
A Novel 1,8-Naphthyridine-2-Carboxamide Derivative Attenuates Inflammatory Responses and Cell Migration in LPS-Treated BV2 Cells via the Suppression of ROS Generation and TLR4/Myd88/NF-κB Signaling Pathway

Abstract
1. Introduction
2. Results
2.1. Effects of 1,8-Naphthyridine-2-Carboxamide Derivatives on the Viability of BV2 Cells
2.2. Effects of 1,8-Naphthyridine-2-Carboxamide Derivatives on the LPS-Stimulated Production of Pro-Inflammatory Mediators in BV2 Cells
2.3. Effects of HSR2104 on the LPS-Induced iNOS and COX2 Expression in BV2 Cells
2.4. Effect of HSR2104 on the LPS-Induced BV2 Cell Migration
2.5. Effect of HSR2104 on the LPS-Stimulated Generation of Intracellular ROS in BV2 Cells
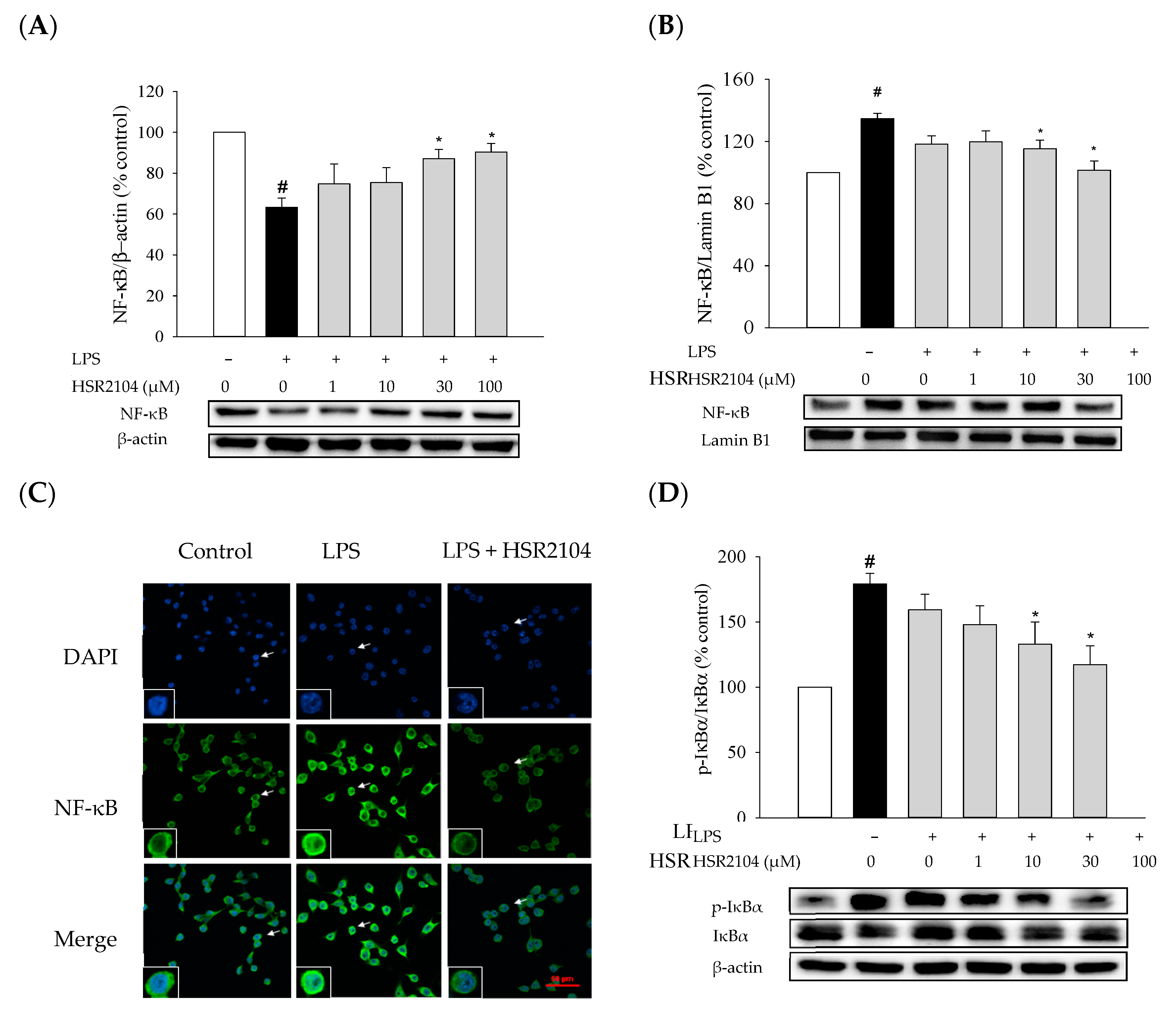
2.6. Effects of HSR2104 on the LPS-Induced IκBα Phosphorylation and NF-κB Translocation in BV2 Cells
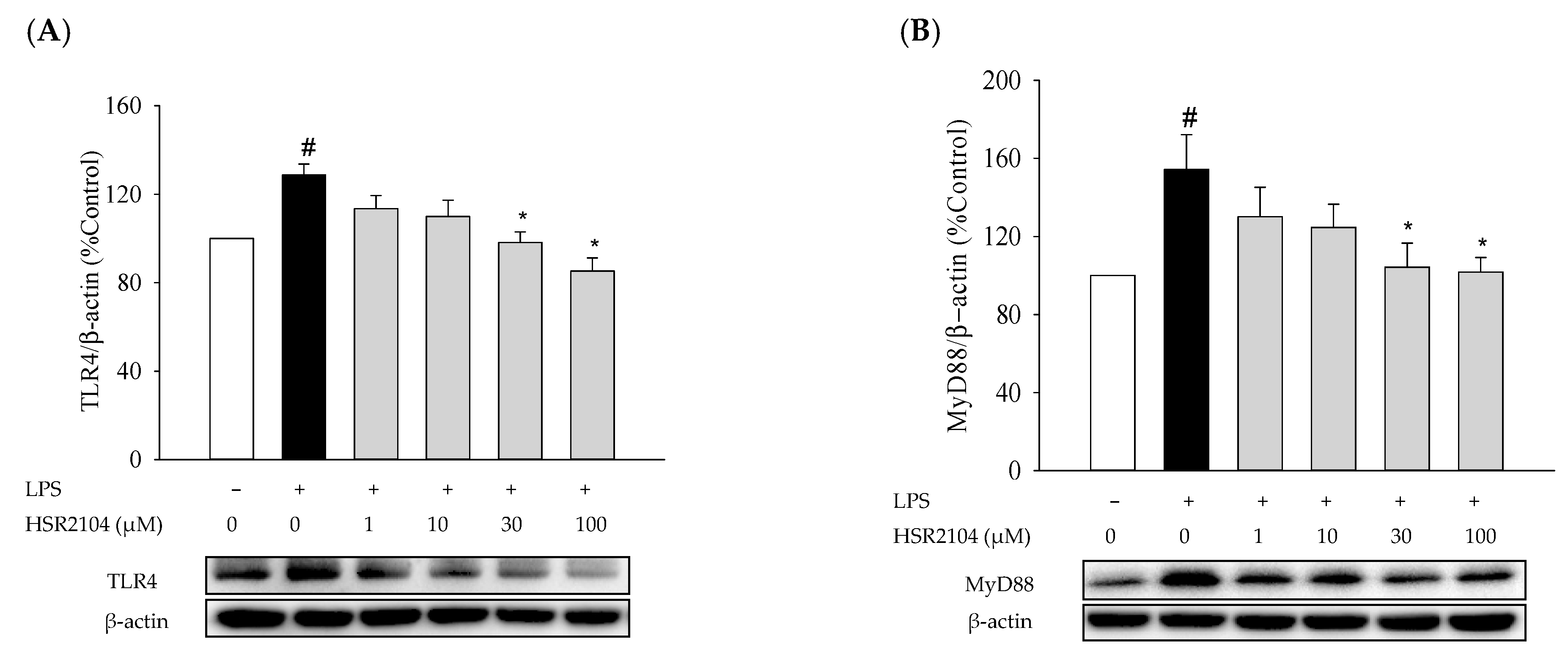
2.7. Effects of HSR2104 on the LPS-Induced Expressions of TLR4 and MyD88 in BV2 Cells
2.8. Effects of TLR4 Inhibitor on the LPS-Induced Pro-Inflammatory Mediators, NF-κB Translocation, Cell Migration, and ROS Production in BV2 Cells
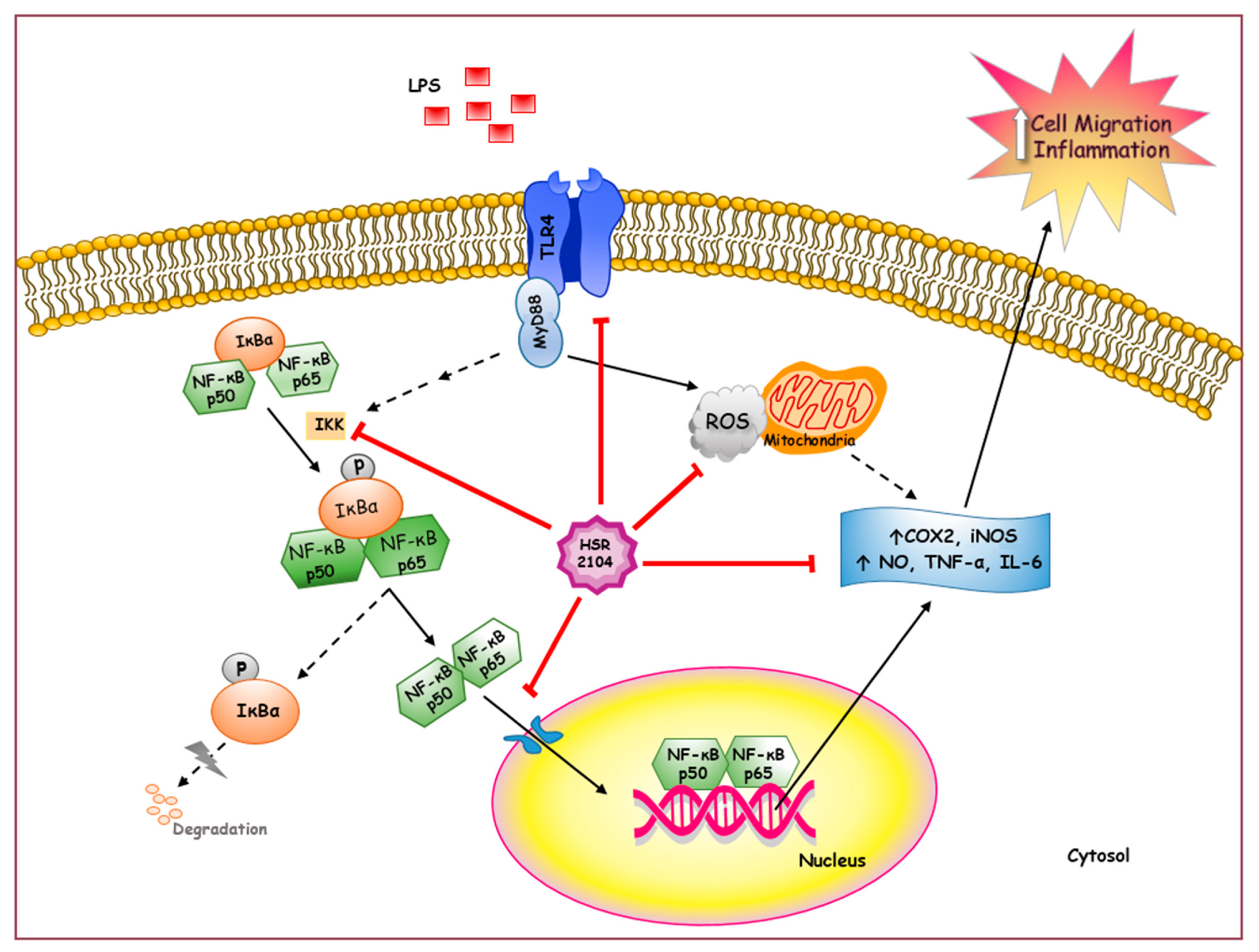
3. Discussion
4. Materials and Methods
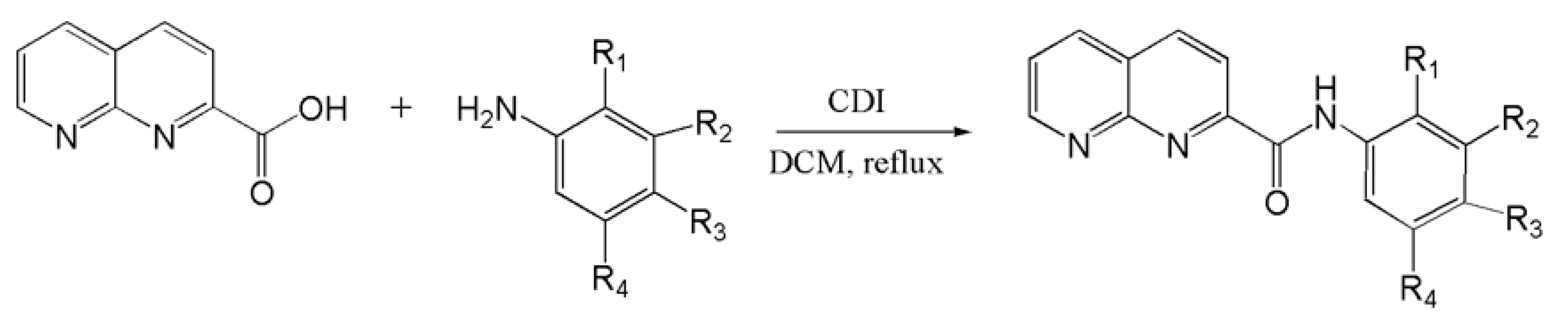
4.1. Synthesis of 1,8-Naphthyridine-2-Carboxamide Derivatives
4.2. Chemicals and Reagents
4.3. BV2 Cell Culture, Treatment of Cells, and Determination of Cell Viability
4.4. Measurements of NO, TNF-α, and IL-6 by Enzyme-Linked Immunosorbent Assays
4.5. Cell Migration Assays
4.5.1. Wound Healing Assay
4.5.2. Transwell Migration Assay
4.6. Western Blotting
4.7. Immunocytochemistry
4.8. Measurements of Reactive Oxygen Species
4.9. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| COX2 | Cyclooxygenase-2 |
| IκBs | Inhibitors of kappa B |
| IL | Interleukin |
| iNOS | Inducible nitric oxide synthase |
| LPS | Lipopolysaccharide |
| MyD88 | Myeloid differentiation factor 88 |
| NF-κB | Nuclear factor-kappa B |
| NO | Nitric oxide |
| ROS | Reactive oxygen species |
| TLR4 | Toll-like receptor 4 |
| TNF-α | Tumor necrosis factor-alpha |
References
- Singh, S.; Swarnkar, S.; Goswami, P.; Nath, C. Astrocytes and microglia: Responses to neuropathological conditions. Int. J. Neurosci. 2011, 121, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Abe, N.; Nishihara, T.; Yorozuya, T.; Tanaka, J. Microglia and Macrophages in the Pathological Central and Peripheral Nervous Systems. Cells 2020, 9, 2132. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Trapp, B.D. Microglia and neuroprotection. J. Neurochem. 2016, 136 (Suppl. 1), 10–17. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C. Microglia and neurodegeneration: The role of systemic inflammation. Glia 2013, 61, 71–90. [Google Scholar] [CrossRef]
- Xu, L.; He, D.; Bai, Y. Microglia-Mediated Inflammation and Neurodegenerative Disease. Mol. Neurobiol. 2016, 53, 6709–6715. [Google Scholar] [CrossRef]
- Duan, Y.; Sahley, C.L.; Muller, K.J. ATP and NO dually control migration of microglia to nerve lesions. Dev. Neurobiol. 2009, 69, 60–72. [Google Scholar] [CrossRef]
- Ho, M.S. Microglia in Parkinson’s Disease. Adv. Exp. Med. Biol. 2019, 1175, 335–353. [Google Scholar] [CrossRef]
- Regen, F.; Hellmann-Regen, J.; Costantini, E.; Reale, M. Neuroinflammation and Alzheimer’s Disease: Implications for Microglial Activation. Curr. Alzheimer. Res. 2017, 14, 1140–1148. [Google Scholar] [CrossRef]
- Réus, G.Z.; Fries, G.R.; Stertz, L.; Badawy, M.; Passos, I.C.; Barichello, T.; Kapczinski, F.; Quevedo, J. The role of inflammation and microglial activation in the pathophysiology of psychiatric disorders. Neuroscience 2015, 300, 141–154. [Google Scholar] [CrossRef]
- Bjelobaba, I.; Savic, D.; Lavrnja, I. Multiple Sclerosis and Neuroinflammation: The Overview of Current and Prospective Therapies. Curr. Pharm. Des. 2017, 23, 693–730. [Google Scholar] [CrossRef]
- Leitner, G.R.; Wenzel, T.J.; Marshall, N.; Gates, E.J.; Klegeris, A. Targeting toll-like receptor 4 to modulate neuroinflammation in central nervous system disorders. Expert Opin. Ther. Targets 2019, 23, 865–882. [Google Scholar] [CrossRef] [PubMed]
- Carty, M.; Bowie, A.G. Evaluating the role of Toll-like receptors in diseases of the central nervous system. Biochem. Pharmacol. 2011, 81, 825–837. [Google Scholar] [CrossRef] [PubMed]
- Von Bernhardi, R.; Eugenín-von Bernhardi, L.; Eugenín, J. Microglial cell dysregulation in brain aging and neurodegeneration. Front. Aging Neurosci. 2015, 7, 124. [Google Scholar] [CrossRef] [PubMed]
- Daulatzai, M.A. Fundamental role of pan-inflammation and oxidative-nitrosative pathways in neuropathogenesis of Alzheimer’s disease in focal cerebral ischemic rats. Am. J. Neurodegener. Dis. 2016, 5, 102–130. [Google Scholar] [PubMed]
- Choi, Y.H. Catalpol attenuates lipopolysaccharide-induced inflammatory responses in BV2 microglia through inhibiting the TLR4-mediated NF-κB pathway. Gen. Physiol. Biophys. 2019, 38, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Akira, S. TLR signaling pathways. Semin. Immunol. 2004, 16, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Zusso, M.; Lunardi, V.; Franceschini, D.; Pagetta, A.; Lo, R.; Stifani, S.; Frigo, A.C.; Giusti, P.; Moro, S. Ciprofloxacin and levofloxacin attenuate microglia inflammatory response via TLR4/NF-kB pathway. J. Neuroinflamm. 2019, 16, 148. [Google Scholar] [CrossRef]
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef]
- Lu, Y.C.; Yeh, W.C.; Ohashi, P.S. LPS/TLR4 signal transduction pathway. Cytokine 2008, 42, 145–151. [Google Scholar] [CrossRef]
- Qin, L.; Wu, X.; Block, M.L.; Liu, Y.; Breese, G.R.; Hong, J.S.; Knapp, D.J.; Crews, F.T. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 2007, 55, 453–462. [Google Scholar] [CrossRef]
- Lively, S.; Schlichter, L.C. Microglia Responses to Pro-inflammatory Stimuli (LPS, IFNγ+TNFα) and Reprogramming by Resolving Cytokines (IL-4, IL-10). Front. Cell Neurosci. 2018, 12, 215. [Google Scholar] [CrossRef]
- Bui, B.P.; Oh, Y.; Lee, H.; Cho, J. Inhibition of inflammatory mediators and cell migration by 1,2,3,4-tetrahydroquinoline derivatives in LPS-stimulated BV2 microglial cells via suppression of NF-κB and JNK pathway. Int. Immunopharmacol. 2020, 80, 106231. [Google Scholar] [CrossRef] [PubMed]
- Do, H.T.T.; Bui, B.P.; Sim, S.; Jung, J.K.; Lee, H.; Cho, J. Anti-Inflammatory and Anti-Migratory Activities of Isoquinoline-1-Carboxamide Derivatives in LPS-Treated BV2 Microglial Cells via Inhibition of MAPKs/NF-κB Pathway. Int. J. Mol. Sci. 2020, 21, 2319. [Google Scholar] [CrossRef] [PubMed]
- Dou, Y.; Wu, H.J.; Li, H.Q.; Qin, S.; Wang, Y.E.; Li, J.; Lou, H.F.; Chen, Z.; Li, X.M.; Luo, Q.M.; et al. Microglial migration mediated by ATP-induced ATP release from lysosomes. Cell Res. 2012, 22, 1022–1033. [Google Scholar] [CrossRef]
- Kawamoto, T.; Ii, M.; Kitazaki, T.; Iizawa, Y.; Kimura, H. TAK-242 selectively suppresses Toll-like receptor 4-signaling mediated by the intracellular domain. Eur. J. Pharmacol. 2008, 584, 40–48. [Google Scholar] [CrossRef]
- Matsunaga, N.; Tsuchimori, N.; Matsumoto, T.; Ii, M. TAK-242 (resatorvid), a small-molecule inhibitor of Toll-like receptor (TLR) 4 signaling, binds selectively to TLR4 and interferes with interactions between TLR4 and its adaptor molecules. Mol. Pharmacol. 2011, 79, 34–41. [Google Scholar] [CrossRef]
- Roma, G.; Di Braccio, M.; Grossi, G.; Piras, D.; Ballabeni, V.; Tognolini, M.; Bertoni, S.; Barocelli, E. 1,8-Naphthyridines VIII. Novel 5-aminoimidazo[1,2-a] [1,8]naphthyridine-6-carboxamide and 5-amino[1,2,4]triazolo[4,3-a] [1,8]naphthyridine-6-carboxamide derivatives showing potent analgesic or anti-inflammatory activity, respectively, and completely devoid of acute gastrolesivity. Eur. J. Med. Chem. 2010, 45, 352–366. [Google Scholar] [CrossRef]
- Dianzani, C.; Collino, M.; Gallicchio, M.; Di Braccio, M.; Roma, G.; Fantozzi, R. Effects of anti-inflammatory [1, 2, 4]triazolo[4, 3-a] [1, 8]naphthyridine derivatives on human stimulated PMN and endothelial cells: An in vitro study. J. Inflamm. (Lond.) 2006, 3, 4. [Google Scholar] [CrossRef]
- Henn, A.; Lund, S.; Hedtjärn, M.; Schrattenholz, A.; Pörzgen, P.; Leist, M. The suitability of BV2 cells as alternative model system for primary microglia cultures or for animal experiments examining brain inflammation. Altex 2009, 26, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Timmerman, R.; Burm, S.M.; Bajramovic, J.J. An Overview of in vitro Methods to Study Microglia. Front. Cell Neurosci. 2018, 12, 242. [Google Scholar] [CrossRef]
- Qin, L.; Li, G.; Qian, X.; Liu, Y.; Wu, X.; Liu, B.; Hong, J.S.; Block, M.L. Interactive role of the toll-like receptor 4 and reactive oxygen species in LPS-induced microglia activation. Glia 2005, 52, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Block, M.L.; Zecca, L.; Hong, J.S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Hanisch, U.K.; Kettenmann, H. Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 2007, 10, 1387–1394. [Google Scholar] [CrossRef]
- Dibaj, P.; Nadrigny, F.; Steffens, H.; Scheller, A.; Hirrlinger, J.; Schomburg, E.D.; Neusch, C.; Kirchhoff, F. NO mediates microglial response to acute spinal cord injury under ATP control in vivo. Glia 2010, 58, 1133–1144. [Google Scholar] [CrossRef]
- Alawieyah Syed Mortadza, S.; Sim, J.A.; Neubrand, V.E.; Jiang, L.H. A critical role of TRPM2 channel in Aβ(42) -induced microglial activation and generation of tumor necrosis factor-α. Glia 2018, 66, 562–575. [Google Scholar] [CrossRef] [PubMed]
- Gruol, D.L.; Nelson, T.E. Physiological and pathological roles of interleukin-6 in the central nervous system. Mol. Neurobiol. 1997, 15, 307–339. [Google Scholar] [CrossRef]
- Thomas, D.D.; Ridnour, L.A.; Isenberg, J.S.; Flores-Santana, W.; Switzer, C.H.; Donzelli, S.; Hussain, P.; Vecoli, C.; Paolocci, N.; Ambs, S.; et al. The chemical biology of nitric oxide: Implications in cellular signaling. Free Radic. Biol. Med. 2008, 45, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Kwon, K.J.; Park, J.Y.; Lee, S.H.; Moon, C.H.; Baik, E.J. Effects of peroxisome proliferator-activated receptor agonists on LPS-induced neuronal death in mixed cortical neurons: Associated with iNOS and COX-2. Brain Res. 2002, 941, 1–10. [Google Scholar] [CrossRef]
- Tsatsanis, C.; Androulidaki, A.; Venihaki, M.; Margioris, A.N. Signalling networks regulating cyclooxygenase-2. Int. J. Biochem. Cell Biol. 2006, 38, 1654–1661. [Google Scholar] [CrossRef]
- Liang, X.; Wu, L.; Wang, Q.; Hand, T.; Bilak, M.; McCullough, L.; Andreasson, K. Function of COX-2 and prostaglandins in neurological disease. J. Mol. Neurosci. 2007, 33, 94–99. [Google Scholar] [CrossRef]
- Ghasemi, M.; Fatemi, A. Pathologic role of glial nitric oxide in adult and pediatric neuroinflammatory diseases. Neurosci. Biobehav. Rev. 2014, 45, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Alhadidi, Q.; Shah, Z.A. Cofilin Mediates LPS-Induced Microglial Cell Activation and Associated Neurotoxicity through Activation of NF-κB and JAK-STAT Pathway. Mol. Neurobiol. 2018, 55, 1676–1691. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Su, H.; Huang, Z.; Feng, L.; Nie, H. Neuroprotective effect of raspberry extract by inhibiting peroxynitrite-induced DNA damage and hydroxyl radical formation. Food Res. Int. 2012, 49, 22–26. [Google Scholar] [CrossRef]
- Closa, D.; Folch-Puy, E. Oxygen free radicals and the systemic inflammatory response. IUBMB Life 2004, 56, 185–191. [Google Scholar] [CrossRef]
- Zeng, K.W.; Zhao, M.B.; Ma, Z.Z.; Jiang, Y.; Tu, P.F. Protosappanin A inhibits oxidative and nitrative stress via interfering the interaction of transmembrane protein CD14 with Toll-like receptor-4 in lipopolysaccharide-induced BV-2 microglia. Int. Immunopharmacol. 2012, 14, 558–569. [Google Scholar] [CrossRef]
- Wang, X.; Wang, C.; Wang, J.; Zhao, S.; Zhang, K.; Wang, J.; Zhang, W.; Wu, C.; Yang, J. Pseudoginsenoside-F11 (PF11) exerts anti-neuroinflammatory effects on LPS-activated microglial cells by inhibiting TLR4-mediated TAK1/IKK/NF-κB, MAPKs and Akt signaling pathways. Neuropharmacology 2014, 79, 642–656. [Google Scholar] [CrossRef] [PubMed]
- Iizumi, T.; Takahashi, S.; Mashima, K.; Minami, K.; Izawa, Y.; Abe, T.; Hishiki, T.; Suematsu, M.; Kajimura, M.; Suzuki, N. A possible role of microglia-derived nitric oxide by lipopolysaccharide in activation of astroglial pentose-phosphate pathway via the Keap1/Nrf2 system. J. Neuroinflamm. 2016, 13, 99. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Wu, P.F.; Zhang, L.; Hu, Z.L.; Wang, W.; Guan, X.L.; Luo, H.; Ni, M.; Yang, J.W.; Li, M.X.; et al. Methionine sulfoxide reductase A negatively controls microglia-mediated neuroinflammation via inhibiting ROS/MAPKs/NF-κB signaling pathways through a catalytic antioxidant function. Antioxid. Redox Signal. 2015, 22, 832–847. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.Y.; Wu, Y.Y.; Huang, H.; He, C.; Li, W.Z.; Wang, H.L.; Chen, H.Q.; Yin, Y.Y. Biochanin A attenuates LPS-induced pro-inflammatory responses and inhibits the activation of the MAPK pathway in BV2 microglial cells. Int. J. Mol. Med. 2015, 35, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wu, Z.; Cao, X.; Ding, L.; Wen, Z.; Bian, J.S. HNO suppresses LPS-induced inflammation in BV-2 microglial cells via inhibition of NF-κB and p38 MAPK pathways. Pharmacol. Res. 2016, 111, 885–895. [Google Scholar] [CrossRef]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Tak, P.P.; Firestein, G.S. NF-kappaB: A key role in inflammatory diseases. J. Clin. Investig. 2001, 107, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, A.S., Jr. The NF-kappa B and I kappa B proteins: New discoveries and insights. Annu. Rev. Immunol. 1996, 14, 649–683. [Google Scholar] [CrossRef]
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [PubMed]
- Labbozzetta, M.; Notarbartolo, M.; Poma, P. Can NF-κB Be Considered a Valid Drug Target in Neoplastic Diseases? Our Point of View. Int. J. Mol. Sci. 2020, 21, 3070. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef]
- O’Neill, L.A.; Bowie, A.G. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat. Rev. Immunol. 2007, 7, 353–364. [Google Scholar] [CrossRef]
- He, Y.; Yao, X.; Taylor, N.; Bai, Y.; Lovenberg, T.; Bhattacharya, A. RNA sequencing analysis reveals quiescent microglia isolation methods from postnatal mouse brains and limitations of BV2 cells. J. Neuroinflamm. 2018, 15, 153. [Google Scholar] [CrossRef]
- Moniruzzaman, M.; Bose, S.; Kim, Y.M.; Chin, Y.W.; Cho, J. The ethyl acetate fraction from Physalis alkekengi inhibits LPS-induced pro-inflammatory mediators in BV2 cells and inflammatory pain in mice. J. Ethnopharmacol. 2016, 181, 26–36. [Google Scholar] [CrossRef]
- Bose, S.; Kim, S.; Oh, Y.; Moniruzzaman, M.; Lee, G.; Cho, J. Effect of CCL2 on BV2 microglial cell migration: Involvement of probable signaling pathways. Cytokine 2016, 81, 39–49. [Google Scholar] [CrossRef]
- Lee, K.; Park, C.; Oh, Y.; Lee, H.; Cho, J. Antioxidant and Neuroprotective Effects of N-((3,4-Dihydro-2H-benzo[h]chromen-2-yl)methyl)-4-methoxyaniline in Primary Cultured Rat Cortical Cells: Involvement of ERK-CREB Signaling. Molecules 2018, 23, 669. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inflammatory Mediators | IC50 Values (µM) * | |||||||
|---|---|---|---|---|---|---|---|---|
| HSR2101 | HSR2102 | HSR2103 | HSR2104 | HSR2105 | HSR2106 | HSR2107 | HSR2113 | |
| NO | >100 | 32.13 | 45.38 | 25.43 | 50.72 | 57.02 | 28.03 | 26.49 |
| TNF-α | 30.47 | 52.09 | 59.36 | 33.24 | 44.61 | 57.21 | 37.26 | 91.93 |
| IL-6 | >100 | 55.74 | 78.91 | 32.03 | 50.50 | 29.16 | 96.06 | 27.98 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, P.L.; Bui, B.P.; Lee, H.; Cho, J. A Novel 1,8-Naphthyridine-2-Carboxamide Derivative Attenuates Inflammatory Responses and Cell Migration in LPS-Treated BV2 Cells via the Suppression of ROS Generation and TLR4/Myd88/NF-κB Signaling Pathway. Int. J. Mol. Sci. 2021, 22, 2527. https://doi.org/10.3390/ijms22052527
Nguyen PL, Bui BP, Lee H, Cho J. A Novel 1,8-Naphthyridine-2-Carboxamide Derivative Attenuates Inflammatory Responses and Cell Migration in LPS-Treated BV2 Cells via the Suppression of ROS Generation and TLR4/Myd88/NF-κB Signaling Pathway. International Journal of Molecular Sciences. 2021; 22(5):2527. https://doi.org/10.3390/ijms22052527
Chicago/Turabian StyleNguyen, Phuong Linh, Bich Phuong Bui, Heesoon Lee, and Jungsook Cho. 2021. "A Novel 1,8-Naphthyridine-2-Carboxamide Derivative Attenuates Inflammatory Responses and Cell Migration in LPS-Treated BV2 Cells via the Suppression of ROS Generation and TLR4/Myd88/NF-κB Signaling Pathway" International Journal of Molecular Sciences 22, no. 5: 2527. https://doi.org/10.3390/ijms22052527
APA StyleNguyen, P. L., Bui, B. P., Lee, H., & Cho, J. (2021). A Novel 1,8-Naphthyridine-2-Carboxamide Derivative Attenuates Inflammatory Responses and Cell Migration in LPS-Treated BV2 Cells via the Suppression of ROS Generation and TLR4/Myd88/NF-κB Signaling Pathway. International Journal of Molecular Sciences, 22(5), 2527. https://doi.org/10.3390/ijms22052527