
Knock-Down of HDAC2 in Human Induced Pluripotent Stem Cell Derived Neurons Improves Neuronal Mitochondrial Dynamics, Neuronal Maturation and Reduces Amyloid Beta Peptides



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
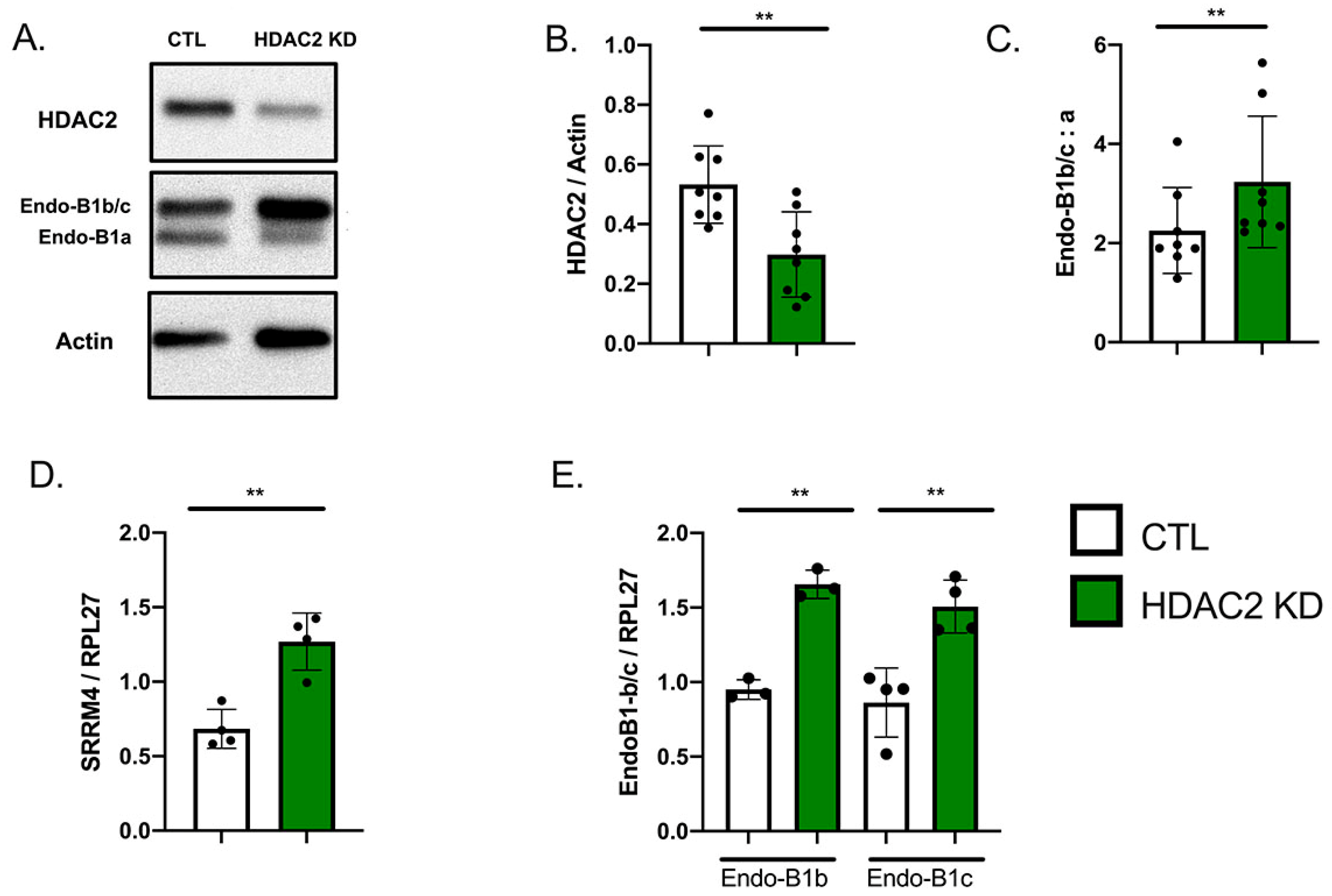
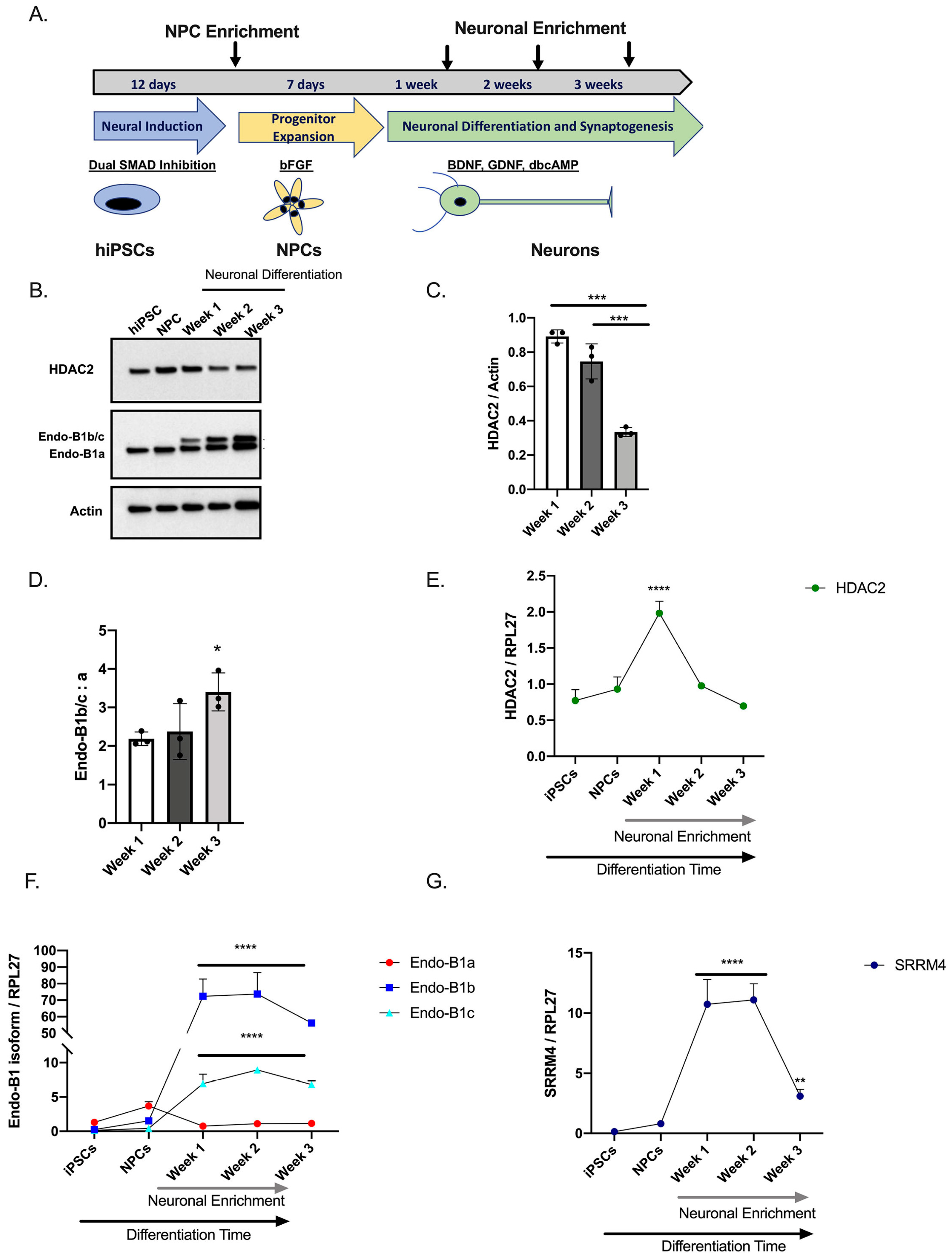
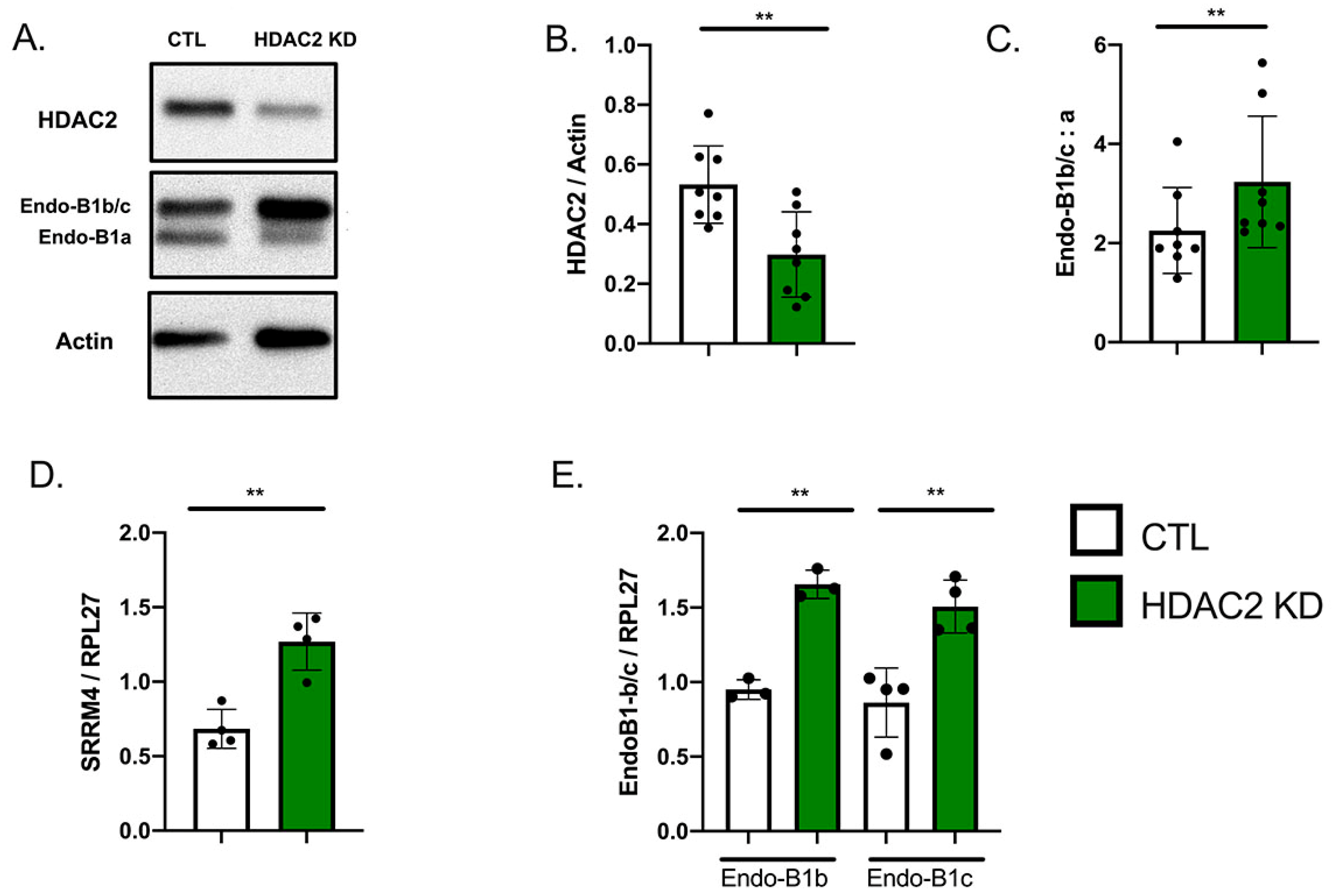
2.1. Expression of HDAC2 and Endophilin-B1 (Endo-B1) Isoforms in hiPSC-Derived Neurons
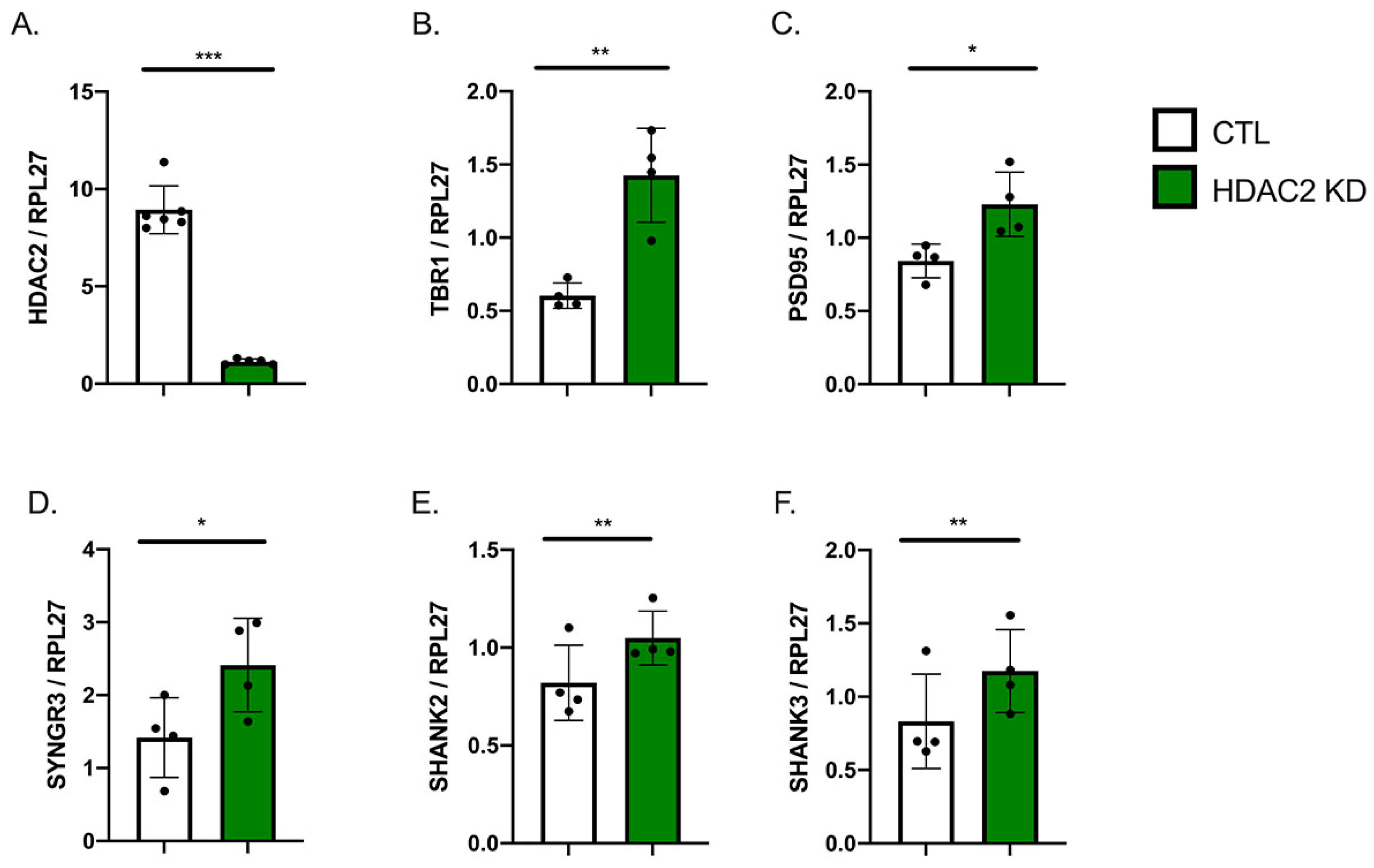
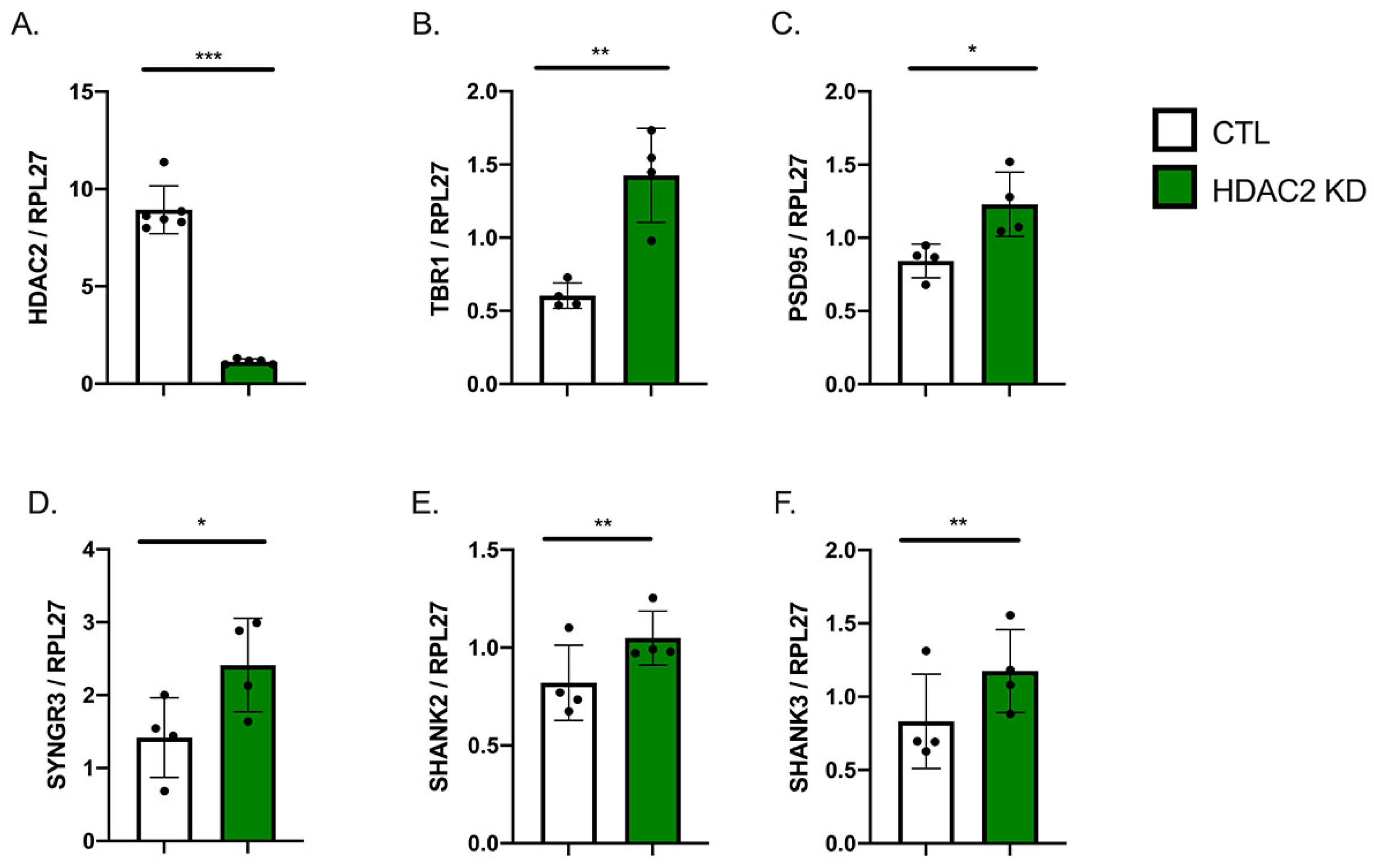
2.2. HDAC2 Knock-Down Influences Neuronal Gene Expression
2.3. HDAC2 Knock-Down Upregulates the Expression of Endo-B1b/c and SRRM4
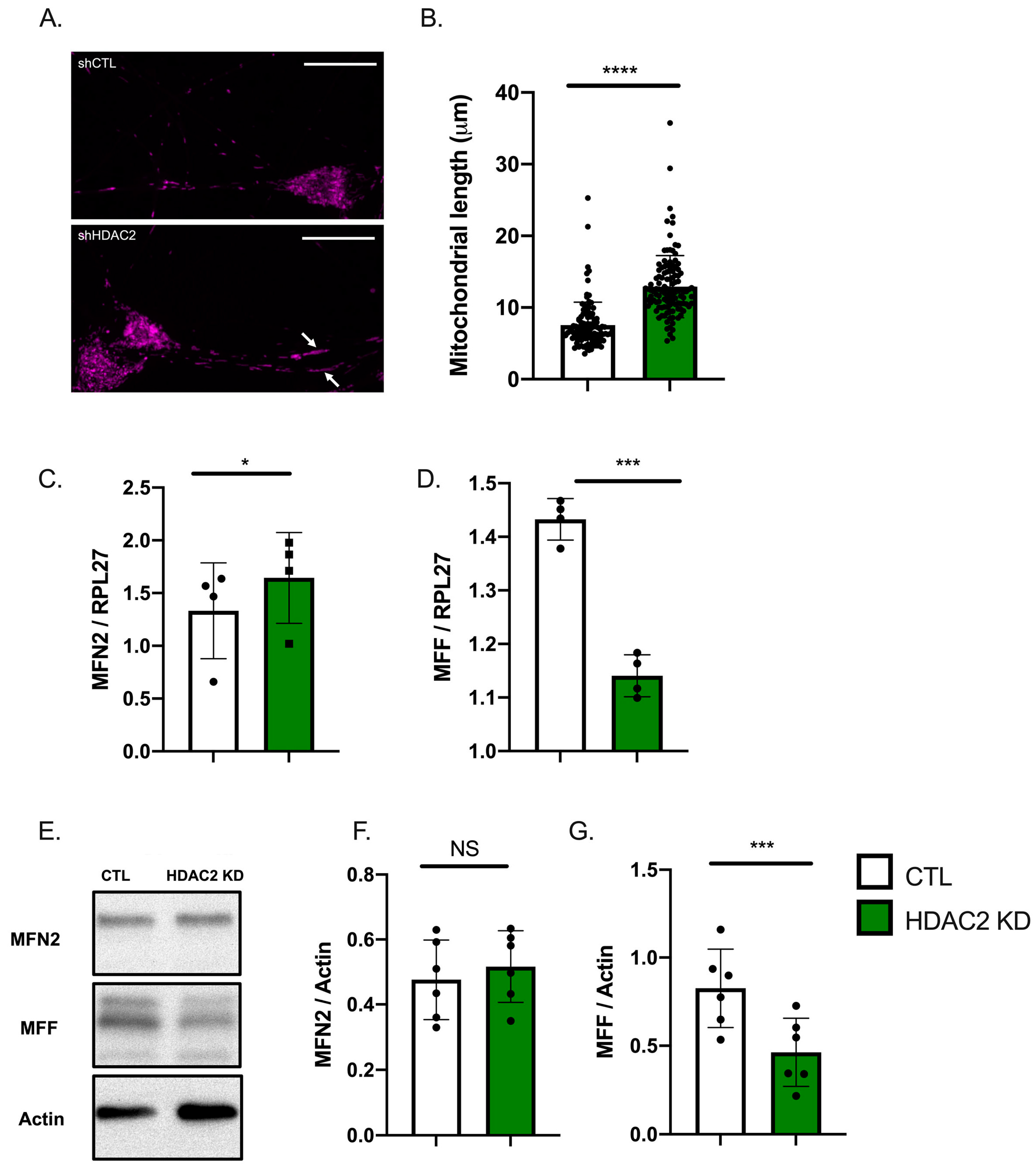
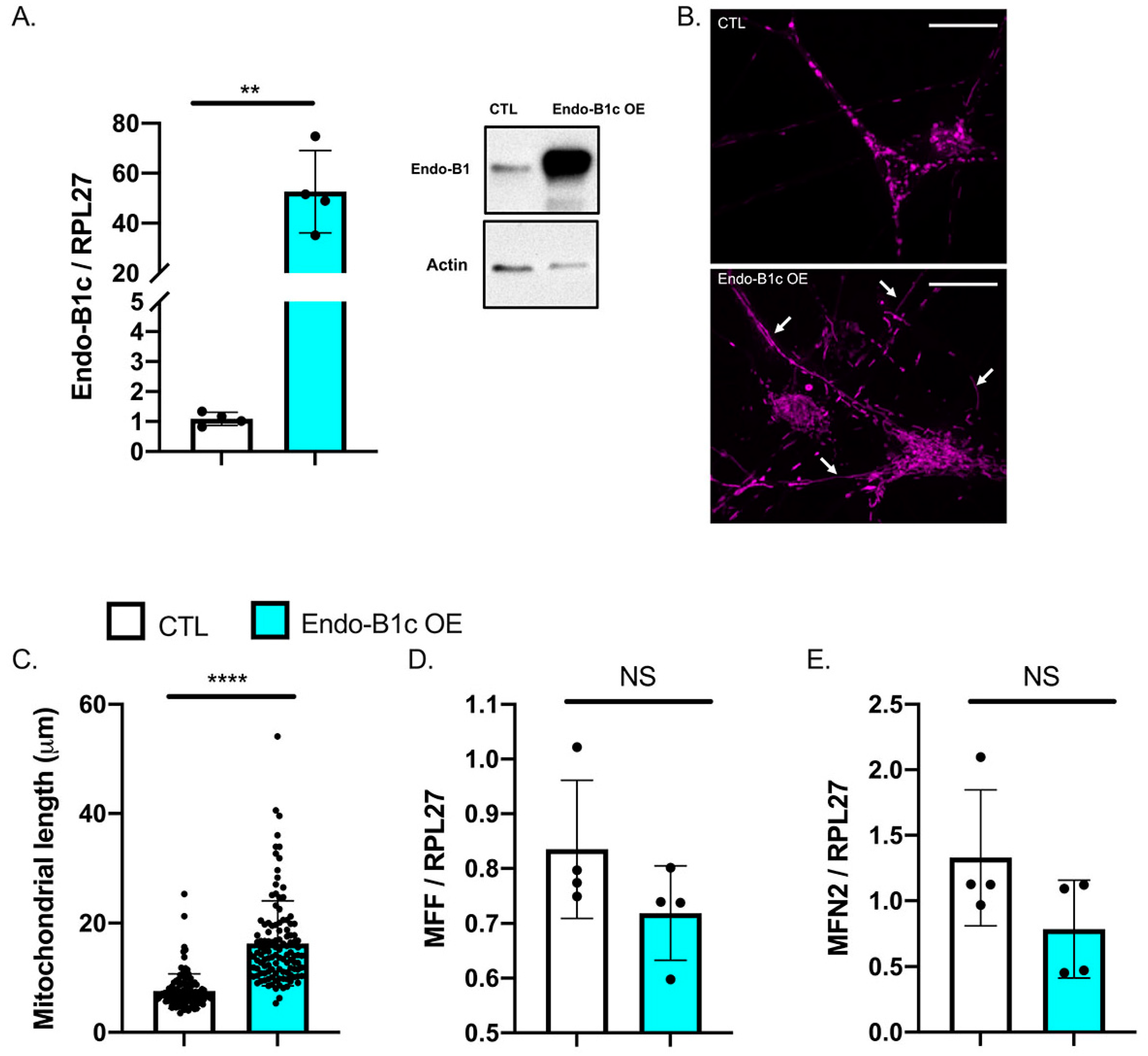
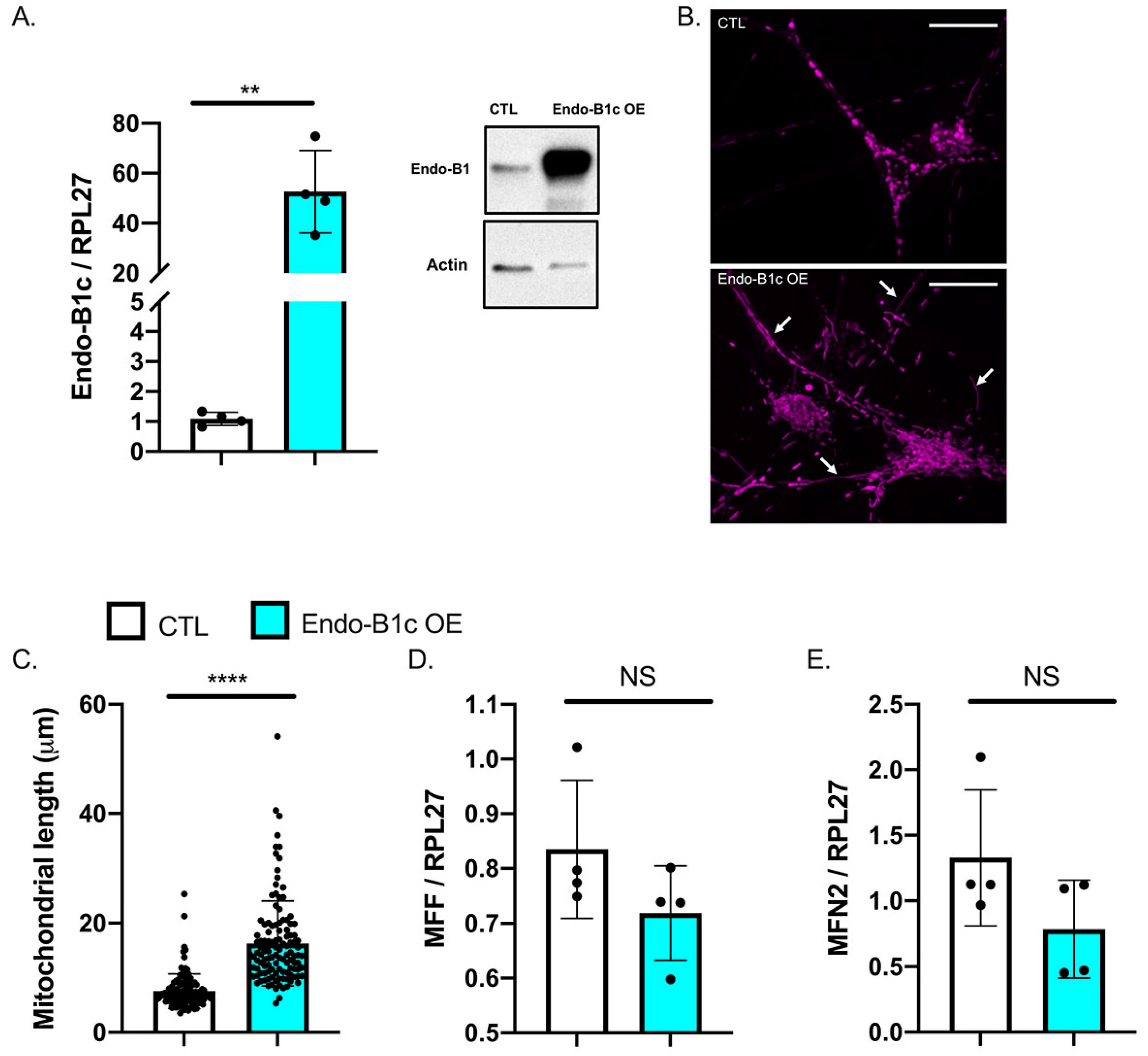
2.4. HDAC2 and Endo-B1c Expression Influences Mitochondrial Length
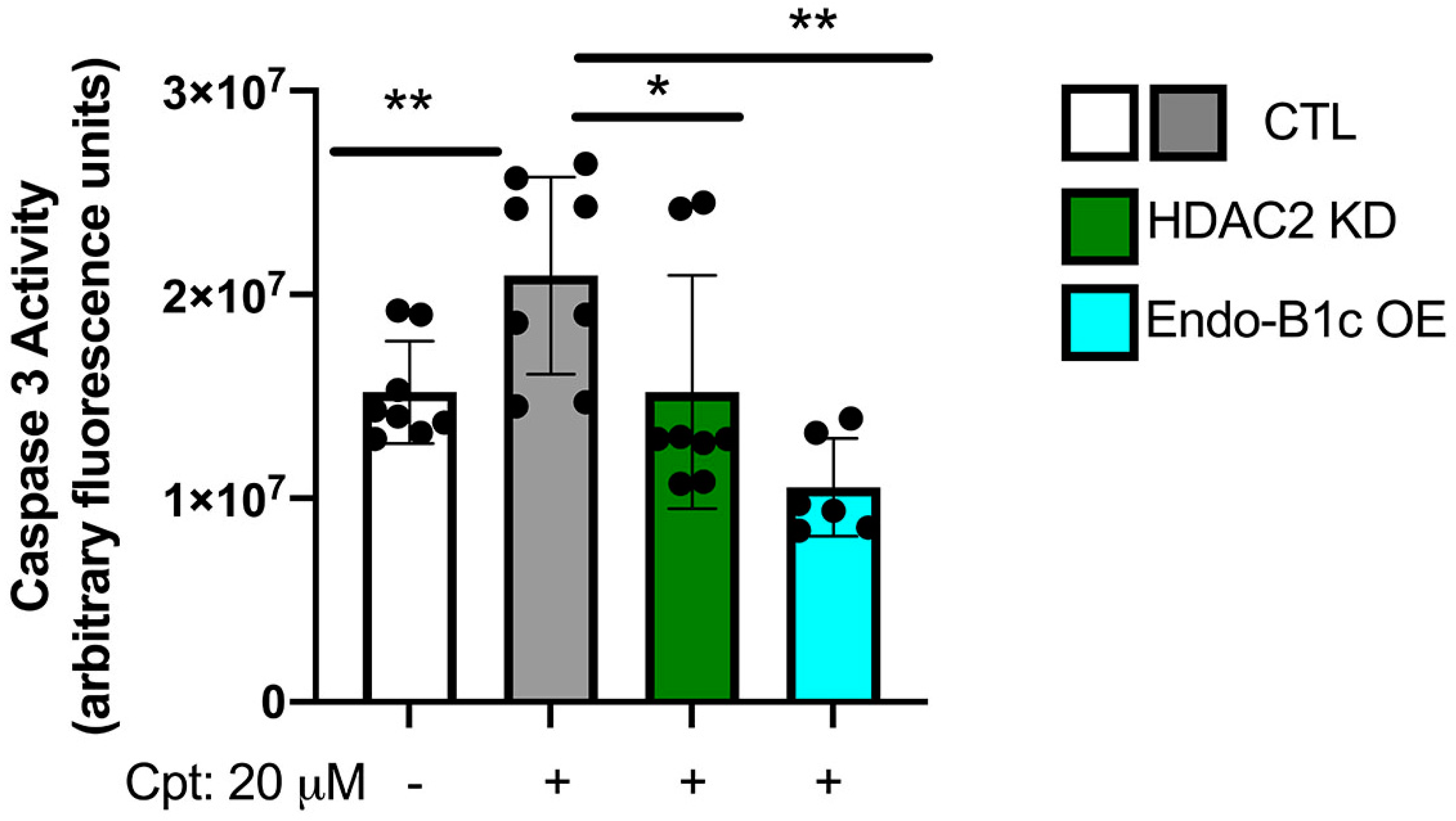
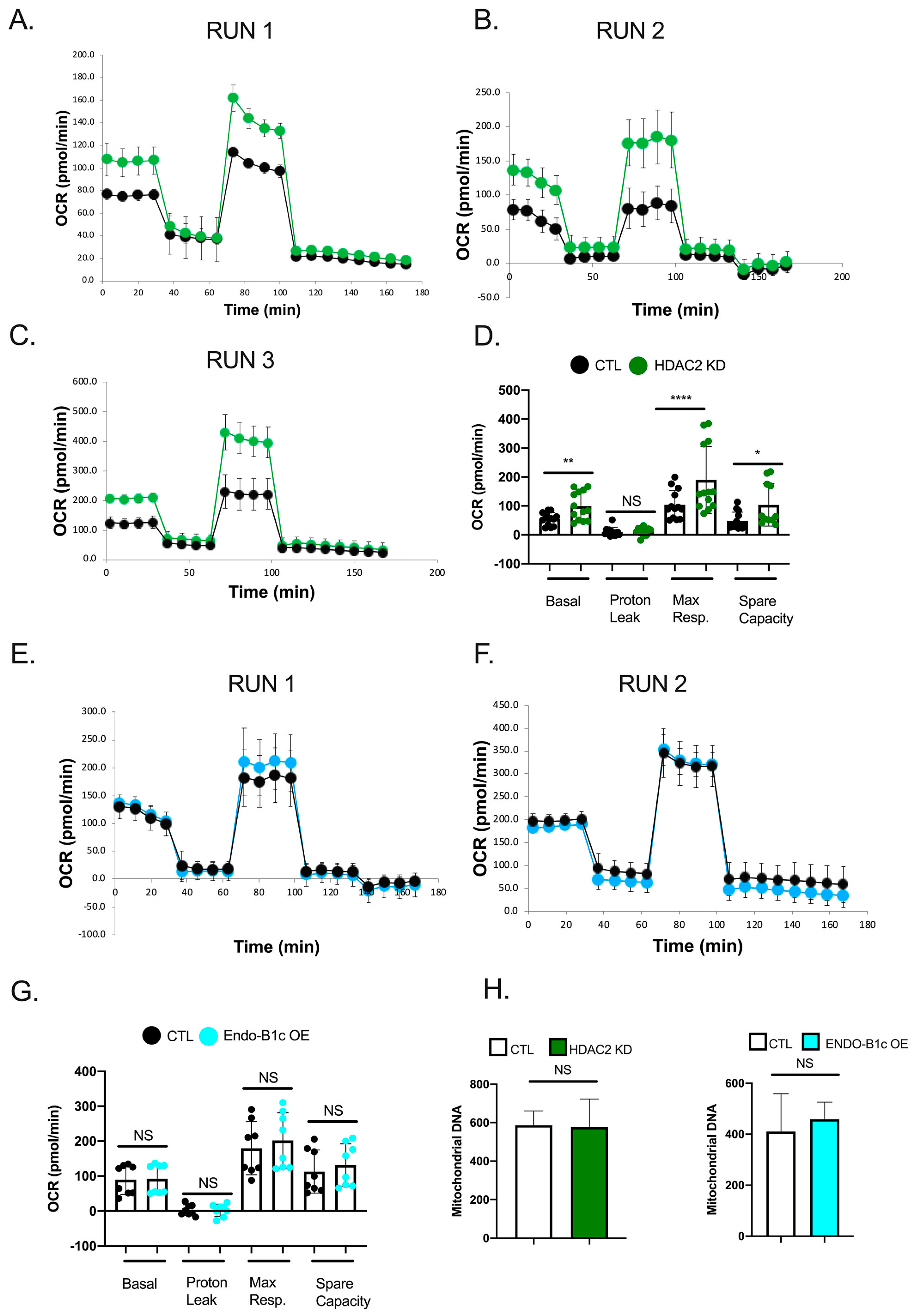
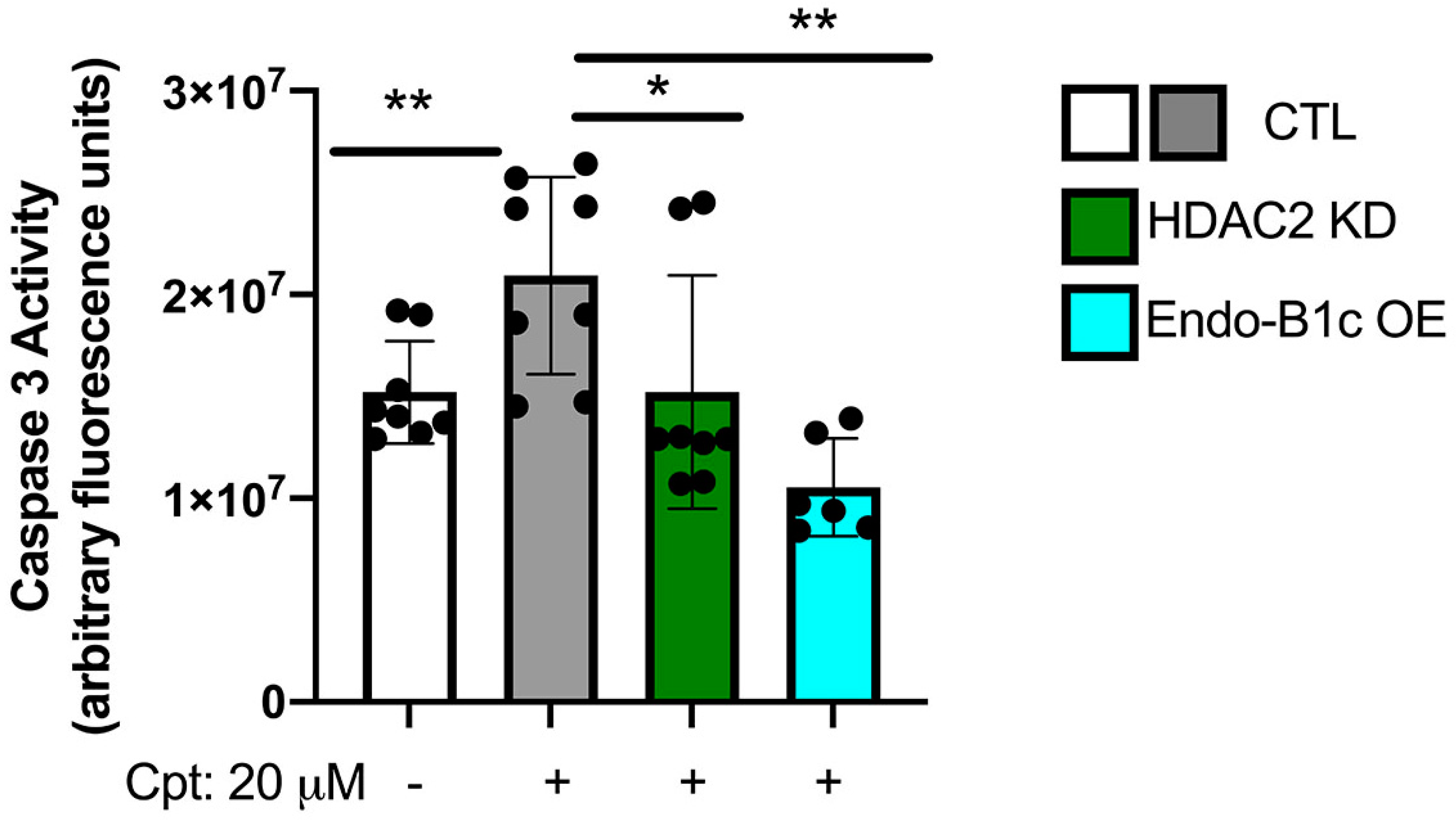
2.5. HDAC2 Expression Influences Neuronal Viability and Mitochondrial Respiration
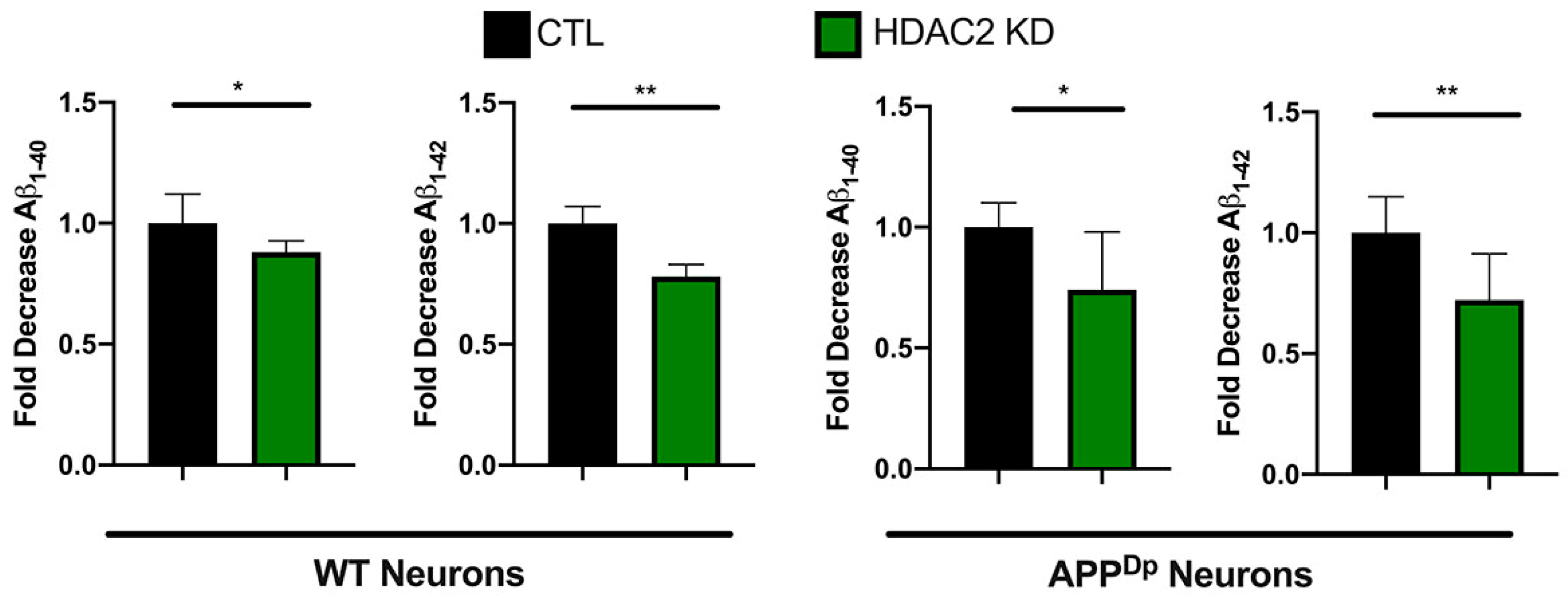
2.6. HDAC2 Knock-Down Reduces Amyloid Beta (Aβ) 1–40 and 1–42 Peptides
3. Discussion
4. Materials and Methods
4.1. Cell Culture and hiPSC Neuronal Differentiation
4.2. Lentivirus Treatment
4.3. RNA Purification and qPCR Analysis
| hHDAC2 | TGAGATTCCCAATGAGTTGCCA | TACTGACATCTGGTCAGACA |
| hMFN2 | CACCCTGATGCAGACGGAAA | TCCATGTACTCGGGCTCTGA |
| hMFF | CAGCTTCACTAAGACGACAGATAA | TTACCTCTAGCGGCGAAACC |
| hPSD95 | CTCAGGGTCAACGACAGCAT | AAGCCAAGACCTTTAGGCCC |
| hRPL27 | TGAGATTCCCAATGAGTTGCCA | TACTGACATCTGGTCAGACA |
| hSH3GLB1 isoform c | CATGTAAAATGGCTGAAGATTTGG | TGGGCATGTGTACTGCTGAT |
| hSHANK2 | CTGGCGAGCTGGGGTGATTA | TCAATGGGTGTGTCAGCTTTG |
| hSHANK3 | CCTCACCTCACACAGCGATT | CCACCGACTCGAGATACTGC |
| hSYNGR3 | CGTCCTGGGTGTTCTCCATC | CTGCTGATTTGCTGGAAGCG |
| hSRMM4 | ATAGCCCATCGCCTGTCAAG | GCCGGCTTCGAGATTGTTTC |
4.4. Western Blot Analysis
| HDAC2 | Sigma H2663 (2 mg/mL) | 1:5000 |
| Bif-1/Endophilin-B1(30A882.1.1) | Novus #NBP2-24733 (0.5 mg/mL) | 1:1000 |
| MFN2 | Abcam #ab124773 (1.549 mg/mL) | 1:2000 |
| MFF | Proteintech #17090-1-AP (45 μg/150 μL) | 1:2000 |
| Actin | Sigma A5441 | 1:2500 |
4.5. Mitochondrial Length Analysis
4.6. Caspase 3 Analysis
4.7. Seahorse Analysis
4.8. Mitochondrial DNA Copy Number Analysis
4.9. Aβ 1–40 and 1–42 Analysis
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lardenoije, R.; Iatrou, A.; Kenis, G.; Kompotis, K.; Steinbusch, H.W.; Mastroeni, D.; Coleman, P.; Lemere, C.A.; Hof, P.R.; van den Hove, D.L.; et al. The epigenetics of aging and neurodegeneration. Prog. Neurobiol. 2015, 131, 21–64. [Google Scholar] [CrossRef]
- Graff, J.; Joseph, N.F.; Horn, M.E.; Samiei, A.; Meng, J.; Seo, J.; Rei, D.; Bero, A.W.; Phan, T.X.; Wagner, F.; et al. Epigenetic priming of memory updating during reconsolidation to attenuate remote fear memories. Cell 2014, 156, 261–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graff, J.; Rei, D.; Guan, J.S.; Wang, W.Y.; Seo, J.; Hennig, K.M.; Nieland, T.J.; Fass, D.M.; Kao, P.F.; Kahn, M.; et al. An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature 2012, 483, 222–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, J.S.; Haggarty, S.J.; Giacometti, E.; Dannenberg, J.H.; Joseph, N.; Gao, J.; Nieland, T.J.; Zhou, Y.; Wang, X.; Mazitschek, R.; et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 2009, 459, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Penney, J.; Tsai, L.H. Histone deacetylases in memory and cognition. Sci. Signal. 2014, 7, re12. [Google Scholar] [CrossRef] [PubMed]
- Yamakawa, H.; Cheng, J.; Penney, J.; Gao, F.; Rueda, R.; Wang, J.; Yamakawa, S.; Kritskiy, O.; Gjoneska, E.; Tsai, L.H. The Transcription Factor Sp3 Cooperates with HDAC2 to Regulate Synaptic Function and Plasticity in Neurons. Cell Rep. 2017, 20, 1319–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacDonald, J.L.; Roskams, A.J. Histone deacetylases 1 and 2 are expressed at distinct stages of neuro-glial development. Dev. Dyn. 2008, 237, 2256–2267. [Google Scholar] [CrossRef]
- Akhtar, M.W.; Raingo, J.; Nelson, E.D.; Montgomery, R.L.; Olson, E.N.; Kavalali, E.T.; Monteggia, L.M. Histone deacetylases 1 and 2 form a developmental switch that controls excitatory synapse maturation and function. J. Neurosci. 2009, 29, 8288–8297. [Google Scholar] [CrossRef] [Green Version]
- Jaworska, J.; Ziemka-Nalecz, M.; Zalewska, T. Histone deacetylases 1 and 2 are required for brain development. Int. J. Dev. Biol. 2015, 59, 171–177. [Google Scholar] [CrossRef] [Green Version]
- Panikker, P.; Xu, S.J.; Zhang, H.; Sarthi, J.; Beaver, M.; Sheth, A.; Akhter, S.; Elefant, F. Restoring Tip60 HAT/HDAC2 Balance in the Neurodegenerative Brain Relieves Epigenetic Transcriptional Repression and Reinstates Cognition. J. Neurosci. 2018, 38, 4569–4583. [Google Scholar] [CrossRef] [PubMed]
- Bonnaud, E.M.; Suberbielle, E.; Malnou, C.E. Histone acetylation in neuronal (dys)function. Biomol. Concepts 2016, 7, 103–116. [Google Scholar] [CrossRef]
- Mahady, L.; Nadeem, M.; Malek-Ahmadi, M.; Chen, K.; Perez, S.E.; Mufson, E.J. HDAC2 dysregulation in the nucleus basalis of Meynert during the progression of Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2019, 45, 380–397. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.B.; Kinoshita, C.; Kinoshita, Y.; Sopher, B.L.; Uo, T.; Lee, R.J.; Kim, J.K.; Murphy, S.P.; Dirk Keene, C.; Garden, G.A.; et al. Neuronal susceptibility to beta-amyloid toxicity and ischemic injury involves histone deacetylase-2 regulation of endophilin-B1. Brain Pathol. 2019, 29, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Marshall, L.; Oh, G.; Jakubowski, J.L.; Groot, D.; He, Y.; Wang, T.; Petronis, A.; Labrie, V. Epigenetic dysregulation of enhancers in neurons is associated with Alzheimer’s disease pathology and cognitive symptoms. Nat. Commun. 2019, 10, 2246. [Google Scholar] [CrossRef] [Green Version]
- Zusso, M.; Barbierato, M.; Facci, L.; Skaper, S.D.; Giusti, P. Neuroepigenetics and Alzheimer’s Disease: An Update. J. Alzheimers Dis. 2018, 64, 671–688. [Google Scholar] [CrossRef]
- Smith, M.A.; Perry, G.; Richey, P.L.; Sayre, L.M.; Anderson, V.E.; Beal, M.F.; Kowall, N. Oxidative damage in Alzheimer’s. Nature 1996, 382, 120–121. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: Implications for neuronal damage. Hum. Mol. Genet. 2011, 20, 2495–2509. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Tammineni, P. Mitochondrial Aspects of Synaptic Dysfunction in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1087–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, K.; Wei, Q.; Liu, F.F.; Hu, F.; Xie, A.J.; Zhu, L.Q.; Liu, D. Synaptic Dysfunction in Alzheimer’s Disease: Abeta, Tau, and Epigenetic Alterations. Mol. Neurobiol. 2018, 55, 3021–3032. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, R.L.; Hsieh, J.; Barbosa, A.C.; Richardson, J.A.; Olson, E.N. Histone deacetylases 1 and 2 control the progression of neural precursors to neurons during brain development. Proc. Natl. Acad. Sci. USA 2009, 106, 7876–7881. [Google Scholar] [CrossRef] [Green Version]
- Gore, A.; Li, Z.; Fung, H.L.; Young, J.E.; Agarwal, S.; Antosiewicz-Bourget, J.; Canto, I.; Giorgetti, A.; Israel, M.A.; Kiskinis, E.; et al. Somatic coding mutations in human induced pluripotent stem cells. Nature 2011, 471, 63–67. [Google Scholar] [CrossRef]
- Woodruff, G.; Young, J.E.; Martinez, F.J.; Buen, F.; Gore, A.; Kinaga, J.; Li, Z.; Yuan, S.H.; Zhang, K.; Goldstein, L.S. The presenilin-1 DeltaE9 mutation results in reduced gamma-secretase activity, but not total loss of PS1 function, in isogenic human stem cells. Cell Rep. 2013, 5, 974–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, J.E.; Boulanger-Weill, J.; Williams, D.A.; Woodruff, G.; Buen, F.; Revilla, A.C.; Herrera, C.; Israel, M.A.; Yuan, S.H.; Edland, S.D.; et al. Elucidating Molecular Phenotypes Caused by the SORL1 Alzheimer’s Disease Genetic Risk Factor Using Human Induced Pluripotent Stem Cells. Cell Stem. Cell 2015, 16, 373–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, J.E.; Fong, L.K.; Frankowski, H.; Petsko, G.A.; Small, S.A.; Goldstein, L.S.B. Stabilizing the Retromer Complex in a Human Stem Cell Model of Alzheimer’s Disease Reduces TAU Phosphorylation Independently of Amyloid Precursor Protein. Stem. Cell Rep. 2018, 10, 1046–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knupp, A.; Mishra, S.; Martinez, R.; Braggin, J.E.; Szabo, M.; Kinoshita, C.; Hailey, D.W.; Small, S.A.; Jayadev, S.; Young, J.E. Depletion of the AD Risk Gene SORL1 Selectively Impairs Neuronal Endosomal Traffic Independent of Amyloidogenic APP Processing. Cell Rep. 2020, 31, 107719. [Google Scholar] [CrossRef] [PubMed]
- Rose, S.E.; Frankowski, H.; Knupp, A.; Berry, B.J.; Martinez, R.; Dinh, S.Q.; Bruner, L.T.; Willis, S.L.; Crane, P.K.; Larson, E.B.; et al. Leptomeninges-Derived Induced Pluripotent Stem Cells and Directly Converted Neurons From Autopsy Cases With Varying Neuropathologic Backgrounds. J. Neuropathol. Exp. Neurol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Zhang, Y.; Wang, Y.; Cai, Z.; Lu, Z.; Li, L.; Huang, R.; Hagelkruys, A.; Matthias, P.; Zhang, H.; et al. HDAC1 and HDAC2 Regulate Intermediate Progenitor Positioning to Safeguard Neocortical Development. Neuron 2019, 101, 1117–1133.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.B.; Uo, T.; Kinoshita, C.; Sopher, B.L.; Lee, R.J.; Murphy, S.P.; Kinoshita, Y.; Garden, G.A.; Wang, H.G.; Morrison, R.S. Bax interacting factor-1 promotes survival and mitochondrial elongation in neurons. J. Neurosci. 2014, 34, 2674–2683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Israel, M.A.; Yuan, S.H.; Bardy, C.; Reyna, S.M.; Mu, Y.; Herrera, C.; Hefferan, M.P.; Van Gorp, S.; Nazor, K.L.; Boscolo, F.S.; et al. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 2012, 482, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.H.; Martin, J.; Elia, J.; Flippin, J.; Paramban, R.I.; Hefferan, M.P.; Vidal, J.G.; Mu, Y.; Killian, R.L.; Israel, M.A.; et al. Cell-surface marker signatures for the isolation of neural stem cells, glia and neurons derived from human pluripotent stem cells. PLoS ONE 2011, 6, e17540. [Google Scholar] [CrossRef] [Green Version]
- Raj, B.; Irimia, M.; Braunschweig, U.; Sterne-Weiler, T.; O’Hanlon, D.; Lin, Z.Y.; Chen, G.I.; Easton, L.E.; Ule, J.; Gingras, A.C.; et al. A global regulatory mechanism for activating an exon network required for neurogenesis. Mol. Cell 2014, 56, 90–103. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.B.; Kinoshita, Y.; Kinoshita, C.; Uo, T.; Sopher, B.L.; Cudaback, E.; Keene, C.D.; Bilousova, T.; Gylys, K.; Case, A.; et al. Loss of endophilin-B1 exacerbates Alzheimer’s disease pathology. Brain 2015, 138, 2005–2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedogni, F.; Hodge, R.D.; Elsen, G.E.; Nelson, B.R.; Daza, R.A.; Beyer, R.P.; Bammler, T.K.; Rubenstein, J.L.; Hevner, R.F. Tbr1 regulates regional and laminar identity of postmitotic neurons in developing neocortex. Proc. Natl. Acad. Sci. USA 2010, 107, 13129–13134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.A.; Hou, X.; Hao, S. Mitochondrial biogenesis in neurodegeneration. J. Neurosci. Res. 2017, 95, 2025–2029. [Google Scholar] [CrossRef] [PubMed]
- Grimm, A.; Eckert, A. Brain aging and neurodegeneration: From a mitochondrial point of view. J. Neurochem. 2017, 143, 418–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.S.; Yoon, Y.G.; Yoo, S.H.; Jeong, N.Y.; Jeong, S.H.; Lee, S.Y.; Jung, D.I.; Jeong, S.Y.; Yoo, Y.H. Histone deacetylase inhibitors induce mitochondrial elongation. J. Cell Physiol. 2012, 227, 2856–2869. [Google Scholar] [CrossRef] [PubMed]
- Schrepfer, E.; Scorrano, L. Mitofusins, from Mitochondria to Metabolism. Mol. Cell 2016, 61, 683–694. [Google Scholar] [CrossRef] [Green Version]
- Morris, E.J.; Geller, H.M. Induction of neuronal apoptosis by camptothecin, an inhibitor of DNA topoisomerase-I: Evidence for cell cycle-independent toxicity. J. Cell Biol. 1996, 134, 757–770. [Google Scholar] [CrossRef] [Green Version]
- Uo, T.; Veenstra, T.D.; Morrison, R.S. Histone deacetylase inhibitors prevent p53-dependent and p53-independent Bax-mediated neuronal apoptosis through two distinct mechanisms. J. Neurosci. 2009, 29, 2824–2832. [Google Scholar] [CrossRef] [Green Version]
- Pich, S.; Bach, D.; Briones, P.; Liesa, M.; Camps, M.; Testar, X.; Palacin, M.; Zorzano, A. The Charcot-Marie-Tooth type 2A gene product, Mfn2, up-regulates fuel oxidation through expression of OXPHOS system. Hum. Mol. Genet. 2005, 14, 1405–1415. [Google Scholar] [CrossRef]
- Fang, D.; Yan, S.; Yu, Q.; Chen, D.; Yan, S.S. Mfn2 is Required for Mitochondrial Development and Synapse Formation in Human Induced Pluripotent Stem Cells/hiPSC Derived Cortical Neurons. Sci. Rep. 2016, 6, 31462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valenti, D.; Rossi, L.; Marzulli, D.; Bellomo, F.; De Rasmo, D.; Signorile, A.; Vacca, R.A. Inhibition of Drp1-mediated mitochondrial fission improves mitochondrial dynamics and bioenergetics stimulating neurogenesis in hippocampal progenitor cells from a Down syndrome mouse model. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 3117–3127. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Tekwani, B.L. Histone Deacetylases Inhibitors in Neurodegenerative Diseases, Neuroprotection and Neuronal Differentiation. Front. Pharmacol. 2020, 11, 537. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Sachse, C.; Richter, W.; Xu, C.; Fandrich, M.; Grigorieff, N. Comparison of Alzheimer Abeta(1-40) and Abeta(1-42) amyloid fibrils reveals similar protofilament structures. Proc. Natl. Acad. Sci. USA 2009, 106, 19813–19818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brownjohn, P.W.; Smith, J.; Portelius, E.; Serneels, L.; Kvartsberg, H.; De Strooper, B.; Blennow, K.; Zetterberg, H.; Livesey, F.J. Phenotypic Screening Identifies Modulators of Amyloid Precursor Protein Processing in Human Stem Cell Models of Alzheimer’s Disease. Stem. Cell Rep. 2017, 8, 870–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Kant, R.; Langness, V.F.; Herrera, C.M.; Williams, D.A.; Fong, L.K.; Leestemaker, Y.; Steenvoorden, E.; Rynearson, K.D.; Brouwers, J.F.; Helms, J.B.; et al. Cholesterol Metabolism Is a Druggable Axis that Independently Regulates Tau and Amyloid-beta in iPSC-Derived Alzheimer’s Disease Neurons. Cell Stem Cell 2019, 24, 363–375.e9. [Google Scholar] [CrossRef] [Green Version]
- Rovelet-Lecrux, A.; Hannequin, D.; Raux, G.; Le Meur, N.; Laquerriere, A.; Vital, A.; Dumanchin, C.; Feuillette, S.; Brice, A.; Vercelletto, M.; et al. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat. Genet. 2006, 38, 24–26. [Google Scholar] [CrossRef] [PubMed]
- D’Mello, S.R. Regulation of Central Nervous System Development by Class I Histone Deacetylases. Dev. Neurosci. 2019, 41, 149–165. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, J.L.; Gin, C.S.; Roskams, A.J. Stage-specific induction of DNA methyltransferases in olfactory receptor neuron development. Dev. Biol. 2005, 288, 461–473. [Google Scholar] [CrossRef] [Green Version]
- Jawerka, M.; Colak, D.; Dimou, L.; Spiller, C.; Lagger, S.; Montgomery, R.L.; Olson, E.N.; Wurst, W.; Gottlicher, M.; Gotz, M. The specific role of histone deacetylase 2 in adult neurogenesis. Neuron Glia Biol. 2010, 6, 93–107. [Google Scholar] [CrossRef]
- Gan, Y.; Li, Y.; Long, Z.; Lee, A.R.; Xie, N.; Lovnicki, J.M.; Tang, Y.; Chen, X.; Huang, J.; Dong, X. Roles of Alternative RNA Splicing of the Bif-1 Gene by SRRM4 During the Development of Treatment-induced Neuroendocrine Prostate Cancer. EBio Med. 2018, 31, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Karbowski, M.; Jeong, S.Y.; Youle, R.J. Endophilin B1 is required for the maintenance of mitochondrial morphology. J. Cell Biol. 2004, 166, 1027–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S.G.; Xiao, X.; Wang, S.; Gao, H.; Rafikov, R.; Black, S.; Huang, S.; Ding, H.F.; Yoon, Y.; Kirken, R.A.; et al. Bif-1 Interacts with Prohibitin-2 to Regulate Mitochondrial Inner Membrane during Cell Stress and Apoptosis. J. Am. Soc. Nephrol 2019, 30, 1174–1191. [Google Scholar] [CrossRef] [PubMed]
- Agostini, M.; Romeo, F.; Inoue, S.; Niklison-Chirou, M.V.; Elia, A.J.; Dinsdale, D.; Morone, N.; Knight, R.A.; Mak, T.W.; Melino, G. Metabolic reprogramming during neuronal differentiation. Cell Death Differ. 2016, 23, 1502–1514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, X.; Boyer, L.; Jin, M.; Mertens, J.; Kim, Y.; Ma, L.; Ma, L.; Hamm, M.; Gage, F.H.; Hunter, T. Metabolic reprogramming during neuronal differentiation from aerobic glycolysis to neuronal oxidative phosphorylation. eLife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Lkhagva, B.; Kao, Y.H.; Lee, T.I.; Lee, T.W.; Cheng, W.L.; Chen, Y.J. Activation of Class I histone deacetylases contributes to mitochondrial dysfunction in cardiomyocytes with altered complex activities. Epigenetics 2018, 13, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Levy, S.; Sutton, G.; Ng, P.C.; Feuk, L.; Halpern, A.L.; Walenz, B.P.; Axelrod, N.; Huang, J.; Kirkness, E.F.; Denisov, G.; et al. The diploid genome sequence of an individual human. PLoS Biol. 2007, 5, e254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyaoka, Y.; Chan, A.H.; Judge, L.M.; Yoo, J.; Huang, M.; Nguyen, T.D.; Lizarraga, P.P.; So, P.L.; Conklin, B.R. Isolation of single-base genome-edited human iPS cells without antibiotic selection. Nat. Methods 2014, 11, 291–293. [Google Scholar] [CrossRef] [Green Version]
- Chambers, S.M.; Fasano, C.A.; Papapetrou, E.P.; Tomishima, M.; Sadelain, M.; Studer, L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 2009, 27, 275–280. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Kirwan, P.; Smith, J.; Robinson, H.P.; Livesey, F.J. Human cerebral cortex development from pluripotent stem cells to functional excitatory synapses. Nat. Neurosci. 2012, 15, 477–486. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frankowski, H.; Yeboah, F.; Berry, B.J.; Kinoshita, C.; Lee, M.; Evitts, K.; Davis, J.; Kinoshita, Y.; Morrison, R.S.; Young, J.E. Knock-Down of HDAC2 in Human Induced Pluripotent Stem Cell Derived Neurons Improves Neuronal Mitochondrial Dynamics, Neuronal Maturation and Reduces Amyloid Beta Peptides. Int. J. Mol. Sci. 2021, 22, 2526. https://doi.org/10.3390/ijms22052526
Frankowski H, Yeboah F, Berry BJ, Kinoshita C, Lee M, Evitts K, Davis J, Kinoshita Y, Morrison RS, Young JE. Knock-Down of HDAC2 in Human Induced Pluripotent Stem Cell Derived Neurons Improves Neuronal Mitochondrial Dynamics, Neuronal Maturation and Reduces Amyloid Beta Peptides. International Journal of Molecular Sciences. 2021; 22(5):2526. https://doi.org/10.3390/ijms22052526
Chicago/Turabian StyleFrankowski, Harald, Fred Yeboah, Bonnie J. Berry, Chizuru Kinoshita, Michelle Lee, Kira Evitts, Joshua Davis, Yoshito Kinoshita, Richard S. Morrison, and Jessica E. Young. 2021. "Knock-Down of HDAC2 in Human Induced Pluripotent Stem Cell Derived Neurons Improves Neuronal Mitochondrial Dynamics, Neuronal Maturation and Reduces Amyloid Beta Peptides" International Journal of Molecular Sciences 22, no. 5: 2526. https://doi.org/10.3390/ijms22052526
APA StyleFrankowski, H., Yeboah, F., Berry, B. J., Kinoshita, C., Lee, M., Evitts, K., Davis, J., Kinoshita, Y., Morrison, R. S., & Young, J. E. (2021). Knock-Down of HDAC2 in Human Induced Pluripotent Stem Cell Derived Neurons Improves Neuronal Mitochondrial Dynamics, Neuronal Maturation and Reduces Amyloid Beta Peptides. International Journal of Molecular Sciences, 22(5), 2526. https://doi.org/10.3390/ijms22052526