-J.; Lee, S.; Lee, K.-Y.; Kim, T.S.; Jung, J.; Shin, J.-M. Cancer Drug Response Profile scan (CDRscan): A Deep Learning Model That Predicts Drug Effectiveness from Cancer Genomic Signature. Sci. Rep. 2018, 8, 8857. [Google Scholar] [CrossRef]
- Chiu, Y.-C.; Chen, H.-I.H.; Zhang, T.; Zhang, S.; Gorthi, A.; Wang, L.-J.; Huang, Y.; Chen, Y. Predicting drug response of tumors from integrated genomic profiles by deep neural networks. BMC Med Genom. 2019, 12, 143–155. [Google Scholar] [CrossRef]
- Sharifi-Noghabi, H.; Zolotareva, O.; Collins, C.C.; Ester, M. MOLI: Multi-Omics Late Integration with Deep Neural Networks for Drug Response Prediction. Bioinformatics 2019, 35, i501–i509. [Google Scholar] [CrossRef]
- Duran-Frigola, M.; Pauls, E.; Guitart-Pla, O.; Bertoni, M.; Alcalde, V.; Amat, D.; Juan-Blanco, T.; Aloy, P. Extending the small-molecule similarity principle to all levels of biology with the Chemical Checker. Nat. Biotechnol. 2020, 38, 1087–1096. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, A.C.; Mehmood, A.; Dai, X.; Wei, D.-Q. A comparative chemogenic analysis for predicting Drug-Target Pair via Machine Learning Approaches. Sci. Rep. 2020, 10, 6870. [Google Scholar] [CrossRef]
- Li, Z.; Han, P.; You, Z.-H.; Li, X.; Zhang, Y.; Yu, H.; Nie, R.; Chen, X. In silico prediction of drug-target interaction networks based on drug chemical structure and protein sequences. Sci. Rep. 2017, 7, 11174. [Google Scholar] [CrossRef]
- Bari, M.G.; Ung, C.Y.; Zhang, C.; Zhu, S.; Li, H. Machine Learning-Assisted Network Inference Approach to Identify a New Class of Genes that Coordinate the Functionality of Cancer Networks. Sci. Rep. 2017, 7, 6993. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.; Lee, H.; Kim, D.; Han, S.K.; Ha, D.; Shin, K.; Kim, S. Network-based machine learning in colorectal and bladder organoid models predicts anti-cancer drug efficacy in patients. Nat. Commun. 2020, 11, 5485. [Google Scholar] [CrossRef]
- Ammad-Ud-Din, M.; Khan, S.A.; Wennerberg, K.; Aittokallio, T. Systematic Identification of Feature Combinations for Predicting Drug Response with Bayesian Multi-View Multi-Task Linear Regression. Bioinformatics 2017, 33, i359–i368. [Google Scholar] [CrossRef]
- Stanfield, Z.; Coşkun, M.; Koyutürk, M. Drug Response Prediction as a Link Prediction Problem. Sci. Rep. 2017, 7, 40321. [Google Scholar] [CrossRef]
- Shaked, I.; Oberhardt, M.A.; Atias, N.; Sharan, R.; Ruppin, E. Metabolic Network Prediction of Drug Side Effects. Cell Syst. 2016, 2, 209–213. [Google Scholar] [CrossRef]
- Zhao, X.; Chen, L.; Lu, J. A similarity-based method for prediction of drug side effects with heterogeneous information. Math. Biosci. 2018, 306, 136–144. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, F.; Luo, L.; Zhang, J. Predicting drug side effects by multi-label learning and ensemble learning. BMC Bioinform. 2015, 16, 365. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Kovács, I.A.; Barabási, A.-L. Network-based prediction of drug combinations. Nat. Commun. 2019, 10, 1197. [Google Scholar] [CrossRef] [PubMed]
- Singh, H.; Rana, P.S.; Singh, U. Prediction of drug synergy in cancer using ensemble-based machine learning techniques. Mod. Phys. Lett. B 2018, 32. [Google Scholar] [CrossRef]
- Wildenhain, J.; Spitzer, M.; Dolma, S.; Jarvik, N.; White, R.; Roy, M.; Griffiths, E.; Bellows, D.S.; Wright, G.D.; Tyers, M. Prediction of Synergism from Chemical-Genetic Interactions by Machine Learning. Cell Syst. 2015, 1, 383–395. [Google Scholar] [CrossRef]
- Zeng, X.; Zhu, S.; Liu, X.; Zhou, Y.; Nussinov, R.; Cheng, F. deepDR: A network-based deep learning approach to in silico drug repositioning. Bioinformatics 2019, 35, 5191–5198. [Google Scholar] [CrossRef]
- Gottlieb, A.; Stein, G.Y.; Ruppin, E.; Sharan, R. PREDICT: A method for inferring novel drug indications with application to personalized medicine. Mol. Syst. Biol. 2011, 7, 496. [Google Scholar] [CrossRef]
- Himmelstein, D.S.; Lizee, A.; Hessler, C.; Brueggeman, L.; Chen, S.L.; Hadley, D.; Green, A.; Khankhanian, P.; E Baranzini, S. Systematic integration of biomedical knowledge prioritizes drugs for repurposing. eLife 2017, 6, e26726. [Google Scholar] [CrossRef] [PubMed]
- Dale, J.M.; Popescu, L.; Karp, P.D. Machine learning methods for metabolic pathway prediction. BMC Bioinform. 2010, 11, 15. [Google Scholar] [CrossRef]
- Baranwal, M.; Magner, A.; Elvati, P.; Saldinger, J.; Violi, A.; Hero, A.O. A deep learning architecture for metabolic pathway prediction. Bioinformatics 2019, 36, 2547–2553. [Google Scholar] [CrossRef] [PubMed]
- Çubuk, C.; Hidalgo, M.R.; Amadoz, A.; Rian, K.; Salavert, F.; Pujana, M.A.; Mateo, F.; Herranz, C.; Carbonell-Caballero, J.; Dopazo, J. Differential metabolic activity and discovery of therapeutic targets using summarized metabolic pathway models. Npj Syst. Biol. Appl. 2019, 5, 7. [Google Scholar] [CrossRef] [PubMed]
- Auslander, N.; Wagner, A.; Oberhardt, M.; Ruppin, E. Data-Driven Metabolic Pathway Compositions Enhance Cancer Survival Prediction. PLoS Comput. Biol. 2016, 12. [Google Scholar] [CrossRef]
- Kim, J.Y.; Lee, H.; Woo, J.; Yue, W.; Kim, K.; Choi, S.; Jang, J.-J.; Kim, Y.; Park, I.A.; Han, D.; et al. Reconstruction of pathway modification induced by nicotinamide using multi-omic network analyses in triple negative breast cancer. Sci. Rep. 2017, 7, 3466. [Google Scholar] [CrossRef]
- Fu, G.; Ding, Y.; Seal, A.; Chen, B.; Sun, Y.; Bolton, E. Predicting drug target interactions using meta-path-based semantic network analysis. BMC Bioinform. 2016, 17, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Simm, J.; Lam, C.C.; Zakeri, P.; Van Westen, G.J.P.; Moreau, Y.; Saez-Rodriguez, J. Linking drug target and pathway activation for effective therapy using multi-task learning. Sci. Rep. 2018, 8, 8322. [Google Scholar] [CrossRef]
- Esteban-Medina, M.; Peña-Chilet, M.; Loucera, C.; Dopazo, J. Exploring the druggable space around the Fanconi anemia pathway using machine learning and mechanistic models. BMC Bioinform. 2019, 20, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Way, G.P.; Sanchez-Vega, F.; Konnor Cancer Genome Atlas Research Network; Armenia, J.; Chatila, W.K.; Luna, A.; Sander, C.; Cherniack, A.D.; Mina, M.; Ciriello, G.; et al. Machine Learning Detects Pan-cancer Ras Pathway Activation in The Cancer Genome Atlas. Cell Rep. 2018, 23, 172–180.e3. [Google Scholar] [CrossRef]
- Huang, E.; Ishida, S.; Pittman, J.; Dressman, H.; Bild, A.; Kloos, M.; D’Amico, M.; Pestell, R.G.; West, M.; Nevins, J.R. Gene expression phenotypic models that predict the activity of oncogenic pathways. Nat. Genet. 2003, 34, 226–230. [Google Scholar] [CrossRef]
- Costello, Z.; Martin, H.G. A machine learning approach to predict metabolic pathway dynamics from time-series multiomics data. NPJ Syst. Biol. Appl. 2018, 4, 1–14. [Google Scholar] [CrossRef]
- Tepeli, Y.I.; Ünal, A.B.; Akdemir, F.M.; Tastan, O. PAMOGK: A pathway graph kernel-based multiomics approach for patient clustering. Bioinformatics 2021, 36, 5237–5246. [Google Scholar] [CrossRef]
- Cho, H.; Berger, B.; Peng, J. Compact Integration of Multi-Network Topology for Functional Analysis of Genes. Cell Syst. 2016, 3, 540–548.e5. [Google Scholar] [CrossRef]
- Auslander, N.; Yizhak, K.; Weinstock, A.; Budhu, A.; Tang, W.; Wang, X.W.; Ambs, S.; Ruppin, E. A joint analysis of transcriptomic and metabolomic data uncovers enhanced enzyme-metabolite coupling in breast cancer. Sci. Rep. 2016, 6, 29662. [Google Scholar] [CrossRef] [PubMed]
- Katzir, R.; Polat, I.H.; Harel, M.; Katz, S.; Foguet, C.; Selivanov, V.A.; Sabatier, P.; Cascante, M.; Geiger, T.; Ruppin, E. The landscape of tiered regulation of breast cancer cell metabolism. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef]
- Lan, L.; Djuric, N.; Guo, Y.; Vucetic, S. MS-k NN: Protein function prediction by integrating multiple data sources. BMC Bioinform. 2013, 14, S8. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Ruzzo, W.L. A Regression-based K nearest neighbor algorithm for gene function prediction from heterogeneous data. BMC Bioinform. 2006, 7, S11. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Michailidis, G. A non-negative matrix factorization method for detecting modules in heterogeneous omics multi-modal data. Bioinformatics 2015, 32, 1–8. [Google Scholar] [CrossRef]
- Kim, S.; Kang, D.; Huo, Z.; Park, Y.; Tseng, G.C. Meta-analytic principal component analysis in integrative omics application. Bioinformatics 2017, 34, 1321–1328. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Shao, W.; Huang, Z.; Tang, H.; Zhang, J.; Ding, Z.; Huang, K. MORONET: Multi-Omics Integration via Graph Convolutional NETworks for Biomedical Data Classification. bioRxiv 2020, 1, 1–10. [Google Scholar] [CrossRef]
- Rappoport, N.; Shamir, R. NEMO: Cancer subtyping by integration of partial multi-omic data. Bioinformatics 2019, 35, 3348–3356. [Google Scholar] [CrossRef]
- Wang, B.; Mezlini, A.M.; Demir, F.; Fiume, M.; Tu, Z.; Brudno, M.; Haibe-Kains, B.; Goldenberg, A. Similarity network fusion for aggregating data types on a genomic scale. Nat. Methods 2014, 11, 333–337. [Google Scholar] [CrossRef]
- Chaudhary, K.; Poirion, O.B.; Lu, L.; Garmire, L.X. Deep Learning–Based Multi-Omics Integration Robustly Predicts Survival in Liver Cancer. Clin. Cancer Res. 2017, 24, 1248–1259. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Dong, Q.; Li, F.; Xu, Y.; Hu, C.; Wang, J.; Shang, D.; Zheng, X.; Yang, H.; Zhang, C.; et al. Identifying subpathway signatures for individualized anticancer drug response by integrating multi-omics data. J. Transl. Med. 2019, 17, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Sathyanarayanan, A.; Gupta, R.; Thompson, E.W.; Nyholt, D.R.; Bauer, D.C.; Nagaraj, S.H. A comparative study of multi-omics integration tools for cancer driver gene identification and tumour subtyping. Briefings Bioinform. 2020, 21, 1920–1936. [Google Scholar] [CrossRef]
- McCabe, S.D.; Lin, D.Y.; Love, M.I. Consistency and Overfitting of Multi-Omics Methods on Experimental Data. Brief. Bioinform. 2019, 21, 1277–1284. [Google Scholar] [CrossRef] [PubMed]
- Haendel, M.A.; Chute, C.G.; Robinson, P.N. Classification, Ontology, and Precision Medicine. New Engl. J. Med. 2018, 379, 1452–1462. [Google Scholar] [CrossRef] [PubMed]
- Hulsen, T.; Jamuar, S.S.; Moody, A.R.; Karnes, J.H.; Varga, O.; Hedensted, S.; Spreafico, R.; Hafler, D.A.; McKinney, E.F. From Big Data to Precision Medicine. Front. Med. 2019, 6, 34. [Google Scholar] [CrossRef]
- Nitsch, D.; Gonçalves, J.P.; Ojeda, F.; De Moor, B.; Moreau, Y. Candidate gene prioritization by network analysis of differential expression using machine learning approaches. BMC Bioinform. 2010, 11, 1–16. [Google Scholar] [CrossRef]
- Li, H.; Zhao, X.; Wang, J.; Zong, M.; Yang, H. Bioinformatics analysis of gene expression profile data to screen key genes involved in pulmonary sarcoidosis. Gene 2017, 596, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Q.; Luo, J.; Liang, C.; Cai, J.; Ding, P. A graph regularized non-negative matrix factorization method for identifying microRNA-disease associations. Bioinformatics 2018, 34, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Asif, M.; Martiniano, H.F.M.C.M.; Vicente, A.M.; Couto, F.M. Identifying disease genes using machine learning and gene functional similarities, assessed through Gene Ontology. PLoS ONE 2018, 13, e0208626. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Bigas, N.; Ouzounis, C.A. Genome-wide identification of genes likely to be involved in human genetic disease. Nucleic Acids Res. 2004, 32, 3108–3114. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Jiang, X.; Hu, X.; Li, G. Visualization of genetic disease-phenotype similarities by multiple maps t-SNE with Laplacian regularization. BMC Med. Genom. 2014, 7, S1. [Google Scholar] [CrossRef]
- Shen, X.; Zhu, X.; Jiang, X.; He, T.; Hu, X. Visualization of Disease Relationships by Multiple Maps T-SNE Regularization Based on Nesterov Accelerated Gradient. In Proceedings of the 2017 IEEE International Conference on Bioinformatics and Biomedicine, BIBM, Kansas City, MO, USA, 13–16 November 2017. [Google Scholar]
- Lage, K.; Karlberg, L.O.E.; Størling, M.Z.; Ólason, P.Í.; Pedersen, A.G.; Rigina, O.; Hinsby, A.M.; Tümer, Z.; Pociot, F.; Tommerup, N.; et al. A human phenome-interactome network of protein complexes implicated in genetic disorders. Nat. Biotechnol. 2007, 25, 309–316. [Google Scholar] [CrossRef]
- Xu, J.; Li, Y. Discovering disease-genes by topological features in human protein–protein interaction network. Bioinformatics 2006, 22, 2800–2805. [Google Scholar] [CrossRef]
- Barman, R.K.; Mukhopadhyay, A.; Maulik, U.; Das, S. Identification of infectious disease-associated host genes using machine learning techniques. BMC Bioinform. 2019, 20, 736. [Google Scholar] [CrossRef]
- Han, Y.; Yang, J.; Qian, X.; Cheng, W.-C.; Liu, S.-H.; Hua, X.; Zhou, L.; Yang, Y.; Wu, Q.; Liu, P.; et al. DriverML: A machine learning algorithm for identifying driver genes in cancer sequencing studies. Nucleic Acids Res. 2019, 47, e45. [Google Scholar] [CrossRef]
- Auslander, N.; Wolf, Y.I.; Koonin, E.V. Interplay between DNA damage repair and apoptosis shapes cancer evolution through aneuploidy and microsatellite instability. Nat. Commun. 2020, 11, 1234. [Google Scholar] [CrossRef]
- Collier, O.; Stoven, V.; Vert, J.-P. LOTUS: A single- and multitask machine learning algorithm for the prediction of cancer driver genes. PLoS Comput. Biol. 2019, 15, e1007381. [Google Scholar] [CrossRef]
- Luo, P.; Ding, Y.; Lei, X.; Wu, F.-X. deepDriver: Predicting Cancer Driver Genes Based on Somatic Mutations Using Deep Convolutional Neural Networks. Front. Genet. 2019, 10, 13. [Google Scholar] [CrossRef]
- Agajanian, S.; Oluyemi, O.; Verkhivker, G.M. Integration of Random Forest Classifiers and Deep Convolutional Neural Networks for Classification and Biomolecular Modeling of Cancer Driver Mutations. Front. Mol. Biosci. 2019, 6, 44. [Google Scholar] [CrossRef]
- Califf, R.M. Biomarker definitions and their applications. Exp. Biol. Med. 2018, 243, 213–221. [Google Scholar] [CrossRef]
- Ray, P.; Manach, Y.L.; Riou, B.; Houle, T.T. Statistical Evaluation of a Biomarker. Anesthesiology 2010, 112, 1023–1040. [Google Scholar] [CrossRef]
- McDermott, J.E.; Wang, J.; Mitchell, H.; Webb-Robertson, B.J.; Hafen, R.; Ramey, J.; Rodland, K.D. Challenges in Biomarker Discovery: Combining Expert Insights with Statistical Analysis of Complex Omics Data. Expert Opin. Med. Diagn. 2013, 7, 37–51. [Google Scholar] [CrossRef]
- Cun, Y.; Fröhlich, H. netClass: An R-package for network based, integrative biomarker signature discovery. Bioinformatics 2014, 30, 1325–1326. [Google Scholar] [CrossRef]
- Yasui, Y.; Pepe, M.; Thompson, M.L.; Adam, B.; Wright, G.L.; Qu, Y.; Potter, J.D.; Winget, M.; Thornquist, M.; Feng, Z. A data-analytic strategy for protein biomarker discovery: Profiling of high-dimensional proteomic data for cancer detection. Biostatistics 2003, 4, 449–463. [Google Scholar] [CrossRef] [PubMed]
- Statnikov, A.; Tsamardinos, I.; Dosbayev, Y.; Aliferis, C.F. GEMS: A system for automated cancer diagnosis and biomarker discovery from microarray gene expression data. Int. J. Med. Inform. 2005, 74, 491–503. [Google Scholar] [CrossRef] [PubMed]
- Abeel, T.; Helleputte, T.; Van de Peer, Y.; Dupont, P.; Saeys, Y. Robust Biomarker Identification for Cancer Diagnosis with Ensemble Feature Selection Methods. Bioinformatics 2010, 26, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Kossenkov, A.V.; Qureshi, R.; Dawany, N.B.; Wickramasinghe, J.; Liu, Q.; Majumdar, R.S.; Chang, C.; Widura, S.; Kumar, T.; Horng, W.-H.; et al. A Gene Expression Classifier from Whole Blood Distinguishes Benign from Malignant Lung Nodules Detected by Low-Dose CT. Cancer Res. 2019, 79, 263–273. [Google Scholar] [CrossRef]
- Gal, O.; Auslander, N.; Fan, Y.; Meerzaman, D. Predicting Complete Remission of Acute Myeloid Leukemia: Machine Learning Applied to Gene Expression. Cancer Inform. 2019, 18. [Google Scholar] [CrossRef] [PubMed]
- Ganti, S.; Weiss, R.H. Urine Metabolomics for Kidney Cancer Detection and Biomarker Discovery. In Urologic Oncology: Seminars and Original Investigations; Elsevier: Amsterdam, The Netherlands, 2011. [Google Scholar]
- Shen, C.; Sun, Z.; Chen, D.; Su, X.; Jiang, J.; Li, G.; Lin, B.; Biaoyang, L. Developing Urinary Metabolomic Signatures as Early Bladder Cancer Diagnostic Markers. OMICS A J. Integr. Biol. 2015, 19, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Leclercq, M.; Vittrant, B.; Martin-Magniette, M.L.; Boyer, M.P.S.; Perin, O.; Bergeron, A.; Fradet, Y.; Droit, A. Large-Scale Automatic Feature Selection for Biomarker Discovery in High-Dimensional OMICs Data. Front. Genet. 2019, 10, 452. [Google Scholar] [CrossRef]
- Wang, J.; Zuo, Y.; Man, Y.G.; Avital, I.; Stojadinovic, A.; Liu, M.; Yang, X.; Varghese, R.S.; Tadesse, M.G.; Ressom, H.W. Pathway and Network Approaches for Identification of Cancer Signature Markers from Omics Data. J. Cancer 2015, 6, 54. [Google Scholar] [CrossRef] [PubMed]
- Long, N.P.; Jung, K.H.; Anh, N.H.; Yan, H.H.; Nghi, T.D.; Park, S.; Yoon, S.J.; Min, J.E.; Kim, H.M.; Lim, J.H.; et al. An Integrative Data Mining and Omics-Based Translational Model for the Identification and Validation of Oncogenic Biomarkers of Pancreatic Cancer. Cancers 2019, 11, 155. [Google Scholar] [CrossRef]
- Rohart, F.; Gautier, B.; Singh, A.; Cao, K.-A.L. mixOmics: An R package for ‘omics feature selection and multiple data integration. PLoS Comput. Biol. 2017, 13, e1005752. [Google Scholar] [CrossRef]
- Guan, X.; Runger, G.; Liu, L. Dynamic incorporation of prior knowledge from multiple domains in biomarker discovery. BMC Bioinform. 2020, 21, 1–10. [Google Scholar] [CrossRef]
- Foroughi Pour, A.; Dalton, L.A. Integrating Prior Information with Bayesian Feature Selection. In Proceedings of the 8th ACM Conference on Bioinformatics, Computational Biology, and Health Informatics (ACM BCB), Boston, MA, USA, 20–23 August 2017; p. 610. [Google Scholar] [CrossRef]
- Liu, L.; Chang, Y.; Yang, T.; Noren, D.P.; Long, B.; Kornblau, S.; Qutub, A.; Ye, J. Evolution-informed modeling improves outcome prediction for cancers. Evol. Appl. 2016, 10, 68–76. [Google Scholar] [CrossRef]
- Johannes, M.; Fröhlich, H.; Sültmann, H.; Beissbarth, T.; Beißbarth, T. pathClass: An R-package for integration of pathway knowledge into support vector machines for biomarker discovery. Bioinformatics 2011, 27, 1442–1443. [Google Scholar] [CrossRef]
- Haider, S.; Yao, C.Q.; Sabine, V.S.; Grzadkowski, M.; Stimper, V.; Starmans, M.H.W.; Wang, J.; Nguyen, F.; Moon, N.C.; Lin, X.; et al. Pathway-based subnetworks enable cross-disease biomarker discovery. Nat. Commun. 2018, 9, 4746. [Google Scholar] [CrossRef]
- Fujita, N.; Mizuarai, S.; Murakami, K.; Nakai, K. Biomarker discovery by integrated joint non-negative matrix factorization and pathway signature analyses. Sci. Rep. 2018, 8, 9743. [Google Scholar] [CrossRef] [PubMed]
- Abbas, M.; Matta, J.; Le, T.; Bensmail, H.; Obafemi-Ajayi, T.; Honavar, V.; El-Manzalawy, Y. Biomarker discovery in inflammatory bowel diseases using network-based feature selection. PLoS ONE 2019, 14, e0225382. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xiang, Y.; Ding, L.; Keen-Circle, K.; Borlawsky, T.B.; Ozer, H.G.; Jin, R.; Payne, P.; Huang, K. Using gene co-expression network analysis to predict biomarkers for chronic lymphocytic leukemia. BMC Bioinform. 2010, 11, S5. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-I.; Celik, S.; Logsdon, B.A.; Lundberg, S.M.; Martins, T.J.; Oehler, V.G.; Estey, E.H.; Miller, C.P.; Chien, S.; Dai, J.; et al. A machine learning approach to integrate big data for precision medicine in acute myeloid leukemia. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Cheerla, N.; Gevaert, O. MicroRNA based Pan-Cancer Diagnosis and Treatment Recommendation. BMC Bioinform. 2017, 18, 1–11. [Google Scholar] [CrossRef]
- Wang, L.; He, X.; Zhang, W.; Zha, H. Supervised Reinforcement Learning with Recurrent Neural Network for Dynamic Treatment Recommendation. In Proceedings of the 24th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, London, UK, 19–23 August 2018. [Google Scholar]
- Samala, R.K.; Chan, H.-P.; Hadjiiski, L.M.; A Helvie, M.; Richter, C.; Cha, K. Evolutionary pruning of transfer learned deep convolutional neural network for breast cancer diagnosis in digital breast tomosynthesis. Phys. Med. Biol. 2018, 63, 095005. [Google Scholar] [CrossRef]
- Asgari, E.; Mofrad, M.R.K. Continuous Distributed Representation of Biological Sequences for Deep Proteomics and Genomics. PLoS ONE 2015, 10, e0141287. [Google Scholar] [CrossRef]
- Du, J.; Jia, P.; Dai, Y.; Tao, C.; Zhao, Z.; Zhi, D. Gene2vec: Distributed representation of genes based on co-expression. BMC Genom. 2019, 20, 7–15. [Google Scholar] [CrossRef]
- Kim, S.; Lee, H.; Kim, K.; Kang, J. Mut2Vec: Distributed representation of cancerous mutations. BMC Med. Genom. 2018, 11, 57–69. [Google Scholar] [CrossRef]
- Xu, Y.; Song, J.; Wilson, C.; Whisstock, J.C. PhosContext2vec: A distributed representation of residue-level sequence contexts and its application to general and kinase-specific phosphorylation site prediction. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Sandve, G.K.; Nekrutenko, A.; Taylor, J.; Hovig, E. Ten Simple Rules for Reproducible Computational Research. PLoS Comput. Biol. 2013, 9, e1003285. [Google Scholar] [CrossRef]
- Patel-Murray, N.L.; Adam, M.; Huynh, N.; Wassie, B.T.; Milani, P.; Fraenkel, E. A Multi-Omics Interpretable Machine Learning Model Reveals Modes of Action of Small Molecules. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef]
- Jha, A.; Aicher, J.K.; Gazzara, M.R.; Singh, D.; Barash, Y. Enhanced Integrated Gradients: Improving interpretability of deep learning models using splicing codes as a case study. Genome Biol. 2020, 21, 1–22. [Google Scholar] [CrossRef]
- Hao, J.; Kosaraju, S.C.; Tsaku, N.Z.; Song, D.H.; Kang, M. PAGE-Net: Interpretable and Integrative Deep Learning for Survival Analysis Using Histopathological Images and Genomic Data. In Proceedings of the Pacific Symposium on Biocomputing, Fairmont Orchid, HI, USA, 3–7 January 2020. [Google Scholar]
- Dey, S.; Luo, H.; Fokoue, A.; Hu, J.; Zhang, P. Predicting adverse drug reactions through interpretable deep learning framework. BMC Bioinform. 2018, 19, 476. [Google Scholar] [CrossRef] [PubMed]
- Mikolov, T.; Sutskever, I.; Chen, K.; Corrado, G.; Dean, J. Distributed Representations Ofwords and Phrases and Their Compositionality. In Advances in Neural Information Processing Systems, Proceedings of the Twenty-Seventh Conference on Neural Information Processing Systems, Lake Tahoe, NV, USA, 5–10 December 2013; NeurIPS: San Diego, CA, USA, 2013. [Google Scholar]
- Henikoff, S.; Henikoff, J.G. Amino Acid Substitution Matrices from Protein Blocks. Proc. Natl. Acad. Sci. USA 1992, 89, 10915–10919. [Google Scholar] [CrossRef]
- Nakamura, Y. Codon Usage Tabulated from International DNA Sequence Databases: Status for the Year. Nucleic Acids Res. 2000, 28, 292. [Google Scholar] [CrossRef] [PubMed]
- Marchler-Bauer, A.; Derbyshire, M.K.; Gonzales, N.R.; Lu, S.; Chitsaz, F.; Geer, L.Y.; Geer, R.C.; He, J.; Gwadz, M.; Hurwitz, D.I.; et al. CDD: NCBI’s conserved domain database. Nucleic Acids Res. 2015, 43, D222–D226. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, D.; Orlando, G.; Vranken, W.F.; Moreau, Y. Exploring the limitations of biophysical propensity scales coupled with machine learning for protein sequence analysis. Sci. Rep. 2019, 9, 16932. [Google Scholar] [CrossRef]
- Yang, K.K.; Wu, Z.; Bedbrook, C.N.; Arnold, F.H. Learned protein embeddings for machine learning. Bioinformatics 2018, 34, 2642–2648. [Google Scholar] [CrossRef]
- Rodríguez, P.; Bautista, M.A.; Gonzàlez, J.; Escalera, S. Beyond one-hot encoding: Lower dimensional target embedding. Image Vis. Comput. 2018, 75, 21–31. [Google Scholar] [CrossRef]
- Zhang, W.; Du, T.; Wang, J. Deep Learning over Multi-field Categorical Data. In Advances in Information Retrieval, Proceedings of the 38th European Conference on IR Research, ECIR 2016, Padua, Italy, 20–23 March 2016; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- Gu, J.; Wang, Z.; Kuen, J.; Ma, L.; Shahroudy, A.; Shuai, B.; Liu, T.; Wang, X.; Wang, G.; Cai, J.; et al. Recent advances in convolutional neural networks. Pattern Recognit. 2018, 77, 354–377. [Google Scholar] [CrossRef]
- Chen, Q.; Britto, R.; Erill, I.; Jeffery, C.J.; Liberzon, A.; Magrane, M.; Onami, J.I.; Robinson-Rechavi, M.; Sponarova, J.; Zobel, J.; et al. Quality Matters: Biocuration Experts on the Impact of Duplication and Other Data Quality Issues in Biological Databases. Genom. Proteom. Bioinform. 2020, 18, 91. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Lapedriza, A.; Xiao, J.; Torralba, A.; Oliva, A. Learning Deep Features for Scene Recognition Using Places Database. In Advances in Neural Information Processing Systems, Proceedings of the 28th Annual Conference on Neural Information Processing Systems 2014 (NIPS), Montreal, QC, Canada, 8–13 December 2014; MIT Press: Cambridge, MA, USA, 2014. [Google Scholar]
- Chen, Y.; Li, Y.; Narayan, R.; Subramanian, A.; Xie, X. Gene expression inference with deep learning. Bioinformatics 2016, 32, 1832–1839. [Google Scholar] [CrossRef]
- Sun, C.; Shrivastava, A.; Singh, S.; Gupta, A. Revisiting Unreasonable Effectiveness of Data in Deep Learning Era. In Proceedings of the IEEE International Conference on Computer Vision, Venice, Italy, 22–29 October 2017. [Google Scholar]
- Roberts, D.R.; Bahn, V.; Ciuti, S.; Boyce, M.S.; Elith, J.; Guillera-Arroita, G.; Hauenstein, S.; Lahoz-Monfort, J.J.; Schröder, B.; Thuiller, W.; et al. Cross-Validation Strategies for Data with Temporal, Spatial, Hierarchical, or Phylogenetic Structure. Ecography 2017, 40, 913–929. [Google Scholar] [CrossRef]
- Shaikhina, T.; Khovanova, N.A. Handling limited datasets with neural networks in medical applications: A small-data approach. Artif. Intell. Med. 2017, 75, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Auslander, N.; Wolf, Y.I.; Koonin, E.V. In Silico Learning of Tumor Evolution through Mutational Time Series. Proc. Natl. Acad. Sci. USA 2019, 116. [Google Scholar] [CrossRef] [PubMed]
- Stodden, V.; McNutt, M.; Bailey, D.H.; Deelman, E.; Gil, Y.; Hanson, B.; Heroux, M.A.; Ioannidis, J.P.A.; Taufer, M. Enhancing Reproducibility for Computational Methods. Science 2016, 354, 1240–1241. [Google Scholar] [CrossRef]
- Arora, S.; Pattwell, S.S.; Holland, E.C.; Bolouri, H. Variability in estimated gene expression among commonly used RNA-seq pipelines. Sci. Rep. 2020, 10, 2734. [Google Scholar] [CrossRef]
- Hong, C.S.; Singh, L.N.; Mullikin, J.C.; Biesecker, L.G. Assessing the reproducibility of exome copy number variations predictions. Genome Med. 2016, 8, 82. [Google Scholar] [CrossRef]
- Sandmann, S.; De Graaf, A.O.; Karimi, M.; Van Der Reijden, B.A.; Hellström-Lindberg, E.; Jansen, J.H.; Dugas, M. Evaluating Variant Calling Tools for Non-Matched Next-Generation Sequencing Data. Sci. Rep. 2017, 7, srep43169. [Google Scholar] [CrossRef]
- Montavon, G.; Samek, W.; Müller, K.R. Methods for Interpreting and Understanding Deep Neural Networks. Digit. Signal Process. A Rev. J. 2018, 73, 1–15. [Google Scholar] [CrossRef]
- Bazen, S.; Joutard, X. The Taylor Decomposition: A Unified Generalization of the Oaxaca Method to Nonlinear Models. In Proceedings of the French Econometrics Conference, Toulouse, France, 14–15 November 2013. [Google Scholar]
- Simonyan, K.; Vedaldi, A.; Zisserman, A. Deep inside Convolutional Networks: Visualising Image Classification Models and Saliency Maps. arXiv 2014, arXiv:1312.6034v2. [Google Scholar]
- Montavon, G.; Binder, A.; Lapuschkin, S.; Samek, W.; Müller, K.R. Layer-Wise Relevance Propagation: An Overview. In Lecture Notes in Computer Science (Including Subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics); Springer: Berlin/Heidelberg, Germany, 2019. [Google Scholar]
- Chicco, D. Ten Quick Tips for Machine Learning in Computational Biology. BioData Min. 2017, 10, 35. [Google Scholar] [CrossRef] [PubMed]


| Bioinformatics Area | Problem Category | Goal | ML Method | Bioinformatic Tools |
|---|---|---|---|---|
| Molecular evolution | Biological sequence clustering | Protein family prediction | CNN | Clusters of Orthologous Groups (COGs) and G protein-coupled receptor (GPCR) dataset [30] |
| Protein function prediction | deep RNN | BLAST and HMMER search [32] | ||
| Anti-CRISPR proteins identification | Random forest | MSA and PSI-BLAST [24] | ||
| EXtreme Gradient Boosting | K-mer based clustering (CD-HIT), BLAST [25] | |||
| Viral pathogenicity feature identification | SVM | MSA, phylogenetic tree construction [20,21] | ||
| Alignment free biological sequence analysis | Identification of viral genomes | RNN | BLAST, Sequence clustering, HHPRED [27] | |
| CNN | BLAST [28] | |||
| protein structure analysis | Post translational modifications | Phosphorylation sites prediction | KNN | Local sequence similarity [53] |
| CNN | K-mer based clustering (CD-HIT), BLAST [55] | |||
| Glycosylation sites prediction | ensemble SVM | curated glycosylated protein database (O-GLYCBASE) [54] | ||
| Protein structure prediction | Protein contact prediction | CNN | MSA [72] | |
| Prediction of distances between pairs of residues | CNN | MSA, HHPRED, PSI-BLAST [77] | ||
| systems biology | inference of biological networks | Gene regulatory network prediction | SVM | GeneNetWeaver, RegulonDB [81] |
| Protein-protein interaction network prediction | SVM | Domain affinity and frequency tables [90] | ||
| Elastic-net regression | Protein descriptors [91] | |||
| Analysis of biological networks | Drug target prediction | K-means | Network analysis tools [98] | |
| Drug side effect prediction | SVM | Genome scale metabolic modeling [112] | ||
| Drug Synergism prediction | Random Forest Ensemble | A chemical-genetic interaction matrix [117] | ||
| Multi-omics integration | Cancer subtype prediction | Neighborhood based clustering | Similarity based integration [141] | |
| Drug response prediction | logistic regression | Cancer hallmarks datasets, pathway data [144] | ||
| biomarker analysis for disease research | Disease-associated genes investigation | Pulmonary sarcoidosis genes identification | Hierarchical clustering | Differential expression analysis [150] |
| Identification of miRNA-disease association | NMF | Disease semantic information and miRNA functional information [151] | ||
| Disease-phenotype visualization | t-SNE | OMIM database and human disease networks [154] | ||
| Biomarker discovery | Cancer diagnosis | SVM | Reference gene selection [170] | |
| Biomarker signature identification | SVM | Network-based gene selection [167] | ||
| Cancer outcome prediction | Random forest | Evolutionary conservation estimation [181] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Auslander, N.; Gussow, A.B.; Koonin, E.V. Incorporating Machine Learning into Established Bioinformatics Frameworks. Int. J. Mol. Sci. 2021, 22, 2903. https://doi.org/10.3390/ijms22062903
Auslander N, Gussow AB, Koonin EV. Incorporating Machine Learning into Established Bioinformatics Frameworks. International Journal of Molecular Sciences. 2021; 22(6):2903. https://doi.org/10.3390/ijms22062903
Chicago/Turabian StyleAuslander, Noam, Ayal B. Gussow, and Eugene V. Koonin. 2021. "Incorporating Machine Learning into Established Bioinformatics Frameworks" International Journal of Molecular Sciences 22, no. 6: 2903. https://doi.org/10.3390/ijms22062903
APA StyleAuslander, N., Gussow, A. B., & Koonin, E. V. (2021). Incorporating Machine Learning into Established Bioinformatics Frameworks. International Journal of Molecular Sciences, 22(6), 2903. https://doi.org/10.3390/ijms22062903