
Impact of SGLT-2 Inhibition on Cardiometabolic Abnormalities in a Rat Model of Polycystic Ovary Syndrome

 , , , and
, , , and
Abstract
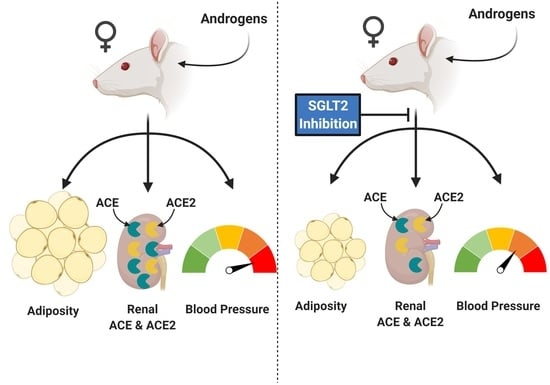
1. Introduction
2. Results
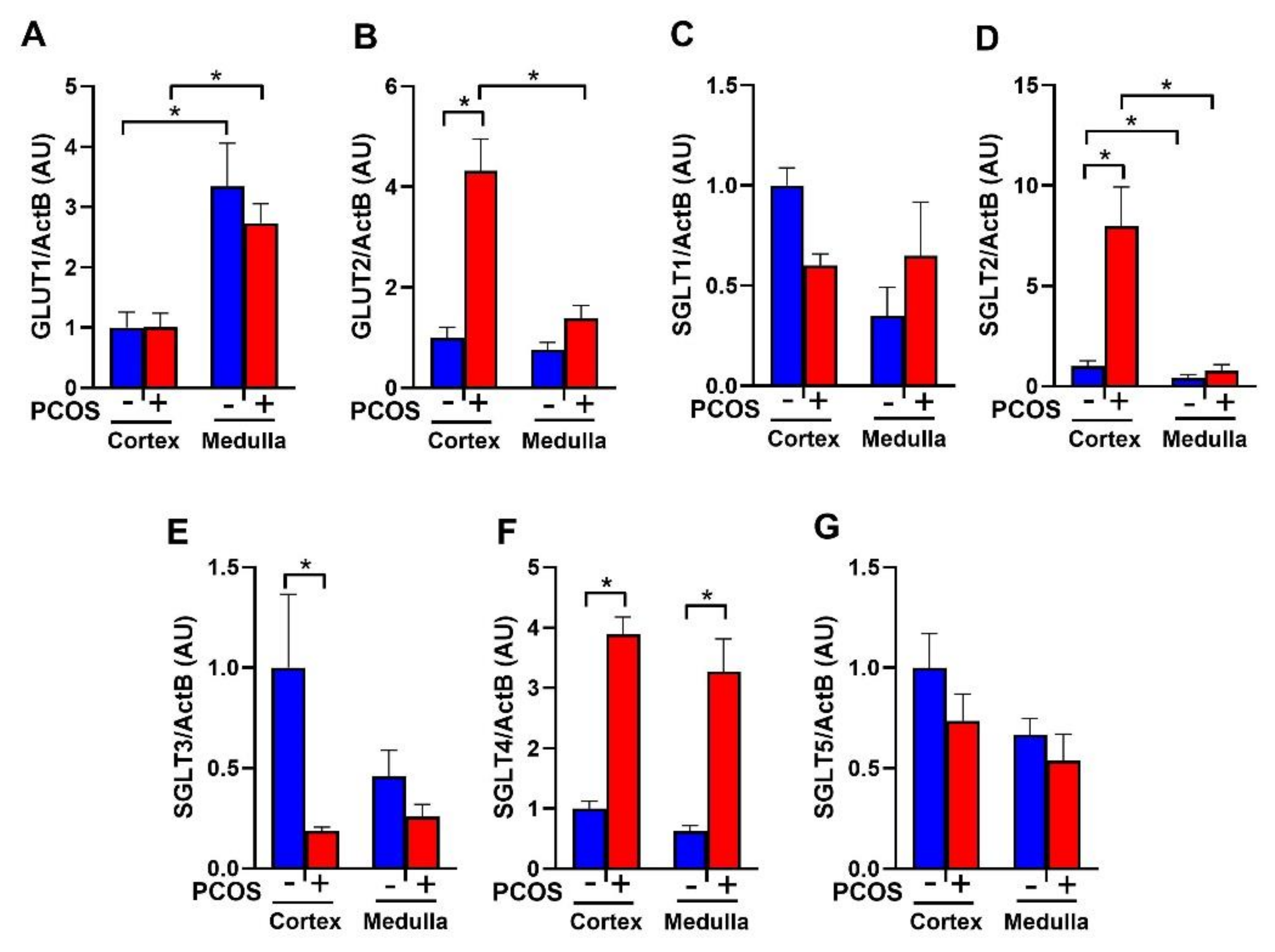
2.1. Effect of Hyperandrogenemia on Renal Glucose Transporter mRNA Expression
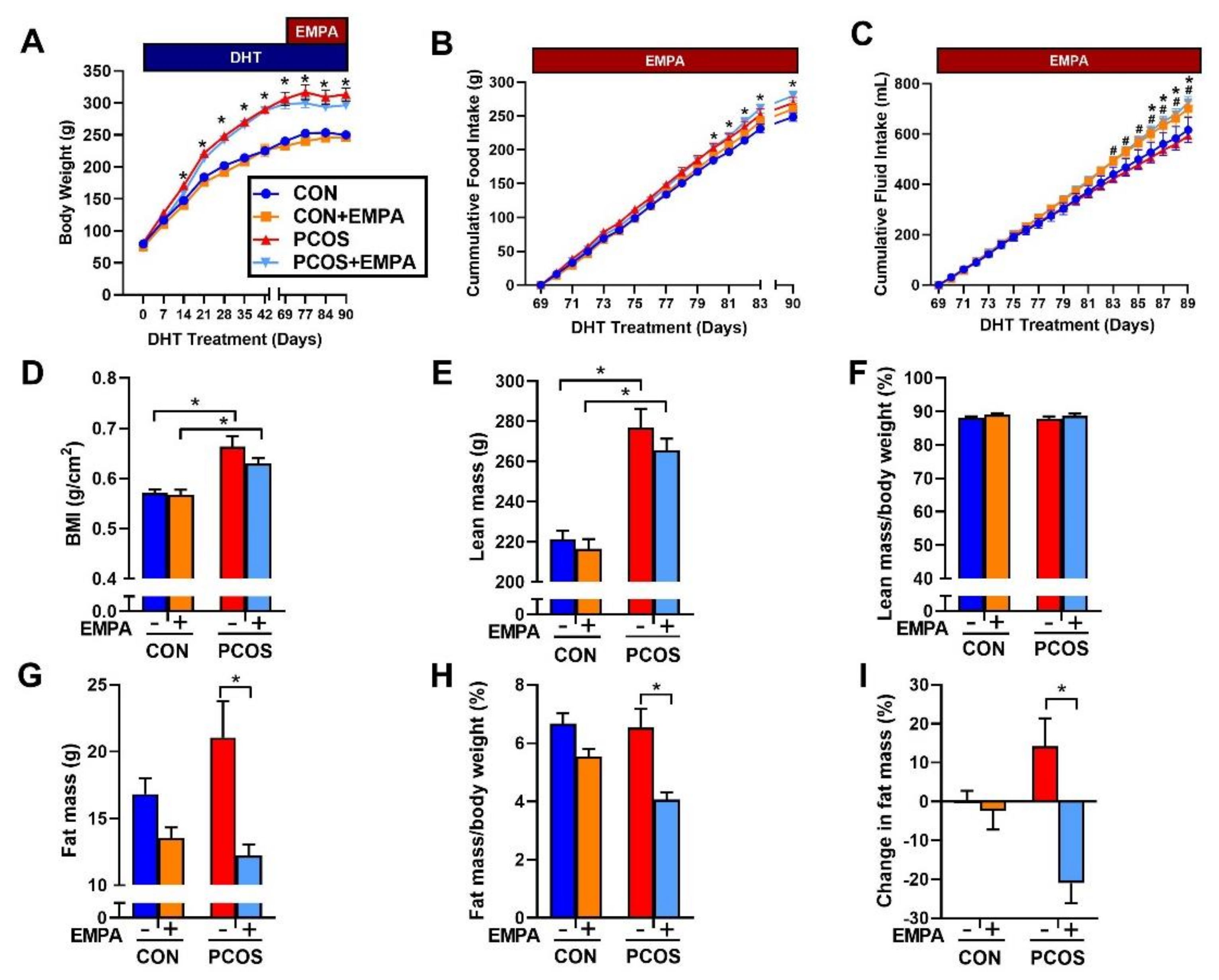
2.2. Effect of EMPA on Body Weight, Food Intake, and Fluid Intake in PCOS Model
2.3. Effect of EMPA on Body Composition in PCOS Model
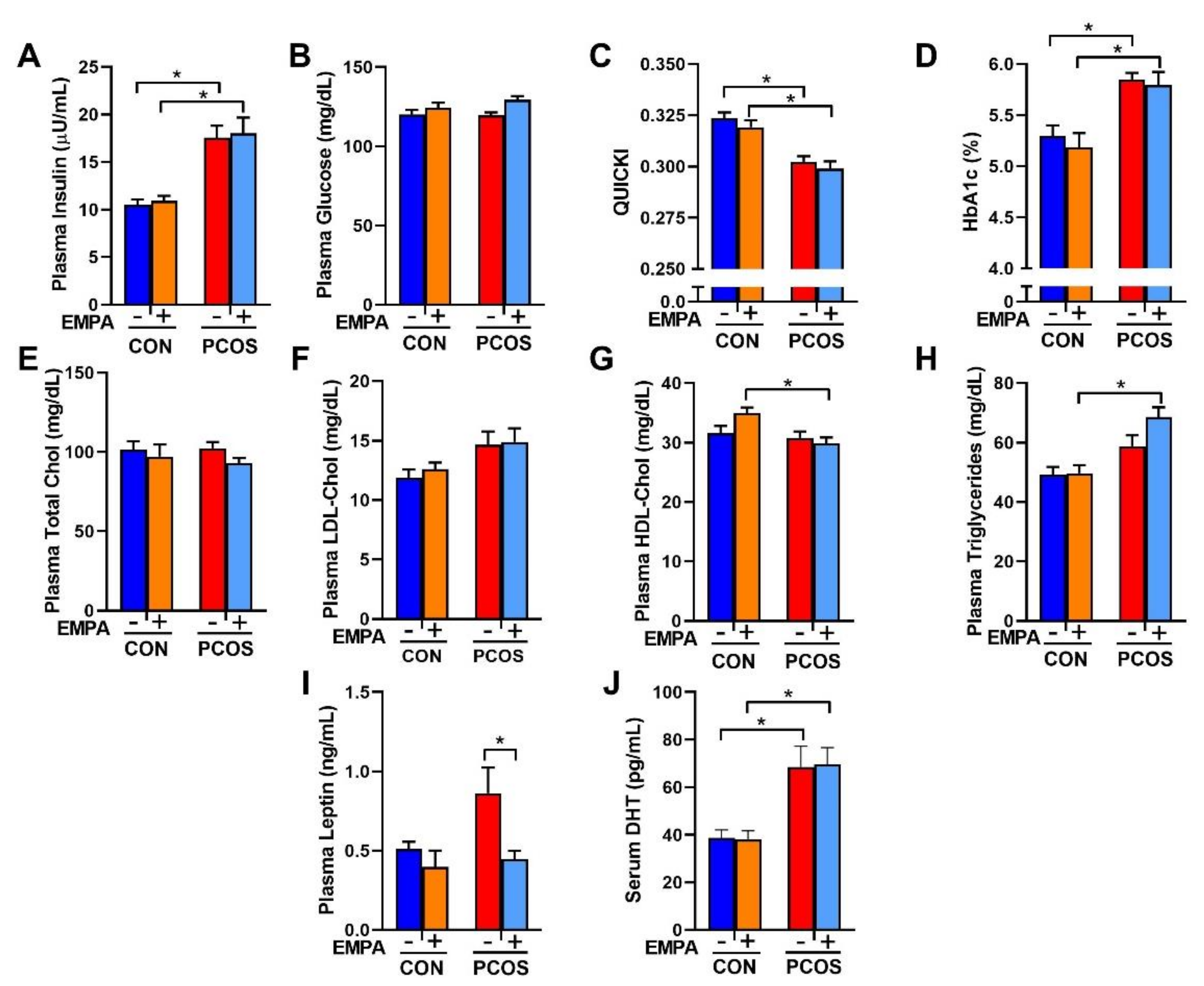
2.4. Effect of EMPA on Insulin Sensitivity, HbA1c, Fasting Lipid Profile, and Leptin in PCOS Model
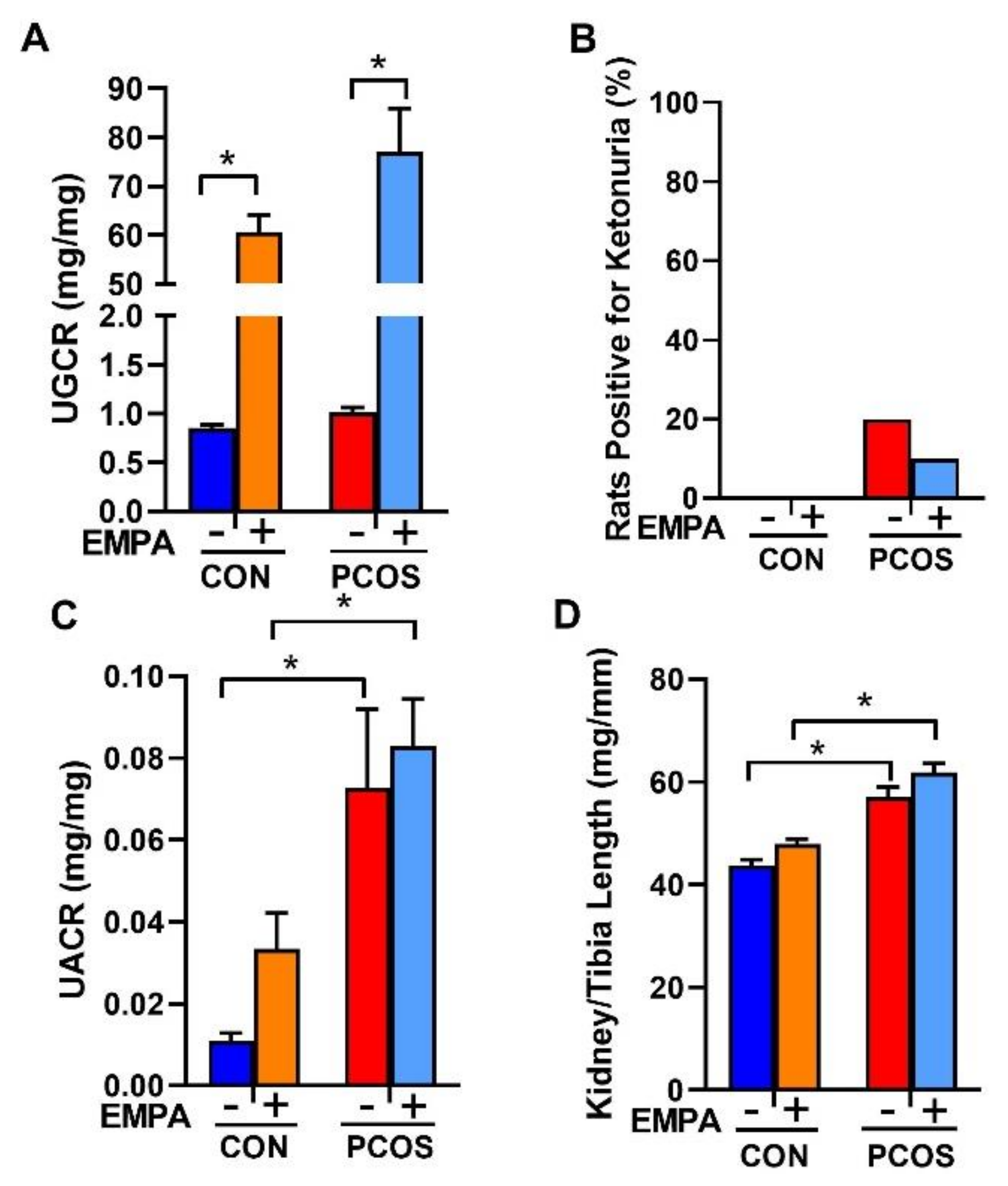
2.5. Effect of EMPA on Urinary Glucose, Ketone, and Albumin Excretion and Kidney Weight in a PCOS Model
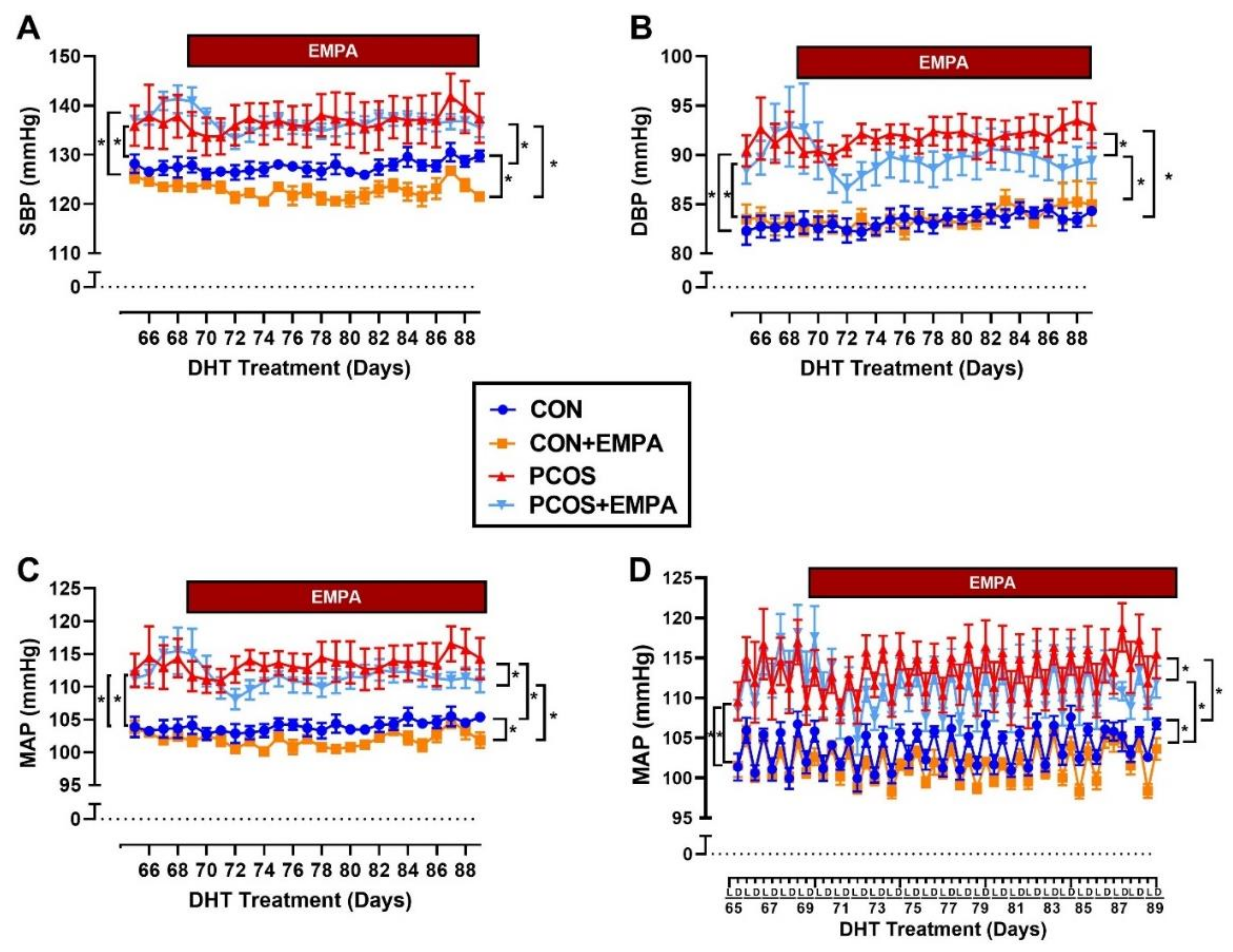
2.6. Effect of EMPA on Blood Pressure in PCOS Model
2.7. Effect of EMPA on Circadian Rhythm of Blood Pressure in PCOS Model
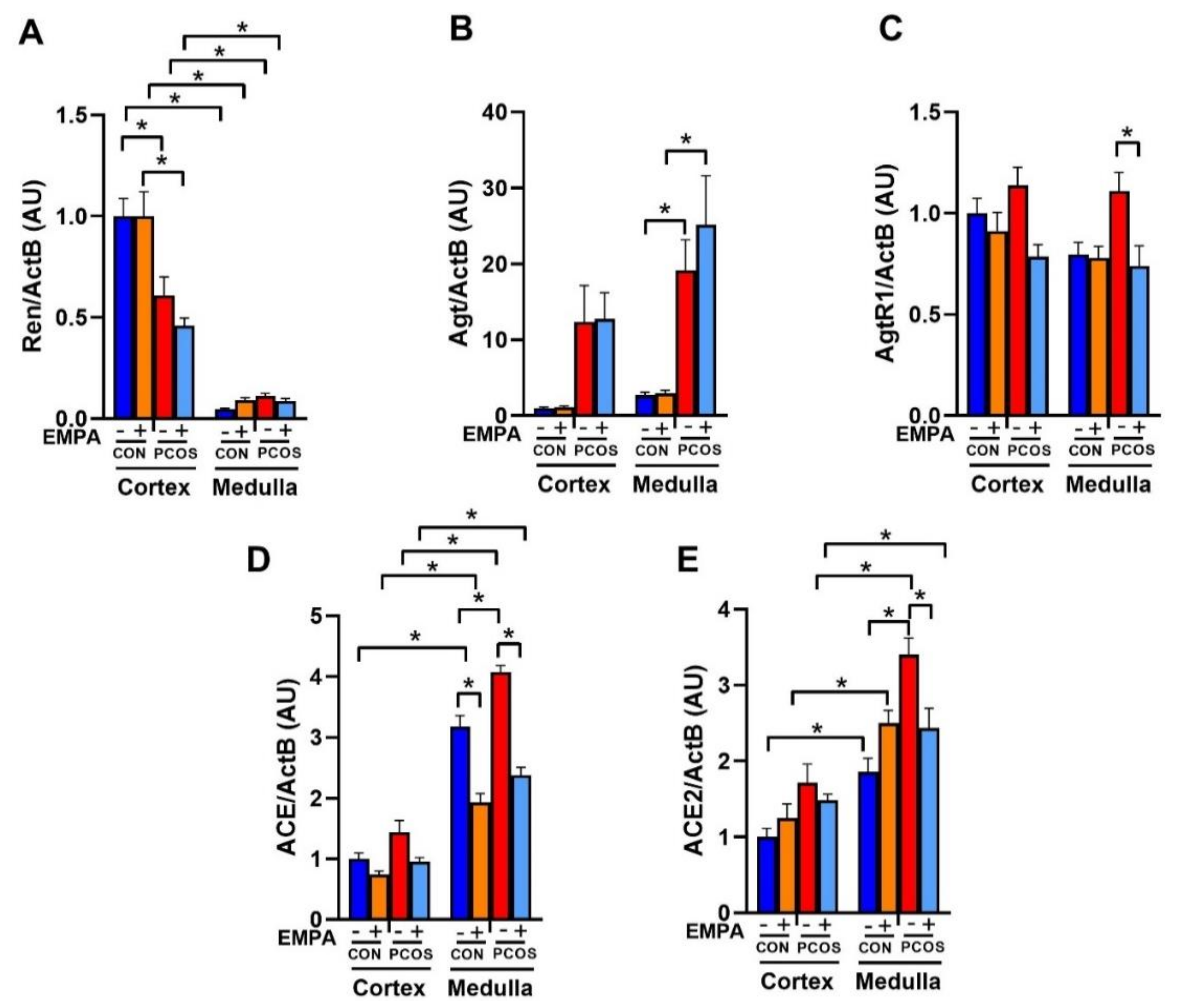
2.8. Effect of EMPA on the mRNA Expression of the Intrarenal Renin-Angiotensin System in PCOS Model
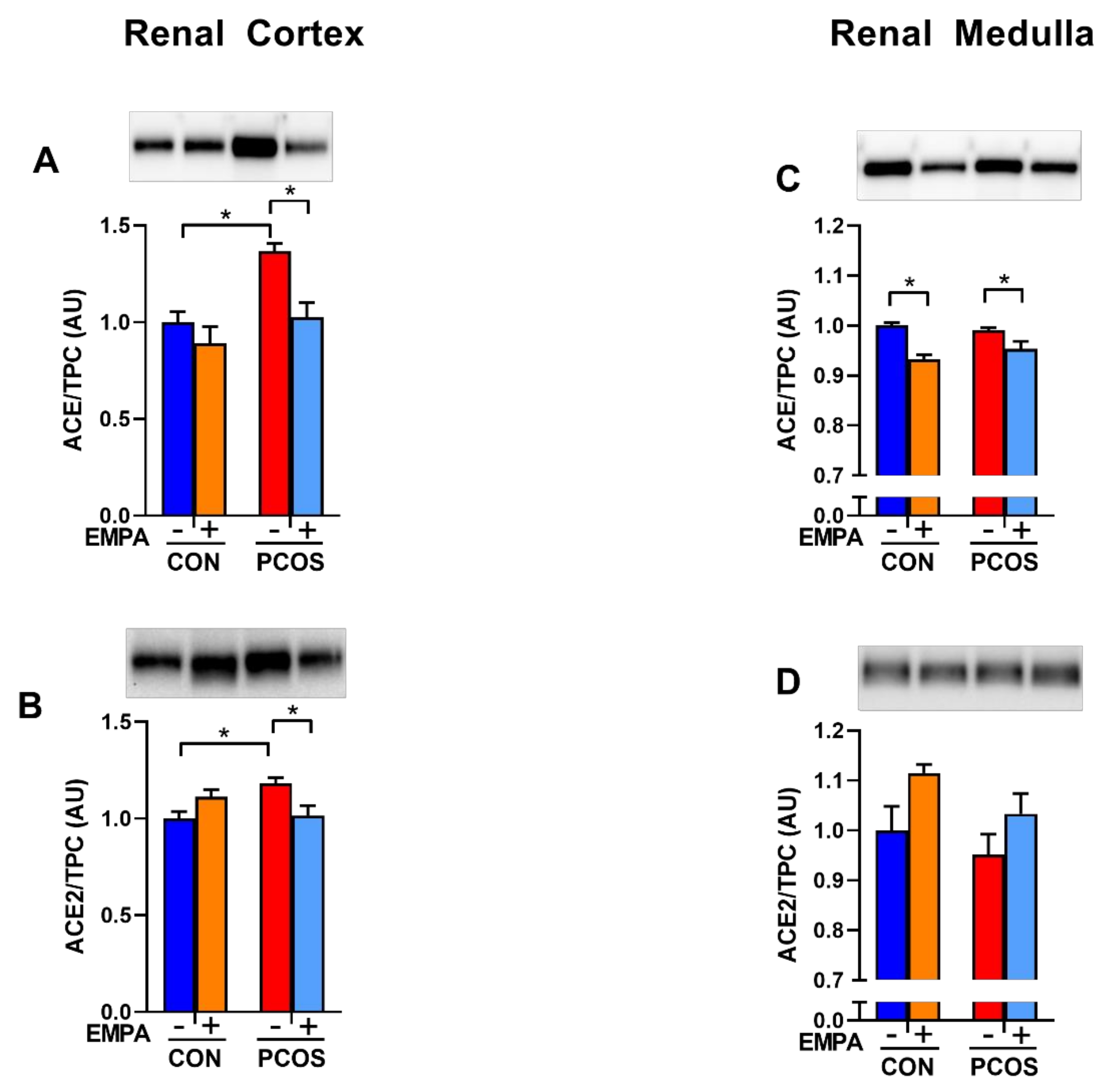
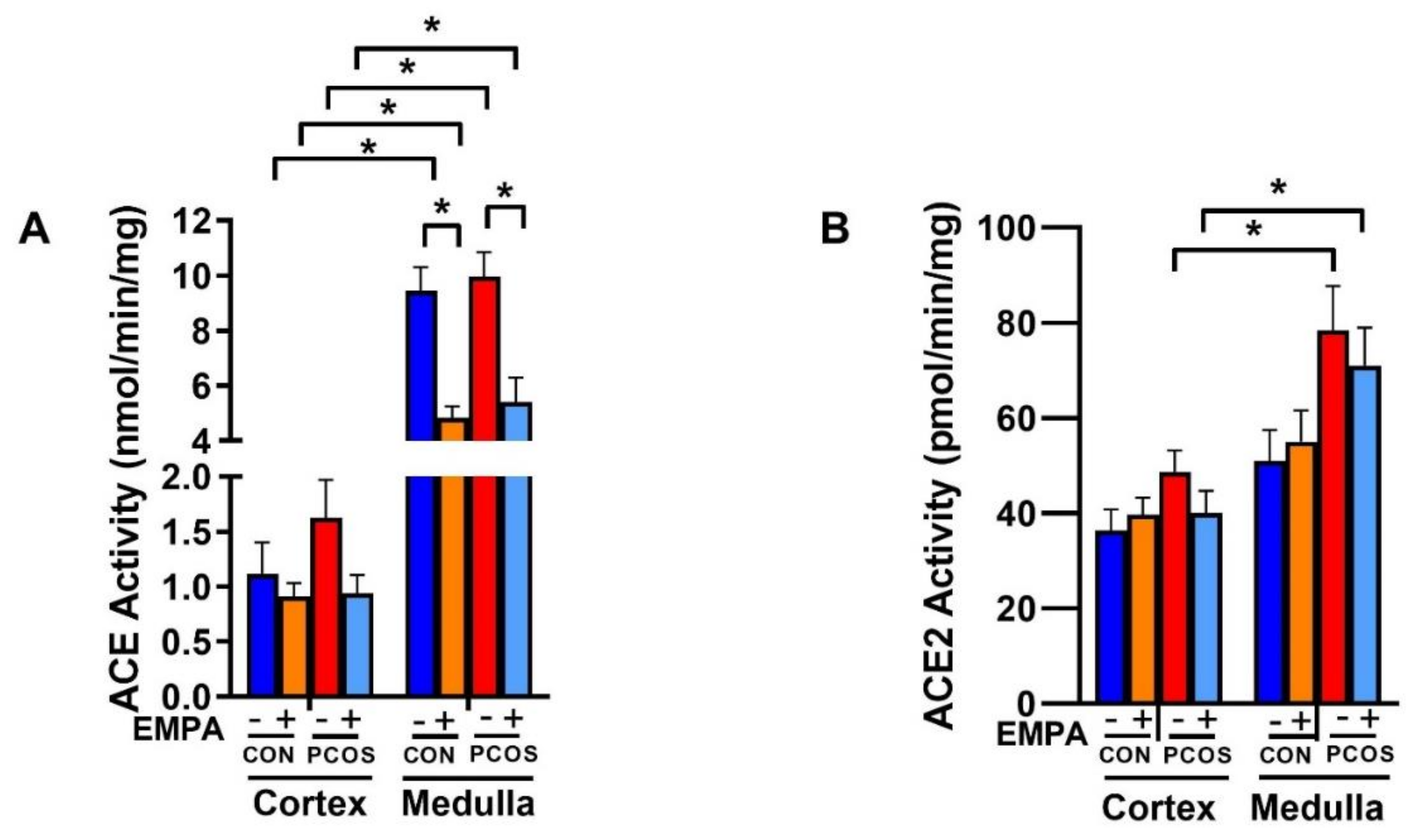
2.9. Effect of EMPA on Intrarenal ACE and ACE2 Protein Expression and Activity in PCOS Model
3. Discussion
4. Material and Methods
4.1. Animals
4.2. Food Intake, Anthropometric Measurements, and Body Composition
4.3. Metabolic Determinations
4.4. Urine Analysis
4.5. Tissue Collection
4.6. mRNA Expression Quantification
4.7. Protein Expression Quantification
4.8. Renal ACE and ACE2 Enzymatic Activity
4.9. Blood Pressure Measurement
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bozdag, G.; Mumusoglu, S.; Zengin, D.; Karabulut, E.; Yildiz, B.O. The prevalence and phenotypic features of polycystic ovary syndrome: A systematic review and meta-analysis. Hum. Reprod. 2016, 31, 2841–2855. [Google Scholar] [CrossRef]
- March, W.A.; Moore, V.M.; Willson, K.J.; Phillips, D.I.; Norman, R.J.; Davies, M.J. The prevalence of polycystic ovary syndrome in a community sample assessed under contrasting diagnostic criteria. Hum. Reprod. 2010, 25, 544–551. [Google Scholar] [CrossRef] [PubMed]
- Knochenhauer, E.S.; Key, T.J.; Kahsar-Miller, M.; Waggoner, W.; Boots, L.R.; Azziz, R. Prevalence of the polycystic ovary syndrome in unselected black and white women of the southeastern United States: A prospective study. J. Clin. Endocrinol. Metab. 1998, 83, 3078–3082. [Google Scholar] [CrossRef] [PubMed]
- Dumesic, D.A.; Oberfield, S.E.; Stener-Victorin, E.; Marshall, J.C.; Laven, J.S.; Legro, R.S. Scientific Statement on the Diagnostic Criteria, Epidemiology, Pathophysiology, and Molecular Genetics of Polycystic Ovary Syndrome. Endocr. Rev. 2015, 36, 487–525. [Google Scholar] [CrossRef] [PubMed]
- Nelson, V.L.; Legro, R.S.; Strauss, J.F., 3rd; McAllister, J.M. Augmented androgen production is a stable steroidogenic phenotype of propagated theca cells from polycystic ovaries. Mol. Endocrinol. 1999, 13, 946–957. [Google Scholar] [CrossRef] [PubMed]
- Carmina, E. Ovarian and adrenal hyperandrogenism. Ann. N. Y. Acad. Sci. 2006, 1092, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Quinkler, M.; Sinha, B.; Tomlinson, J.W.; Bujalska, I.J.; Stewart, P.M.; Arlt, W. Androgen generation in adipose tissue in women with simple obesity--a site-specific role for 17beta-hydroxysteroid dehydrogenase type 5. J. Endocrinol. 2004, 183, 331–342. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, M.; Gathercole, L.; Capper, F.; Arlt, W.; Tomlinson, J. Effect of insulin on AKR1C3 expression in female adipose tissue: In-vivo and in-vitro study of adipose androgen generation in polycystic ovary syndrome. Lancet 2015, 385, S16. [Google Scholar] [CrossRef]
- Lo, J.C.; Feigenbaum, S.L.; Yang, J.; Pressman, A.R.; Selby, J.V.; Go, A.S. Epidemiology and adverse cardiovascular risk profile of diagnosed polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2006, 91, 1357–1363. [Google Scholar] [CrossRef]
- Elting, M.W.; Korsen, T.J.; Bezemer, P.D.; Schoemaker, J. Prevalence of diabetes mellitus, hypertension and cardiac complaints in a follow-up study of a Dutch PCOS population. Hum. Reprod. 2001, 16, 556–560. [Google Scholar] [CrossRef]
- Song, Y.; Ye, W.; Ye, H.; Xie, T.; Shen, W.; Zhou, L. Serum testosterone acts as a prognostic indicator in polycystic ovary syndrome-associated kidney injury. Physiol. Rep. 2019, 7, e14219. [Google Scholar] [CrossRef] [PubMed]
- Rojas, J.; Chavez, M.; Olivar, L.; Rojas, M.; Morillo, J.; Mejias, J.; Calvo, M.; Bermudez, V. Polycystic ovary syndrome, insulin resistance, and obesity: Navigating the pathophysiologic labyrinth. Int. J. Reprod. Med. 2014, 2014, 719050. [Google Scholar] [CrossRef]
- McCartney, C.R.; Marshall, J.C. CLINICAL PRACTICE. Polycystic Ovary Syndrome. N. Engl. J. Med. 2016, 375, 54–64. [Google Scholar] [CrossRef]
- Javed, Z.; Papageorgiou, M.; Deshmukh, H.; Rigby, A.S.; Qamar, U.; Abbas, J.; Khan, A.Y.; Kilpatrick, E.S.; Atkin, S.L.; Sathyapalan, T. Effects of empagliflozin on metabolic parameters in polycystic ovary syndrome: A randomized controlled study. Clin. Endocrinol. 2019, 90, 805–813. [Google Scholar] [CrossRef]
- Sabolic, I.; Vrhovac, I.; Eror, D.B.; Gerasimova, M.; Rose, M.; Breljak, D.; Ljubojevic, M.; Brzica, H.; Sebastiani, A.; Thal, S.C.; et al. Expression of Na+-D-glucose cotransporter SGLT2 in rodents is kidney-specific and exhibits sex and species differences. Am. J. Physiol. Cell Physiol. 2012, 302, C1174–C1188. [Google Scholar] [CrossRef]
- Alicic, R.Z.; Neumiller, J.J.; Johnson, E.J.; Dieter, B.; Tuttle, K.R. Sodium-Glucose Cotransporter 2 Inhibition and Diabetic Kidney Disease. Diabetes 2019, 68, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Butler, J.; Handelsman, Y.; Bakris, G.; Verma, S. Use of sodium-glucose co-transporter-2 inhibitors in patients with and without type 2 diabetes: Implications for incident and prevalent heart failure. Eur. J. Heart Fail. 2020, 22, 604–617. [Google Scholar] [CrossRef] [PubMed]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef]
- Mudaliar, S.; Polidori, D.; Zambrowicz, B.; Henry, R.R. Sodium-Glucose Cotransporter Inhibitors: Effects on Renal and Intestinal Glucose Transport: From Bench to Bedside. Diabetes Care 2015, 38, 2344–2353. [Google Scholar] [CrossRef]
- Kojima, N.; Williams, J.M.; Slaughter, T.N.; Kato, S.; Takahashi, T.; Miyata, N.; Roman, R.J. Renoprotective effects of combined SGLT2 and ACE inhibitor therapy in diabetic Dahl S rats. Physiol. Rep. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Gallo, L.A.; Ward, M.S.; Fotheringham, A.K.; Zhuang, A.; Borg, D.J.; Flemming, N.B.; Harvie, B.M.; Kinneally, T.L.; Yeh, S.M.; McCarthy, D.A.; et al. Once daily administration of the SGLT2 inhibitor, empagliflozin, attenuates markers of renal fibrosis without improving albuminuria in diabetic db/db mice. Sci. Rep. 2016, 6, 26428. [Google Scholar] [CrossRef] [PubMed]
- Takasu, T.; Hayashizaki, Y.; Hirosumi, J.; Minoura, H.; Amino, N.; Kurosaki, E.; Takakura, S. The Sodium Glucose Cotransporter 2 Inhibitor Ipragliflozin Promotes Preferential Loss of Fat Mass in Non-obese Diabetic Goto-Kakizaki Rats. Biol. Pharm. Bull. 2017, 40, 675–680. [Google Scholar] [CrossRef][Green Version]
- Diamanti-Kandarakis, E.; Economou, F.N.; Livadas, S.; Tantalaki, E.; Piperi, C.; Papavassiliou, A.G.; Panidis, D. Hyperreninemia characterizing women with polycystic ovary syndrome improves after metformin therapy. Kidney Blood Press. Res. 2009, 32, 24–31. [Google Scholar] [CrossRef]
- Jensterle, M.; Janez, A.; Vrtovec, B.; Meden-Vrtovec, H.; Pfeifer, M.; Prezelj, J.; Kocjan, T. Decreased androgen levels and improved menstrual pattern after angiotensin II receptor antagonist telmisartan treatment in four hypertensive patients with polycystic ovary syndrome: Case series. Croat. Med. J. 2007, 48, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Yanes, L.L.; Romero, D.G.; Moulana, M.; Lima, R.; Davis, D.D.; Zhang, H.; Lockhart, R.; Racusen, L.C.; Reckelhoff, J.F. Cardiovascular-renal and metabolic characterization of a rat model of polycystic ovary syndrome. Gend. Med. 2011, 8, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Bautista, R.; Manning, R.; Martinez, F.; Avila-Casado Mdel, C.; Soto, V.; Medina, A.; Escalante, B. Angiotensin II-dependent increased expression of Na+-glucose cotransporter in hypertension. Am. J. Physiol. Renal Physiol. 2004, 286, F127–F133. [Google Scholar] [CrossRef] [PubMed]
- Woods, T.C.; Satou, R.; Miyata, K.; Katsurada, A.; Dugas, C.M.; Klingenberg, N.C.; Fonseca, V.A.; Navar, L.G. Canagliflozin Prevents Intrarenal Angiotensinogen Augmentation and Mitigates Kidney Injury and Hypertension in Mouse Model of Type 2 Diabetes Mellitus. Am. J. Nephrol. 2019, 49, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Navar, L.G.; Kobori, H.; Prieto, M.C.; Gonzalez-Villalobos, R.A. Intratubular renin-angiotensin system in hypertension. Hypertension 2011, 57, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Torres Fernandez, E.D.; Adams, K.V.; Syed, M.; Maranon, R.O.; Romero, D.G.; Yanes Cardozo, L.L. Long-Lasting Androgen-Induced Cardiometabolic Effects in Polycystic Ovary Syndrome. J. Endocr. Soc. 2018, 2, 949–964. [Google Scholar] [CrossRef]
- Torres Fernandez, E.D.; Huffman, A.M.; Syed, M.; Romero, D.G.; Yanes Cardozo, L.L. Effect of GLP-1 Receptor Agonists in the Cardiometabolic Complications in a Rat Model of Postmenopausal PCOS. Endocrinology 2019, 160, 2787–2799. [Google Scholar] [CrossRef] [PubMed]
- Yanes Cardozo, L.L.; Romero, D.G.; Reckelhoff, J.F. Cardiometabolic Features of Polycystic Ovary Syndrome: Role of Androgens. Physiology 2017, 32, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Novikov, A.; Vallon, V. Sodium glucose cotransporter 2 inhibition in the diabetic kidney: An update. Curr. Opin. Nephrol. Hypertens 2016, 25, 50–58. [Google Scholar] [CrossRef]
- Song, P.; Onishi, A.; Koepsell, H.; Vallon, V. Sodium glucose cotransporter SGLT1 as a therapeutic target in diabetes mellitus. Expert Opin. Ther. Targets 2016, 20, 1109–1125. [Google Scholar] [CrossRef] [PubMed]
- Sabolic, I.; Skarica, M.; Gorboulev, V.; Ljubojevic, M.; Balen, D.; Herak-Kramberger, C.M.; Koepsell, H. Rat renal glucose transporter SGLT1 exhibits zonal distribution and androgen-dependent gender differences. Am. J. Physiol. Renal Physiol. 2006, 290, F913–F926. [Google Scholar] [CrossRef]
- Sotak, M.; Marks, J.; Unwin, R.J. Putative tissue location and function of the SLC5 family member SGLT3. Exp. Physiol. 2017, 102, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Wright, E.M. Glucose transport families SLC5 and SLC50. Mol. Asp. Med. 2013, 34, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Ansary, T.M.; Nakano, D.; Nishiyama, A. Diuretic Effects of Sodium Glucose Cotransporter 2 Inhibitors and Their Influence on the Renin-Angiotensin System. Int. J. Mol. Sci. 2019, 20, 629. [Google Scholar] [CrossRef]
- Alreja, G.; Joseph, J. Renin and cardiovascular disease: Worn-out path, or new direction. World J. Cardiol. 2011, 3, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Yanes Cardozo, L.L.; Rezq, S.; Pruett, J.E.; Romero, D.G. Androgens, the kidney, and COVID-19: An opportunity for translational research. Am. J. Physiol. Renal Physiol. 2021, 320, F243–F248. [Google Scholar] [CrossRef]
- Shin, S.J.; Chung, S.; Kim, S.J.; Lee, E.M.; Yoo, Y.H.; Kim, J.W.; Ahn, Y.B.; Kim, E.S.; Moon, S.D.; Kim, M.J.; et al. Effect of Sodium-Glucose Co-Transporter 2 Inhibitor, Dapagliflozin, on Renal Renin-Angiotensin System in an Animal Model of Type 2 Diabetes. PLoS ONE 2016, 11, e0165703. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Nagata, N.; Nagashimada, M.; Zhuge, F.; Ni, Y.; Chen, G.; Mayoux, E.; Kaneko, S.; Ota, T. SGLT2 inhibition by empagliflozin promotes fat utilization and browning and attenuates inflammation and insulin resistance by polarizing M2 macrophages in diet-induced obese mice. EBioMedicine 2017, 20, 137–149. [Google Scholar] [CrossRef]
- Hall, J.E.; do Carmo, J.M.; da Silva, A.A.; Wang, Z.; Hall, M.E. Obesity, kidney dysfunction and hypertension: Mechanistic links. Nat. Rev. Nephrol. 2019, 15, 367–385. [Google Scholar] [CrossRef] [PubMed]
- Herat, L.Y.; Magno, A.L.; Rudnicka, C.; Hricova, J.; Carnagarin, R.; Ward, N.C.; Arcambal, A.; Kiuchi, M.G.; Head, G.A.; Schlaich, M.P.; et al. SGLT2 Inhibitor-Induced Sympathoinhibition: A Novel Mechanism for Cardiorenal Protection. JACC Basic Transl. Sci. 2020, 5, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Derelanko, M.J. Determination of erythrocyte life span in F-344, Wistar, and Sprague-Dawley rats using a modification of the [3H]diisopropylfluorophosphate ([3H]DFP) method. Fundam. Appl. Toxicol. 1987, 9, 271–276. [Google Scholar] [CrossRef]
- Abdul-Ghani, M.; Al Jobori, H.; Daniele, G.; Adams, J.; Cersosimo, E.; Triplitt, C.; DeFronzo, R.A. Inhibition of Renal Sodium-Glucose Cotransport With Empagliflozin Lowers Fasting Plasma Glucose and Improves beta-Cell Function in Subjects with Impaired Fasting Glucose. Diabetes 2017, 66, 2495–2502. [Google Scholar] [CrossRef]
- Gembardt, F.; Bartaun, C.; Jarzebska, N.; Mayoux, E.; Todorov, V.T.; Hohenstein, B.; Hugo, C. The SGLT2 inhibitor empagliflozin ameliorates early features of diabetic nephropathy in BTBR ob/ob type 2 diabetic mice with and without hypertension. Am. J. Physiol. Renal Physiol. 2014, 307, F317–F325. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Fu, Y.; Ge, Y.; Juncos, L.A.; Reckelhoff, J.F.; Liu, R. The vasodilatory effect of testosterone on renal afferent arterioles. Gend. Med. 2012, 9, 103–111. [Google Scholar] [CrossRef]
- Zanoli, L.; Granata, A.; Lentini, P.; Rastelli, S.; Fatuzzo, P.; Rapisarda, F.; Castellino, P. Sodium-glucose linked transporter-2 inhibitors in chronic kidney disease. Sci. World J. 2015, 2015, 317507. [Google Scholar] [CrossRef]
- Gibson-Helm, M.; Teede, H.; Dunaif, A.; Dokras, A. Delayed diagnosis and a lack of information associated with dissatisfaction in women with polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2017, 102, 604–612. [Google Scholar] [CrossRef]
- Steven, S.; Oelze, M.; Hanf, A.; Kroller-Schon, S.; Kashani, F.; Roohani, S.; Welschof, P.; Kopp, M.; Godtel-Armbrust, U.; Xia, N.; et al. The SGLT2 inhibitor empagliflozin improves the primary diabetic complications in ZDF rats. Redox Biol. 2017, 13, 370–385. [Google Scholar] [CrossRef] [PubMed]
- Gopal, N.M.; Sridhar, C. A Validated Stability Indicating Ultra-Performance Liquid Chromatographic Method for Simultaneous Determination of Metformin Hydrochloride and Empagliflozin in Bulk Drug and Tablet Dosage Form. Int. J. Appl. Pharm. 2017, 9, 45–50. [Google Scholar] [CrossRef]
- Syed, M.; Ball, J.P.; Mathis, K.W.; Hall, M.E.; Ryan, M.J.; Rothenberg, M.E.; Yanes Cardozo, L.L.; Romero, D.G. MicroRNA-21 ablation exacerbates aldosterone-mediated cardiac injury, remodeling, and dysfunction. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E1154–E1167. [Google Scholar] [CrossRef] [PubMed]
- Gilda, J.E.; Gomes, A.V. Stain-Free total protein staining is a superior loading control to β-actin for Western blots. Anal. Biochem. 2013, 440, 186–188. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Zor, T.; Selinger, Z. Linearization of the Bradford protein assay increases its sensitivity: Theoretical and experimental studies. Anal. Biochem. 1996, 236, 302–308. [Google Scholar] [CrossRef]
- Friedland, J.; Silverstein, E. A sensitive fluorimetric assay for serum angiotensin-converting enzyme. Am. J. Clin. Pathol. 1976, 66, 416–424. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.A.; Krieger, E.M.; Greene, L.J. An improved fluorometric assay of rat serum and plasma converting enzyme. Hypertension 1985, 7, 244–252. [Google Scholar] [CrossRef]
- Oliveira, E.M.; Santos, R.A.; Krieger, J.E. Standardization of a fluorimetric assay for the determination of tissue angiotensin-converting enzyme activity in rats. Braz. J. Med. Biol. Res. 2000, 33, 755–764. [Google Scholar] [CrossRef]
- Vickers, C.; Hales, P.; Kaushik, V.; Dick, L.; Gavin, J.; Tang, J.; Godbout, K.; Parsons, T.; Baronas, E.; Hsieh, F.; et al. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J. Biol. Chem. 2002, 277, 14838–14843. [Google Scholar] [CrossRef] [PubMed]
- Guy, J.L.; Jackson, R.M.; Acharya, K.R.; Sturrock, E.D.; Hooper, N.M.; Turner, A.J. Angiotensin-converting enzyme-2 (ACE2): Comparative modeling of the active site, specificity requirements, and chloride dependence. Biochemistry 2003, 42, 13185–13192. [Google Scholar] [CrossRef]
- Pedersen, K.B.; Sriramula, S.; Chhabra, K.H.; Xia, H.; Lazartigues, E. Species-specific inhibitor sensitivity of angiotensin-converting enzyme 2 (ACE2) and its implication for ACE2 activity assays. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301, 1293–1299. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.; Wysocki, J.; Gonzalez-Pacheco, F.R.; Salem, M.; Evora, K.; Garcia-Halpin, L.; Poglitsch, M.; Schuster, M.; Batlle, D. Murine recombinant angiotensin-converting enzyme 2: Effect on angiotensin II-dependent hypertension and distinctive angiotensin-converting enzyme 2 inhibitor characteristics on rodent and human angiotensin-converting enzyme 2. Hypertension 2012, 60, 730–740. [Google Scholar] [CrossRef] [PubMed]
- Xiao, F.; Burns, K.D. Measurement of Angiotensin Converting Enzyme 2 Activity in Biological Fluid (ACE2). In Hypertension: Methods and Protocols; Touyz, R.M., Schiffrin, E.L., Eds.; Springer: New York, NY, USA, 2017. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Gene Symbol | TaqMan Assay ID |
|---|---|---|
| Glucose Transporter-1 (GLUT1) | Slc2a1 | Rn01417099_m1 |
| Glucose Transporter-2 (GLUT2) | Slc2a2 | Rn00563565_m1 |
| Sodium-Glucose Cotransporter-1 (SGLT1) | Slc5a1 | Rn01640634_m1 |
| Sodium-Glucose Cotransporter-2 (SGLT2) | Slc5a2 | Rn00574917_m1 |
| Sodium-Glucose Cotransporter-3 (SGLT3) | Slc5a4 | Rn01429310_m1 |
| Sodium-Glucose Cotransporter-4 (SGLT4) | Slc5a9 | Rn01761671_m1 |
| Sodium-Glucose Cotransporter-5 (SGLT5) | Slc5a10 | Rn01773089_m1 |
| Renin | Ren | Rn00561847_m1 |
| Angiotensinogen | Agt | Rn00593114_m1 |
| Angiotensin-Converting Enzyme | Ace | Rn00561094_m1 |
| Angiotensin-Converting Enzyme 2 | Ace2 | Rn01416293_m1 |
| Angiotensin II Receptor Type 1a | Agtr1a | Rn02758772_s1 |
| β-Actin | Actb | Rn00667869_m1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pruett, J.E.; Torres Fernandez, E.D.; Everman, S.J.; Vinson, R.M.; Davenport, K.; Logan, M.K.; Ye, S.A.; Romero, D.G.; Yanes Cardozo, L.L. Impact of SGLT-2 Inhibition on Cardiometabolic Abnormalities in a Rat Model of Polycystic Ovary Syndrome. Int. J. Mol. Sci. 2021, 22, 2576. https://doi.org/10.3390/ijms22052576
Pruett JE, Torres Fernandez ED, Everman SJ, Vinson RM, Davenport K, Logan MK, Ye SA, Romero DG, Yanes Cardozo LL. Impact of SGLT-2 Inhibition on Cardiometabolic Abnormalities in a Rat Model of Polycystic Ovary Syndrome. International Journal of Molecular Sciences. 2021; 22(5):2576. https://doi.org/10.3390/ijms22052576
Chicago/Turabian StylePruett, Jacob E., Edgar D. Torres Fernandez, Steven J. Everman, Ruth M. Vinson, Kacey Davenport, Madelyn K. Logan, Stephanie A. Ye, Damian G. Romero, and Licy L. Yanes Cardozo. 2021. "Impact of SGLT-2 Inhibition on Cardiometabolic Abnormalities in a Rat Model of Polycystic Ovary Syndrome" International Journal of Molecular Sciences 22, no. 5: 2576. https://doi.org/10.3390/ijms22052576
APA StylePruett, J. E., Torres Fernandez, E. D., Everman, S. J., Vinson, R. M., Davenport, K., Logan, M. K., Ye, S. A., Romero, D. G., & Yanes Cardozo, L. L. (2021). Impact of SGLT-2 Inhibition on Cardiometabolic Abnormalities in a Rat Model of Polycystic Ovary Syndrome. International Journal of Molecular Sciences, 22(5), 2576. https://doi.org/10.3390/ijms22052576