
Urinary Exosomes Identify Inflammatory Pathways in Vancomycin Associated Acute Kidney Injury

 , , and
, , and
Abstract
1. Introduction
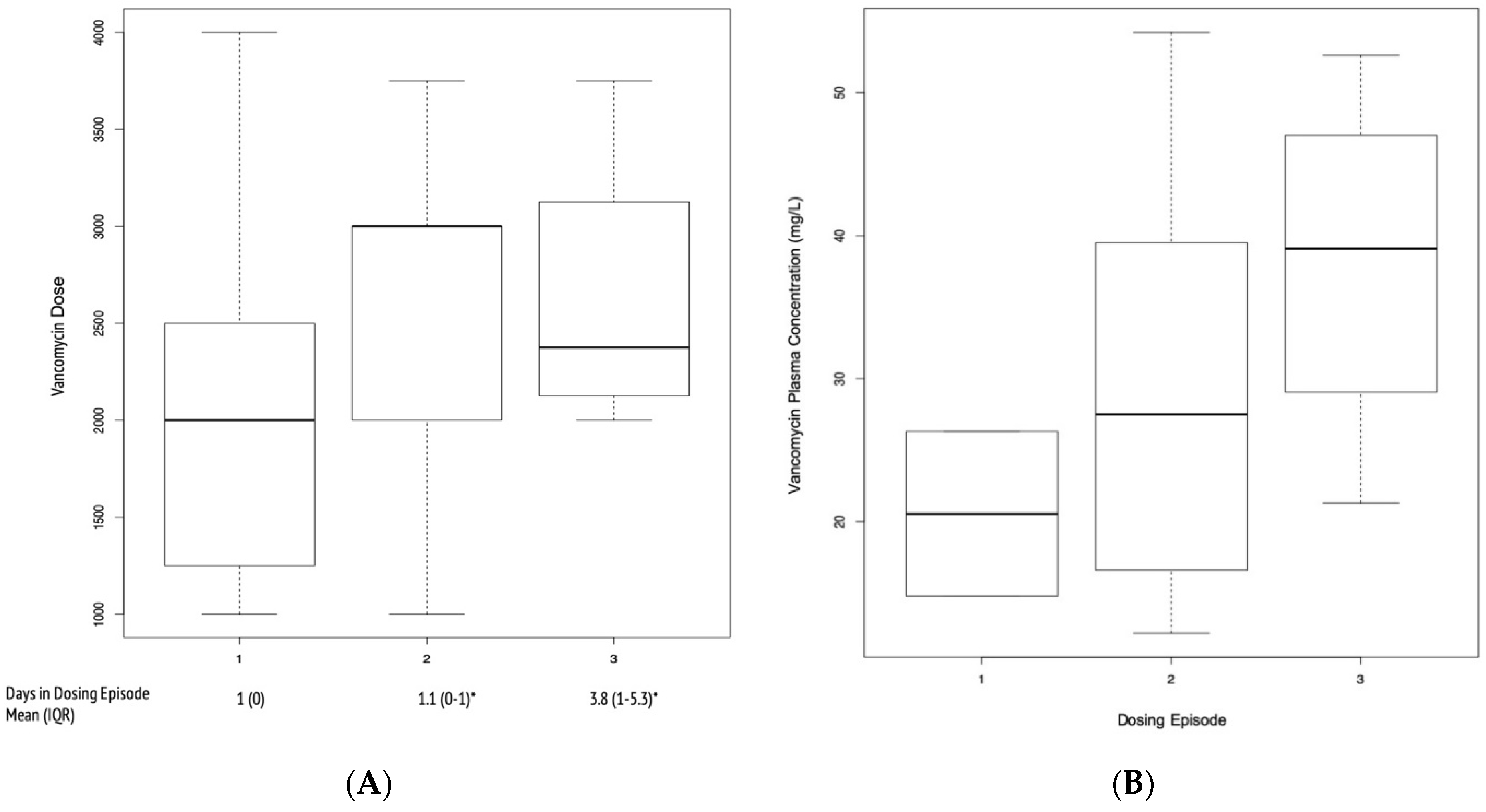
2. Results
Urine Exosome Proteomic Analysis Showed Upregulation of Inflammatory Pathways
3. Discussion
4. Materials and Methods
4.1. Patient Selection
4.2. Exosome Preparation and Proteomic Data Collection
4.3. Analysis of Proteomics Data
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jorgensen, S.C.J.; Murray, K.P.; Lagnf, A.M.; Melvin, S.; Bhatia, S.; Shamim, M.D.; Smith, J.R.; Brade, K.D.; Simon, S.P.; Nagel, J.; et al. A Multicenter Evaluation of Vancomycin-Associated Acute Kidney Injury in Hospitalized Patients with Acute Bacterial Skin and Skin Structure Infections. Infect. Dis. Ther. 2020, 9, 89–106. [Google Scholar] [CrossRef]
- Filippone, E.J.; Kraft, W.K.; Farber, J.L. The Nephrotoxicity of Vancomycin. Clin. Pharmacol. Ther. 2017, 102, 459–469. [Google Scholar] [CrossRef]
- AbdulHameed, M.D.; Ippolito, D.L.; Stallings, J.D.; Wallqvist, A. Mining kidney toxicogenomic data by using gene co-expression modules. BMC Genom. 2016, 17, 790. [Google Scholar] [CrossRef]
- Amin, R.P.; Vickers, A.E.; Sistare, F.; Thompson, K.L.; Roman, R.J.; Lawton, M.; Kramer, J.; Hamadeh, H.K.; Collins, J.; Grissom, J.; et al. Identification of putative gene based markers of renal toxicity. Environ. Health Perspect. 2004, 112, 465–479. [Google Scholar] [CrossRef]
- Dieterich, C.; Puey, A.; Lin, S.; Swezey, R.; Furimsky, A.; Fairchild, D.; Mirsalis, J.C.; Ng, H.H. Gene expression analysis reveals new possible mechanisms of vancomycin-induced nephrotoxicity and identifies gene markers candidates. Toxicol. Sci. 2009, 107, 258–269. [Google Scholar] [CrossRef] [PubMed]
- Nishino, Y.; Takemura, S.; Minamiyama, Y.; Hirohashi, K.; Tanaka, H.; Inoue, M.; Okada, S.; Kinoshita, H. Inhibition of vancomycin-induced nephrotoxicity by targeting superoxide dismutase to renal proximal tubule cells in the rat. Redox Rep. 2002, 7, 317–319. [Google Scholar] [CrossRef] [PubMed]
- Oktem, F.; Arslan, M.K.; Ozguner, F.; Candir, O.; Yilmaz, H.R.; Ciris, M.; Uz, E. In vivo evidences suggesting the role of oxidative stress in pathogenesis of vancomycin-induced nephrotoxicity: Protection by erdosteine. Toxicology 2005, 215, 227–233. [Google Scholar] [CrossRef]
- Sokol, P.P. Mechanism of vancomycin transport in the kidney: Studies in rabbit renal brush border and basolateral membrane vesicles. J. Pharmacol. Exp. Ther. 1991, 259, 1283–1287. [Google Scholar] [PubMed]
- Lin, J.; Liu, J.; Huang, B.; Chen, X.; Chen, X.-M.; Xu, Y.-M.; Huang, L.-F.; Wang, X.-Z. Exosomes: Novel biomarkers for clinical diagnosis. Sci. World J. 2015, 2015, 657086. [Google Scholar] [CrossRef]
- Awdishu, L.; Nievergelt, C.M.; Davenport, A.; Murray, P.T.; Macedo, E.; Cerda, J.; Chakaravarthi, R.; Rao, S.P.R.; Holden, A.; Goldstein, S.L.; et al. Rationale and Design of the Genetic Contribution to Drug Induced Renal Injury (DIRECT) Study. Kidney Int. Rep. 2016, 1, 288–298. [Google Scholar] [CrossRef][Green Version]
- Ostermann, M.; Bellomo, R.; Burdmann, E.A.; Doi, K.; Endre, Z.H.; Goldstein, S.L.; Kane-Gill, S.L.; Liu, K.D.; Prowle, J.R.; Shaw, A.D.; et al. Controversies in acute kidney injury: Conclusions from a Kidney Disease: Improving Global Outcomes (KDIGO) Conference. Kidney Int. 2020, 98, 294–309. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Hsiao, T.-H.; Suresh, U.; Chen, H.-I.H.; Wu, X.; Wolf, S.E.; Chen, Y. A novel significance score for gene selection and ranking. Bioinformatics 2014, 30, 801–807. [Google Scholar] [CrossRef]
- Lodise, T.P.; Lomaestro, B.; Graves, J.; Drusano, G.L. Larger vancomycin doses (at least four grams per day) are associated with an increased incidence of nephrotoxicity. Antimicrob. Agents Chemother. 2008, 52, 1330–1336. [Google Scholar] [CrossRef]
- van der Pol, E.; Boing, A.N.; Harrison, P.; Sturk, A.; Nieuwland, R. Classification, functions, and clinical relevance of extracellular vesicles. Pharmacol. Rev. 2012, 64, 676–705. [Google Scholar] [CrossRef] [PubMed]
- Lv, L.-L.; Feng, Y.; Wu, M.; Wang, B.; Li, Z.-L.; Zhong, X.; Wu, W.-J.; Chen, J.; Ni, H.-F.; Tang, T.-T.; et al. Exosomal miRNA-19b-3p of tubular epithelial cells promotes M1 macrophage activation in kidney injury. Cell Death Differ. 2020, 27, 210–226. [Google Scholar] [CrossRef]
- Mathern, D.R.; Heeger, P.S. Molecules Great and Small: The Complement System. Clin. J. Am. Soc. Nephrol. 2015, 10, 1636–1650. [Google Scholar] [CrossRef] [PubMed]
- McCullough, J.W.; Renner, B.; Thurman, J.M. The role of the complement system in acute kidney injury. Semin. Nephrol. 2013, 33, 543–556. [Google Scholar] [CrossRef]
- Siedlecki, A.; Irish, W.; Brennan, D.C. Delayed graft function in the kidney transplant. Am. J. Transplant. 2011, 11, 2279–2296. [Google Scholar] [CrossRef]
- Peng, Q.; Li, K.; Smyth, L.A.; Xing, G.; Wang, N.; Meader, L.; Lu, B.; Sacks, S.H.; Zhou, W. C3a and C5a promote renal ischemia-reperfusion injury. J. Am. Soc. Nephrol. 2012, 23, 1474–1485. [Google Scholar] [CrossRef]
- Farrar, C.A.; Zhou, W.; Lin, T.; Sacks, S.H. Local extravascular pool of C3 is a determinant of postischemic acute renal failure. FASEB J. 2006, 20, 217–226. [Google Scholar] [CrossRef]
- Naik, A.; Sharma, S.; Quigg, R.J. Complement regulation in renal disease models. Semin. Nephrol. 2013, 33, 575–585. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kooijmans, S.A.; Vader, P.; van Dommelen, S.M.; van Solinge, W.W.; Schiffelers, R.M. Exosome mimetics: A novel class of drug delivery systems. Int. J. Nanomed. 2012, 7, 1525–1541. [Google Scholar]
- Karasu, E.; Eisenhardt, S.U.; Harant, J.; Huber-Lang, M. Extracellular Vesicles: Packages Sent WITH Complement. Front. Immunol. 2018, 9, 721. [Google Scholar] [CrossRef]
- Chen, S.C.; Kuo, P.L. The Role of Galectin-3 in the Kidneys. Int. J. Mol. Sci. 2016, 17, 565. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, J.; Kobayashi, S.; Ishida, A.; Nakabayashi, I.; Tajima, O.; Miura, S.; Katayama, M.; Nogami, H. Up-regulation of galectin-3 in acute renal failure of the rat. Am. J. Pathol. 2000, 157, 815–823. [Google Scholar] [CrossRef]
- O’Seaghdha, C.M.; Hwang, S.J.; Ho, J.E.; Vasan, R.S.; Levy, D.; Fox, C.S. Elevated galectin-3 precedes the development of CKD. J. Am. Soc. Nephrol. 2013, 24, 1470–1477. [Google Scholar] [CrossRef]
- Henderson, N.C.; Sethi, T. The regulation of inflammation by galectin-3. Immunol. Rev. 2009, 230, 160–171. [Google Scholar] [CrossRef]
- Tsuchiyama, Y.; Wada, J.; Zhang, H.; Morita, Y.; Hiragushi, K.; Hida, K.; Shikata, K.; Yamamura, M.; Kanwar, Y.S.; Makino, H. Efficacy of galectins in the amelioration of nephrotoxic serum nephritis in Wistar Kyoto rats. Kidney Int. 2000, 58, 1941–1952. [Google Scholar] [CrossRef]
- Hoffmann, D.; Bijol, V.; Krishnamoorthy, A.; Gonzalez, V.R.; Frendl, G.; Zhang, Q.; Goering, P.L.; Brown, R.P.; Waikar, S.S.; Vaidya, V.S. Fibrinogen excretion in the urine and immunoreactivity in the kidney serves as a translational biomarker for acute kidney injury. Am. J. Pathol. 2012, 181, 818–828. [Google Scholar] [CrossRef]
- Adams, R.A.; Schachtrup, C.; Davalos, D.; Tsigelny, I.; Akassoglou, K. Fibrinogen signal transduction as a mediator and therapeutic target in inflammation: Lessons from multiple sclerosis. Curr. Med. Chem. 2007, 14, 2925–2936. [Google Scholar] [CrossRef]
- Schultz, D.R.; Arnold, P.I. Properties of four acute phase proteins: C-reactive protein, serum amyloid A protein, alpha 1-acid glycoprotein, and fibrinogen. Semin. Arthritis Rheum. 1990, 20, 129–147. [Google Scholar] [CrossRef]
- Reinhart, W.H. Fibrinogen—Marker or mediator of vascular disease? Vasc. Med. 2003, 8, 211–216. [Google Scholar] [CrossRef]
- Ajay, A.K.; Saikumar, J.; Bijol, V.; Vaidya, V.S. Heterozygosity for fibrinogen results in efficient resolution of kidney ischemia reperfusion injury. PLoS ONE 2012, 7, e45628. [Google Scholar] [CrossRef]
- Sörensen-Zender, I.; Rong, S.; Susnik, N.; Lange, J.; Gueler, F.; Degen, J.L.; Melk, A.; Haller, H.; Schmitt, R. Role of fibrinogen in acute ischemic kidney injury. Am. J. Physiol. Renal Physiol. 2013, 305, F777–F785. [Google Scholar] [CrossRef]
- Hosohata, K. Role of Oxidative Stress in Drug-Induced Kidney Injury. Int. J. Mol. Sci. 2016, 17, 1826. [Google Scholar] [CrossRef]
- Murray, P.T.; Mehta, R.L.; Shaw, A.; Ronco, C.; Endre, Z.; Kellum, J.A.; Chawla, L.S.; Cruz, D.; Ince, C.; Okusa, M.D. Potential use of biomarkers in acute kidney injury: Report and summary of recommendations from the 10th Acute Dialysis Quality Initiative consensus conference. Kidney Int. 2014, 85, 513–521. [Google Scholar] [CrossRef]
- Mishra, J.; Mori, K.; Ma, Q.; Kelly, C.; Barasch, J.; Devarajan, P. Neutrophil gelatinas x 10associated lipocalin: A novel early urinary biomarker for cisplatin nephrotoxicity. Am. J. Nephrol. 2004, 24, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Prozialeck, W.C.; Vaidya, V.S.; Liu, J.; Waalkes, M.P.; Edwards, J.R.; Lamar, P.C.; Bernard, A.M.; Dumont, X.; Bonventre, J.V. Kidney injury molecul x 101 is an early biomarker of cadmium nephrotoxicity. Kidney Int. 2007, 72, 985–993. [Google Scholar] [CrossRef] [PubMed]
- George, B.; Wen, X.; Mercke, N.; Gomez, M.; O’Bryant, C.; Bowles, D.W.; Hu, Y.; Hogan, S.L.; Joy, M.S.; Aleksunes, L.M. Profiling of Kidney Injury Biomarkers in Patients Receiving Cisplatin: Tim x 10dependent Changes in the Absence of Clinical Nephrotoxicity. Clin. Pharmacol. Ther. 2017, 101, 510–518. [Google Scholar] [CrossRef] [PubMed]
- George, B.; Wen, X.; Mercke, N.; Gomez, M.; O’Bryant, C.; Bowles, D.W.; Hu, Y.; Hogan, S.L.; Joy, M.S.; Aleksunes, L.M. Tim x 10dependent changes in kidney injury biomarkers in patients receiving multiple cycles of cisplatin chemotherapy. Toxicol. Rep. 2020, 7, 571–576. [Google Scholar] [CrossRef]
- Chawla, L.S.; Bellomo, R.; Bihorac, A.; Goldstein, S.L.; Siew, E.D.; Bagshaw, S.M.; Fitzgerald, D.B.R.L.; Fitzgerald, D.B.R.L.; Mehta, D.C.E.M.R.; Mehta, D.C.E.M.R.; et al. Acute kidney disease and renal recovery: Consensus report of the Acute Disease Quality Initiative (ADQI) 16 Workgroup. Nat. Rev. Nephrol. 2017, 13, 241–257. [Google Scholar] [CrossRef]
- RamachandraRao, S.P.; Matthias, M.A.; Kokoy-Mondragon, C.; Aghania, E.; Park, C.; Kong, C.; Ishaya, M.; Madrigal, A.; Horng, J.; Khoshaba, R.; et al. Proteomic analysis of urine exosomes reveals renal tubule response to leptospiral colonization in experimentally infected rats. PLoS Negl. Trop. Dis. 2015, 9, e0003640. [Google Scholar]
- Haas, W.; Faherty, B.K.; Gerber, S.A.; Elias, J.E.; Beausoleil, S.A.; Bakalarski, C.E.; Li, X.; Villén, J.; Gygi, S.P. Optimization and use of peptide mass measurement accuracy in shotgun proteomics. Mol. Cell Proteom. 2006, 5, 1326–1337. [Google Scholar] [CrossRef]
- Tolonen, A.C.; Haas, W. Quantitative proteomics using reductive dimethylation for stable isotope labeling. J. Vis. Exp. 2014, 51416. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Mills, R.H.; Wozniak, J.M.; Vrbanac, A.; Campeau1, A.; Chassaing, B.; Gewirtz, A.; Knight, R.; Gonzalez1, D.J. Organ-level protein networks as a reference for the host effects of the microbiome. Genome Res. 2020, 30, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Tan, Q.; Kir, J.; Liu, D.; Bryant, D.; Guo, Y.; Stephens, R.; Baseler, M.W.; Lane, H.C.; et al. DAVID Bioinformatics Resources: Expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007, 35, W169–W175. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, J.M.; Mills, R.H.; Olson, J.; Caldera, J.; Sepich-Poore, G.D.; Carrillo-Terrazas, M.; Tsai, C.-M.; Vargas, F.; Knight, R.; Dorrestein, P.C.; et al. Mortality Risk Profiling of Staphylococcus aureus Bacteremia by Multi-omic Serum Analysis Reveals Early Predictive and Pathogenic Signatures. Cell 2020, 182, 1311–1327.e14. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genom x 10wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | V-AKI Cases N = 10 | Controls N = 12 | p-Value |
|---|---|---|---|
| Age (years), mean ± SD | 40.8 ± 18.3 | 32.7 ± 14.0 | 0.25 |
| Male gender, N (%) | 10 (100) | 12 (100) | 1 |
| Race/ethnicity, N (%) Caucasian African American Asian Hispanic American Indian or Alaska Native | 6 (60) 2 (20) 1 (10) 1 (10) 0 (0) | 2 (17) 2 (17) 3 (25) 0 (0) 5 (42) | 0.04 0.03 0.37 0.27 0.02 |
| Body mass index, mean ± SD | 26.6 ± 7.0 | 25.5 ± 4.7 | 0.67 |
| Comorbidities, N (%) Hypertension Diabetes mellitus Chronic obstructive pulmonary disease Coronary artery disease Heart Failure Liver disease Kidney disease Malignancy | 3 (30) 3 (30) 1 (10) 2 (20) 3 (30) 1 (10) 2 (20) 1 (10) | 0 (0) 0 (0) 0 (0) 0 (0) 0 (0) 0 (0) 0 (0) 0 (0) | 0.05 0.05 0.27 0.11 0.05 0.27 0.11 0.27 |
| Systolic Blood Pressure (mm Hg), mean ± SD | 121.1 ± 26.9 | 137.1 ± 10.8 | 0.07 |
| Diastolic blood pressure (mm Hg), mean ± SD | 78.5 ± 15.1 | 76 ± 11.1 | 0.66 |
| Outcome | V-AKI Cases N = 10 |
|---|---|
| Mortality, N(%) | 1 (10) |
| Need for RRT, N(%) | 2 (20) |
| AKI at Hospital Discharge | 9 (90) |
| AKD at Day 28 * | 2 (33) |
| AKD at Day 90 * | 0 (0) |
| Kidney Biopsy, N(%) | 2 (20) |
| AIN | 1 (10) |
| ATN | 2 (20) |
| GN | 1 (10) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Awdishu, L.; Le, A.; Amato, J.; Jani, V.; Bal, S.; Mills, R.H.; Carrillo-Terrazas, M.; Gonzalez, D.J.; Tolwani, A.; Acharya, A.; et al. Urinary Exosomes Identify Inflammatory Pathways in Vancomycin Associated Acute Kidney Injury. Int. J. Mol. Sci. 2021, 22, 2784. https://doi.org/10.3390/ijms22062784
Awdishu L, Le A, Amato J, Jani V, Bal S, Mills RH, Carrillo-Terrazas M, Gonzalez DJ, Tolwani A, Acharya A, et al. Urinary Exosomes Identify Inflammatory Pathways in Vancomycin Associated Acute Kidney Injury. International Journal of Molecular Sciences. 2021; 22(6):2784. https://doi.org/10.3390/ijms22062784
Chicago/Turabian StyleAwdishu, Linda, Amy Le, Jordan Amato, Vidhyut Jani, Soma Bal, Robert H. Mills, Marvic Carrillo-Terrazas, David J. Gonzalez, Ashita Tolwani, Anjali Acharya, and et al. 2021. "Urinary Exosomes Identify Inflammatory Pathways in Vancomycin Associated Acute Kidney Injury" International Journal of Molecular Sciences 22, no. 6: 2784. https://doi.org/10.3390/ijms22062784
APA StyleAwdishu, L., Le, A., Amato, J., Jani, V., Bal, S., Mills, R. H., Carrillo-Terrazas, M., Gonzalez, D. J., Tolwani, A., Acharya, A., Cerda, J., Joy, M. S., Nicoletti, P., Macedo, E., Vaingankar, S., Mehta, R., RamachandraRao, S. P., & on behalf of the Direct Investigators. (2021). Urinary Exosomes Identify Inflammatory Pathways in Vancomycin Associated Acute Kidney Injury. International Journal of Molecular Sciences, 22(6), 2784. https://doi.org/10.3390/ijms22062784