
Muscle Diversity, Heterogeneity, and Gradients: Learning from Sarcoglycanopathies



 and
and 
Abstract
1. Introduction
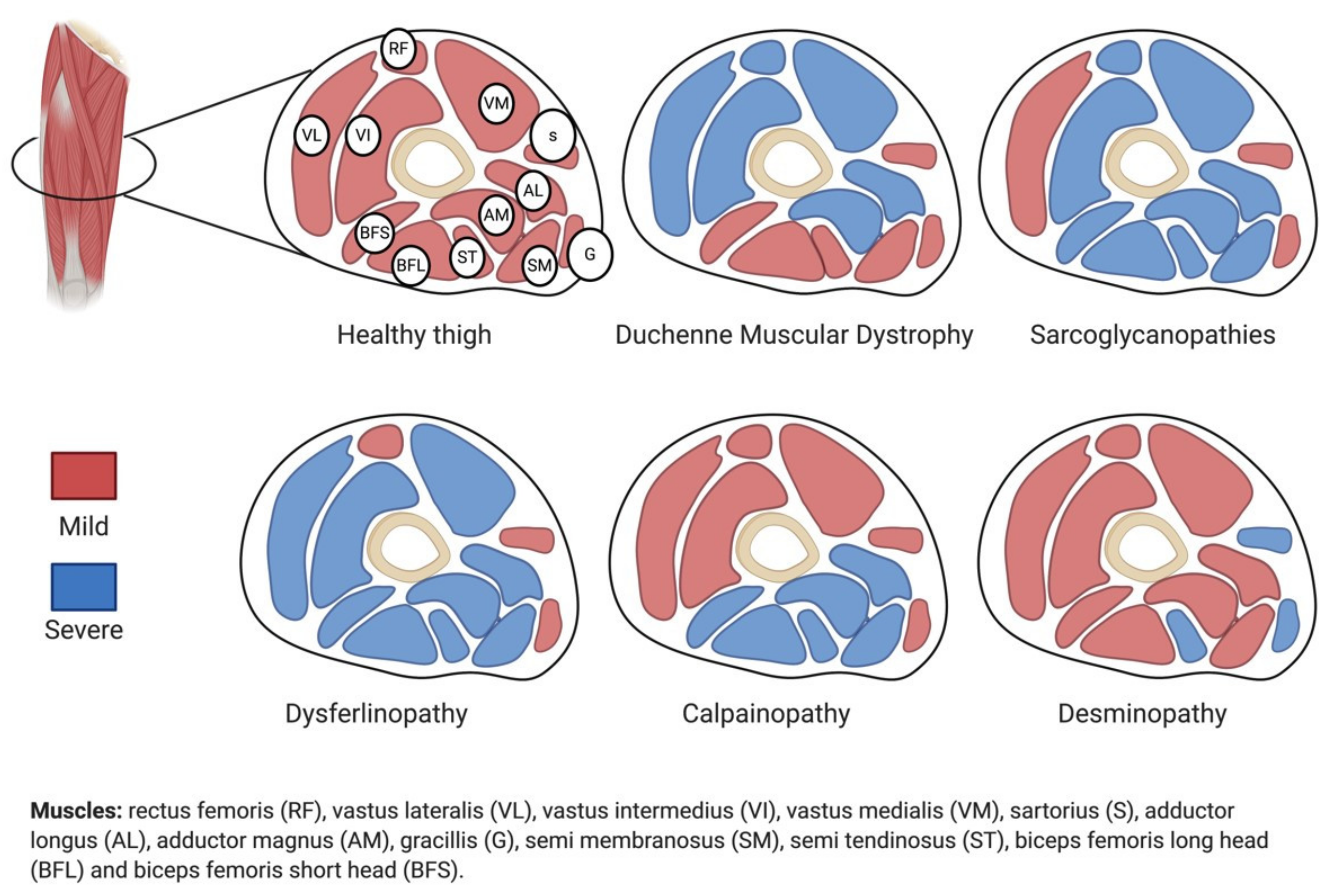
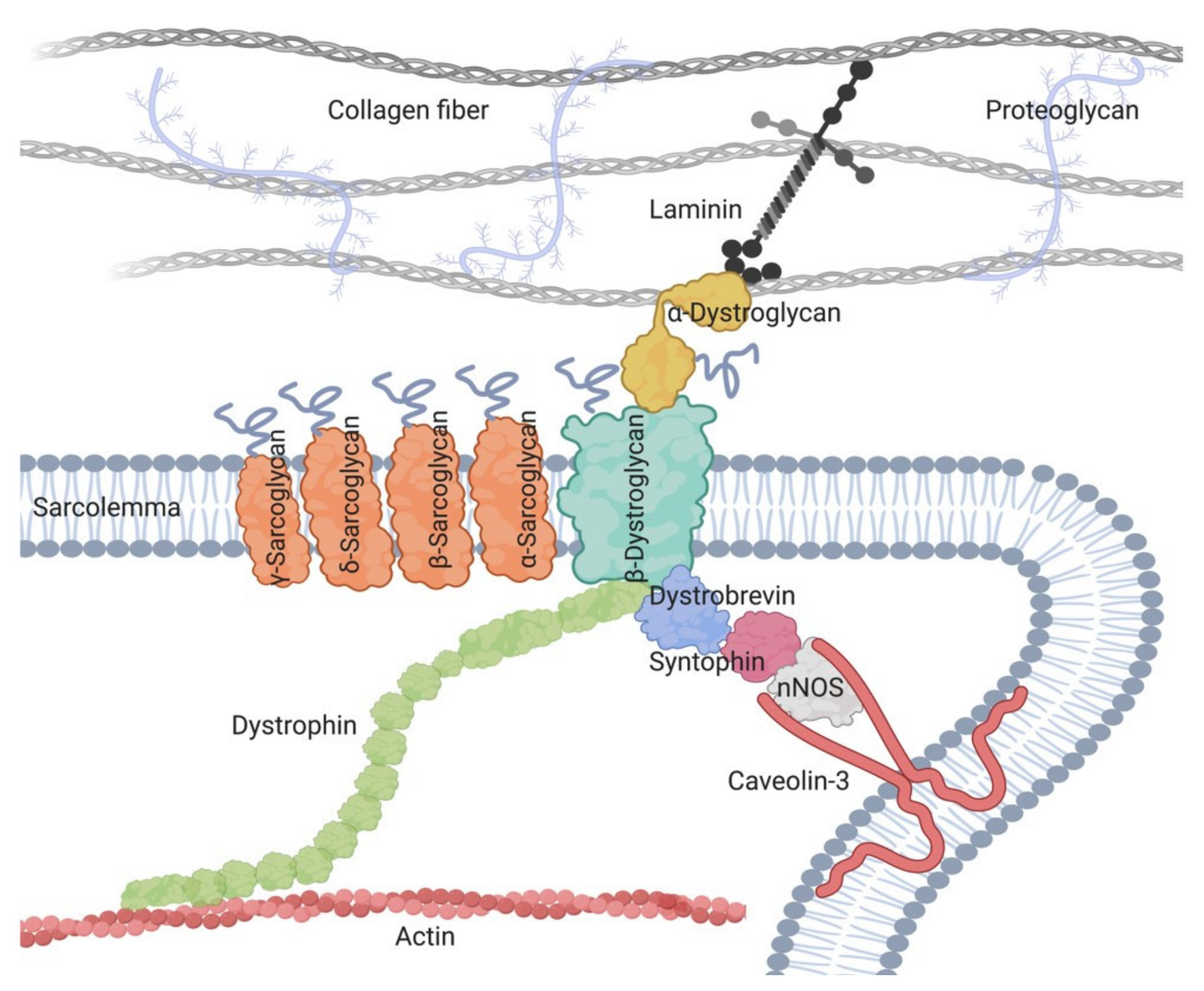
2. Sarcoglycanopathies
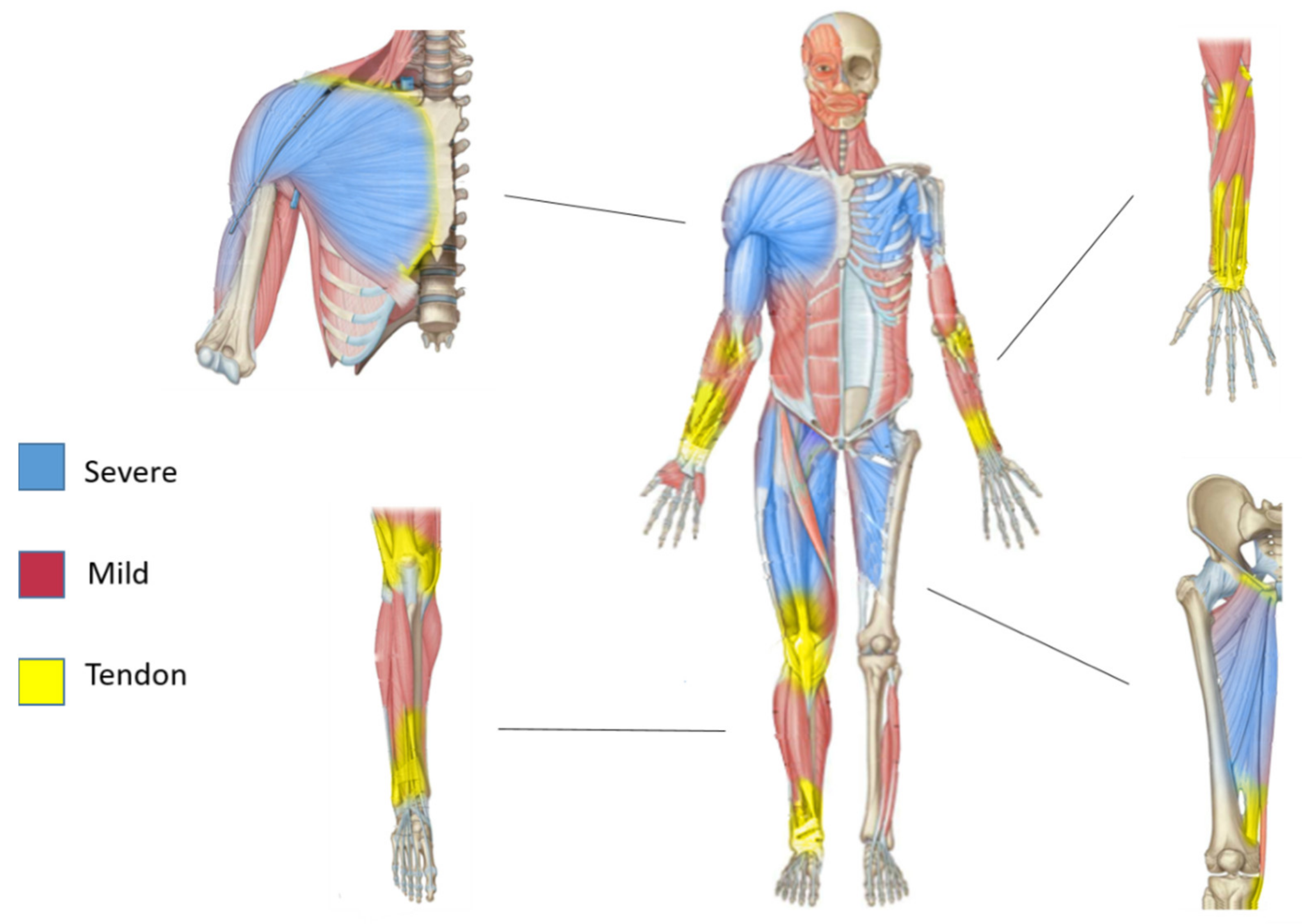
3. Contractile and Non-Contractile Tissue Heterogeneity
4. Cell Population Heterogeneity
5. Gradients and Sarcoglycanopathies
6. Muscle Diversity: Evidence from Murine Models

{kind=link}
{kind=link}
{kind=link}
| Name | Mutation | Target Sarcoglycan | References | Phenotype | Heart Involvement |
|---|---|---|---|---|---|
| Alpha-Sarcoglycan Knock Out | Missense mutation (H77C) exon 3 | Alpha | Kobuke et al. 2008 [88] | Progressive muscle degeneration | No |
| Alpha-Sarcoglycan Knock Out | Sgca (H77C) + transgene human Sgce | Alpha | Kobayashi et al. 2008 [89] | No dystrophic symptoms but fatigue | No |
| Alpha-Sarcoglycan Knock Out | Deletion Exon 2–3 | Alpha | Duclos et al. 1998 [90] | Progressive muscle degeneration | No |
| Alpha-Sarcoglycan Knock Out | Deletion Exon 1–2 | Alpha | Liu et al. 1999 [91] | Progressive muscle degeneration | No |
| Alpha-Sarcoglycan Knock Out Immuno Deficient | Sgca/Rag1/il2rg Knock out | Alpha | Duclos et al. 1998 [90] Mombaerts et al. 1992 [92] Cao et al. 1995 [93] | Progressive muscle degeneration | No |
| Beta-Sarcoglycan Knock Out | Deletion exon 3–6 | Beta | Durbeej et al. 2000 [94] | Progressive muscle degeneration | Yes |
| Beta-Sarcoglycan Knock Out | Disruption exon 2 | Beta | Araishi et al. 1999 [95] | Muscle degeneration and hind limb hypertrophy | Yes—Old age |
| Beta-Sarcoglycan Knock Out | Sgcb/Rag2/γc Knock out | Beta | Giovannelli et al. [70] | Exacerbated dystrophic phenotype | Yes |
| Beta-Sarcoglycan Knock In | Missense mutation T153R KI Exon 4 | Beta | Henriques et al. 2018 [96] | No symptoms | No |
| Gamma-Sarcoglycan Knock Out | Deletion exon 2 | Gamma | Hack AA et al. 1998 [97] | Progressive muscle degeneration | Yes |
| Gamma-Sarcoglycan Knock Out | 521ΔT Single nucleotide deletion Exon 6 | Gamma | Demonbreun et al. 2020 [98] | Progressive muscle degeneration | Yes |
7. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CK | Creatine kinase |
| DAGC | Dystrophin-associated glycoprotein complex |
| ECM | Extracellular matrix |
| FAPs | Fibro-adipogenic progenitors |
| LGMD | Limb girdle muscular dystrophy |
| LGMD-R4 | Beta-sarcoglycanopathy |
| MRI | Magnetic resonance imaging |
| MYH16 | Myosin heavy chain 16 |
| NADH | Nicotinamide adenine dinucleotide reduced form |
| PICs | PW1+ interstitial cells |
| SCs | Satellite cells |
| SG | Sarcoglycan |
| SHD | Succinate dehydrogenase |
| SCGE | Sarcoglycan subtype E |
| SCGZ | Sarcoglycan subtype Z |
References
- Biferali, B.; Proietti, D.; Mozzetta, C.; Madaro, L. Fibro-Adipogenic Progenitors Cross-Talk in Skeletal Muscle: The Social Network. Front. Physiol. 2019, 10, 1074. [Google Scholar] [CrossRef] [PubMed]
- Lieber, R.L.; Roberts, T.J.; Blemker, S.S.; Lee, S.S.M.; Herzog, W. Skeletal muscle mechanics, energetics and plasticity. J. Neuroeng. Rehabil. 2017, 14, 1–16. [Google Scholar] [CrossRef]
- Mukund, K.; Subramaniam, S. Skeletal muscle: A review of molecular structure and function, in health and disease. Wiley Interdiscip. Rev. Syst. Biol. Med. 2019, 12, e1462. [Google Scholar] [CrossRef]
- Yin, H.; Price, F.; Rudnicki, M.A. Satellite Cells and the Muscle Stem Cell Niche. Physiol. Rev. 2013, 93, 23–67. [Google Scholar] [CrossRef]
- Hepple, R.T.; Rice, C.L. Innervation and neuromuscular control in ageing skeletal muscle. J. Physiol. 2016, 594, 1965–1978. [Google Scholar] [CrossRef] [PubMed]
- Ciciliot, S.; Rossi, A.C.; Dyar, K.A.; Blaauw, B.; Schiaffino, S. Muscle type and fiber type specificity in muscle wasting. Int. J. Biochem. Cell Biol. 2013, 45, 2191–2199. [Google Scholar] [CrossRef] [PubMed]
- Bottinelli, R.; Pellegrino, M.; Canepari, M.; Rossi, R.; Reggiani, C. Specific contributions of various muscle fibre types to human muscle performance: An in vitro study. J. Electromyogr. Kinesiol. 1999, 9, 87–95. [Google Scholar] [CrossRef]
- Pierotti, D.J.; Roy, R.R.; Hodgson, J.A.; Edgerton, V.R. Level of independence of motor unit properties from neuromuscular activity. Muscle Nerve 1994, 17, 1324–1335. [Google Scholar] [CrossRef] [PubMed]
- Zardini, D.M.; Parry, D.J. Physiological characteristics of identified motor units in the mouse extensor digitorum longus muscle: An in vitro approach. Can. J. Physiol. Pharmacol. 1998, 76, 68–71. [Google Scholar] [CrossRef] [PubMed]
- Egan, B.; Zierath, J.R. Exercise Metabolism and the Molecular Regulation of Skeletal Muscle Adaptation. Cell Metab. 2013, 17, 162–184. [Google Scholar] [CrossRef] [PubMed]
- Bassel-Duby, R.; Olson, E.N. Signaling Pathways in Skeletal Muscle Remodeling. Annu. Rev. Biochem. 2006, 75, 19–37. [Google Scholar] [CrossRef]
- Egerman, M.A.; Glass, D.J. Signaling pathways controlling skeletal muscle mass. Crit. Rev. Biochem. Mol. Biol. 2013, 49, 59–68. [Google Scholar] [CrossRef]
- Kuo, I.Y.; Ehrlich, B.E. Signaling in Muscle Contraction. Cold Spring Harb. Perspect. Biol. 2015, 7, a006023. [Google Scholar] [CrossRef] [PubMed]
- Larsson, L.; Salviati, G. Effects of age on calcium transport activity of sarcoplasmic reticulum in fast- and slow-twitch rat muscle fibres. J. Physiol. 1989, 419, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Schiaffino, S.; Reggiani, C. Fiber Types in Mammalian Skeletal Muscles. Physiol. Rev. 2011, 91, 1447–1531. [Google Scholar] [CrossRef] [PubMed]
- Gacesa, J.Z.P.; Klasnja, A.V.; Grujic, N.G. Changes in Strength, Endurance, and Fatigue During a Resistance-Training Program for the Triceps Brachii Muscle. J. Athl. Train. 2013, 48, 804–809. [Google Scholar] [CrossRef] [PubMed]
- Chrzanowski, S.M.; Baligand, C.; Willcocks, R.J.; Deol, J.; Schmalfuss, I.; Lott, N.J.; Daniels, M.J.; Senesac, C.; Walter, G.A.; Vandenborne, K. Multi-slice MRI reveals heterogeneity in disease distribution along the length of muscle in Duchenne muscular dystrophy. Acta Myol. 2017, 36, 151–162. [Google Scholar] [PubMed]
- Díaz-Manera, J.; Llauger, J.; Gallardo, E.; Illa, I. Muscle MRI in muscular dystrophies. Acta Myol. 2015, 34, 95–108. [Google Scholar] [PubMed]
- Zatz, M.; Vainzof, M.; Passos-Bueno, M.R. Limb-girdle muscular dystrophy: One gene with different phenotypes, one phenotype with different genes. Curr. Opin. Neurol. 2000, 13, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Manera, J.; Fernandez-Torron, R.; Llauger, J.; James, M.K.; Mayhew, A.; Smith, F.E.; Moore, U.R.; Blamire, A.M.; Carlier, P.G.; Rufibach, L.; et al. Muscle MRI in patients with dysferlinopathy: Pattern recognition and implications for clinical trials. J. Neurol. Neurosurg. Psychiatry 2018, 89, 1071–1081. [Google Scholar] [CrossRef]
- Tasca, G.; Monforte, M.; Díaz-Manera, J.; Brisca, G.; Semplicini, C.; D’Amico, A.; Fattori, F.; Pichiecchio, A.; Berardinelli, A.; Maggi, L.; et al. MRI in sarcoglycanopathies: A large international cohort study. J. Neurol. Neurosurg. Psychiatry 2018, 89, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Murphy, A.P.; Straub, V. The Classification, Natural History and Treatment of the Limb Girdle Muscular Dystrophies. J. Neuromuscul. Dis. 2015, 2, S7–S19. [Google Scholar] [CrossRef] [PubMed]
- Marsolier, J.; Laforet, P.; Pegoraro, E.; Vissing, J.; Richard, I.; Barnerias, C.; Carlier, R.-Y.; Díaz-Manera, J.; Fayssoil, A.; Galy, A.; et al. 1st International Workshop on Clinical trial readiness for sarcoglycanopathies 15–16 November 2016, Evry, France. Neuromuscul. Disord. 2017, 27, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Liewluck, T.; Milone, M. Untangling the complexity of limb-girdle muscular dystrophies. Muscle Nerve 2018, 58, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Pérez, J.; González-Quereda, L.; Bello, L.; Guglieri, M.; Straub, V.; Gallano, P.; Semplicini, C.; Pegoraro, E.; Zangaro, V.; Nascimento, A.; et al. New genotype-phenotype correlations in a large European cohort of patients with sarcoglycanopathy. Brain 2020, 143, 2696–2708. [Google Scholar] [CrossRef] [PubMed]
- Terry, E.E.; Zhang, X.; Hoffmann, C.; Hughes, L.D.; Lewis, S.A.; Li, J.; Wallace, M.J.; Riley, L.A.; Douglas, C.M.; Gutierrez-Montreal, M.A.; et al. Transcriptional profiling reveals extraordinary diversity among skeletal muscle tissues. eLife 2018, 7, 1–27. [Google Scholar] [CrossRef]
- Lim, L.E.; Duclos, F.; Broux, O.; Bourg, N.; Sunada, Y.; Allamand, V.; Meyer, J.; Richard, I.; Moomaw, C.; Slaughter, C.; et al. β-sarcoglycan: Characterization and role in limb-girdle muscular dystrophy linked to 4q12. Nat. Genet. 1995, 11, 257–265. [Google Scholar] [CrossRef]
- Moore, S.A.; Shilling, C.J.; Westra, S.; Wall, C.; Wicklund, M.P.; Stolle, C.; Brown, C.A.; Michele, D.E.; Piccolo, F.; Winder, T.L.; et al. Limb-Girdle Muscular Dystrophy in the United States. J. Neuropathol. Exp. Neurol. 2006, 65, 995–1003. [Google Scholar] [CrossRef]
- Liu, W.; Pajusalu, S.; Lake, N.J.; Zhou, G.; Ioannidis, N.; Mittal, P.; Msc, N.E.J.; Weihl, C.C.; Williams, B.A.; Albrecht, D.E.; et al. Estimating prevalence for limb-girdle muscular dystrophy based on public sequencing databases. Genet. Med. 2019, 21, 2512–2520. [Google Scholar] [CrossRef]
- Noguchi, S.; Wakabayashi, E.; Imamura, M.; Yoshida, M.; Ozawa, E. Formation of sarcoglycan complex with differentiation in cultured myocytes. JBIC J. Biol. Inorg. Chem. 2000, 267, 640–648. [Google Scholar] [CrossRef]
- Hack, A.A.; Groh, M.E.; McNally, E.M. Sarcoglycans in muscular dystrophy. Microsc. Res. Tech. 2000, 48, 167–180. [Google Scholar] [CrossRef]
- Groh, S.; Zong, H.; Goddeeris, M.M.; Lebakken, C.S.; Venzke, D.; Pessin, J.E.; Campbell, K.P. Sarcoglycan Complex. J. Biol. Chem. 2009, 284, 19178–19182. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, E.; Mizuno, Y.; Hagiwara, Y.; Sasaoka, T.; Yoshida, M. Molecular and cell biology of the sarcoglycan complex. Muscle Nerve 2005, 32, 563–576. [Google Scholar] [CrossRef] [PubMed]
- Hack, A.A.; Lam, M.Y.; Cordier, L.; Shoturma, D.I.; Ly, C.T.; Hadhazy, M.A.; Sweeney, H.L.; McNally, E.M. Differential requirement for individual sarcoglycans and dystrophin in the assembly and function of the dystrophin-glycoprotein complex. J. Cell Sci. 2000, 113, 2535–2544. [Google Scholar] [PubMed]
- Lim, L.E.; Campbell, K.P. The sarcoglycan complex in limb-girdle muscular dystrophy. Curr. Opin. Neurol. 1998, 11, 443–452. [Google Scholar] [CrossRef]
- Hack, A.A.; Cordier, L.; Shoturma, D.I.; Lam, M.Y.; Sweeney, H.L.; McNally, E.M. Muscle degeneration without mechanical injury in sarcoglycan deficiency. Proc. Natl. Acad. Sci. USA 1999, 96, 10723–10728. [Google Scholar] [CrossRef] [PubMed]
- Siciliano, G.; Simoncini, C.; Giannotti, S.; Zampa, V.; Angelini, C.; Ricci, G. Muscle exercise in limb girdle muscular dystrophies: Pitfall and advantages. Acta Myol. 2015, 34, 3–8. [Google Scholar] [PubMed]
- Boulay, A.-C.; Saubaméa, B.; Cisternino, S.; Mignon, V.; Mazeraud, A.; Jourdren, L.; Blugeon, C.; Cohen-Salmon, M. The Sarcoglycan complex is expressed in the cerebrovascular system and is specifically regulated by astroglial Cx30 channels. Front. Cell. Neurosci. 2015, 9. [Google Scholar] [CrossRef] [PubMed]
- Fanin, M.; Melacini, P.; Boito, C.; Pegoraro, E.; Angelini, C. LGMD2E patients risk developing dilated cardiomyopathy. Neuromuscul. Disord. 2003, 13, 303–309. [Google Scholar] [CrossRef]
- Sveen, M.-L.; Thune, J.J.; Køber, L.; Vissing, J. Cardiac Involvement in Patients With Limb-Girdle Muscular Dystrophy Type 2 and Becker Muscular Dystrophy. Arch. Neurol. 2008, 65, 1196–1201. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.; Straub, V. Limb-girdle muscular dystrophies—International collaborations for translational research. Nat. Rev. Neurol. 2016, 12, 294–309. [Google Scholar] [CrossRef]
- Mitsuhashi, S.; Kang, P.B. Update on the Genetics of Limb Girdle Muscular Dystrophy. Semin. Pediatr. Neurol. 2012, 19, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Hou, Y.; Yu, M.; Liu, Y.; Fan, Y.; Zhang, W.; Wang, Z.; Xiong, H.; Yuan, Y. Clinical and genetic spectrum of sarcoglycanopathies in a large cohort of Chinese patients. Orphanet J. Rare Dis. 2019, 14, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Berry, S.E. Concise Review: Mesoangioblast and Mesenchymal Stem Cell Therapy for Muscular Dystrophy: Progress, Challenges, and Future Directions. STEM CELLS Transl. Med. 2015, 4, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Wong-Kisiel, L.C.; Kuntz, N.L. Two siblings with limb-girdle muscular dystrophy type 2E responsive to deflazacort. Neuromuscul. Disord. 2010, 20, 122–124. [Google Scholar] [CrossRef] [PubMed]
- Pozsgai, E.R.; Griffin, D.A.; Heller, K.N.; Mendell, J.R.; Rodino-Klapac, L.R. Systemic AAV-Mediated β-Sarcoglycan Delivery Targeting Cardiac and Skeletal Muscle Ameliorates Histological and Functional Deficits in LGMD2E Mice. Mol. Ther. 2017, 25, 855–869. [Google Scholar] [CrossRef] [PubMed]
- Pozsgai, E.; Griffin, D.; Heller, K.N.; Mendell, J.R.; Rodinoklapac, L.R. β-Sarcoglycan gene transfer decreases fibrosis and restores force in LGMD2E mice. Gene Ther. 2015, 23, 57–66. [Google Scholar] [CrossRef]
- Mepham, B.L. Theory and practice of histological techniques, 3rd ed. J. D. Bancroft, A. Stevens (Eds). Churchill Livingstone, Edinburgh, 1990. No. of pages: 740. Price: £55. ISBN: 0 443 03559 8. J. Pathol. 1991, 164, 281. [Google Scholar] [CrossRef]
- Noden, D.M.; Francis-West, P. The differentiation and morphogenesis of craniofacial muscles. Dev. Dyn. 2006, 235, 1194–1218. [Google Scholar] [CrossRef]
- Schiaffino, S.; Reggiani, C. Myosin isoforms in mammalian skeletal muscle. J. Appl. Physiol. 1994, 77, 493–501. [Google Scholar] [CrossRef]
- Haddad, F.; Qin, A.X.; Bodell, P.W.; Jiang, W.; Giger, J.M.; Baldwin, K.M. Intergenic transcription and developmental regulation of cardiac myosin heavy chain genes. Am. J. Physiol. Circ. Physiol. 2008, 294, H29–H40. [Google Scholar] [CrossRef]
- Porter, J.D.; Khanna, S.; Kaminski, H.J.; Rao, J.S.; Merriam, A.P.; Richmonds, C.R.; Leahy, P.; Li, J.; Andrade, F.H. Extraocular muscle is defined by a fundamentally distinct gene expression profile. Proc. Natl. Acad. Sci. USA 2001, 98, 12062–12067. [Google Scholar] [CrossRef] [PubMed]
- Khanna, S.; Merriam, A.P.; Gong, B.; Leahy, P.; Porter, J.D.; Zwaka, T.P.; Torzewski, J.; Hoeflich, A.; Déjosez, M.; Kaiser, S.; et al. Comprehensive expression profiling by muscle tissue class and identification of the molecular niche of extraocular muscle. FASEB J. 2003, 17, 1370–1372. [Google Scholar] [CrossRef]
- Bs, A.R.G.; Lieber, R.L. Structure and function of the skeletal muscle extracellular matrix. Muscle Nerve 2011, 44, 318–331. [Google Scholar] [CrossRef]
- Mann, C.J.; Perdiguero, E.; Kharraz, Y.; Aguilar, S.; Pessina, P.; Serrano, A.L.; Muñoz-Cánoves, P. Aberrant repair and fibrosis development in skeletal muscle. Skelet. Muscle 2011, 1, 21. [Google Scholar] [CrossRef] [PubMed]
- Lemos, D.R.; Babaeijandaghi, F.; Low, M.; Chang, C.-K.; Lee, S.T.; Fiore, D.; Zhang, R.-H.; Natarajan, A.; Nedospasov, S.A.; Rossi, F.M.V. Nilotinib reduces muscle fibrosis in chronic muscle injury by promoting TNF-mediated apoptosis of fibro/adipogenic progenitors. Nat. Med. 2015, 21, 786–794. [Google Scholar] [CrossRef]
- Gelse, K. Collagens—Structure, function, and biosynthesis. Adv. Drug Deliv. Rev. 2003, 55, 1531–1546. [Google Scholar] [CrossRef]
- Light, N.; Champion, A.E. Characterization of muscle epimysium, perimysium and endomysium collagens. Biochem. J. 1984, 219, 1017–1026. [Google Scholar] [CrossRef] [PubMed]
- Farup, J.; Madaro, L.; Puri, P.L.; Mikkelsen, U.R. Interactions between muscle stem cells, mesenchymal-derived cells and immune cells in muscle homeostasis, regeneration and disease. Cell Death Dis. 2015, 6, e1830. [Google Scholar] [CrossRef] [PubMed]
- Biewener, A.A.; Roberts, T.J. Muscle and tendon contributions to force, work, and elastic energy savings: A comparative perspective. Exerc. Sport Sci. Rev. 2000, 28, 99–107. [Google Scholar] [PubMed]
- Borg, T.K.; Caulfield, J.B. Morphology of connective tissue in skeletal muscle. Tissue Cell 1980, 12, 197–207. [Google Scholar] [CrossRef]
- Järvinen, T.A.H.; Józsa, L.; Kannus, P.; Järvinen, T.L.N.; Järvinen, M. Organization and distribution of intramuscular connective tissue in normal and immobilized skeletal muscles. J. Muscle Res. Cell Motil. 2002, 23, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Mauro, A. Satellite cell of skeletal muscle fibers. J. Cell Biol. 1961, 9, 493–495. [Google Scholar] [CrossRef]
- LaBarge, M.A.; Blau, H.M. Biological Progression from Adult Bone Marrow to Mononucleate Muscle Stem Cell to Multinucleate Muscle Fiber in Response to Injury. Cell 2002, 111, 589–601. [Google Scholar] [CrossRef]
- Uezumi, A.; Fukada, S.-I.; Yamamoto, N.; Takeda, S.; Tsuchida, K. Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nat. Cell Biol. 2010, 12, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Angelis, M.G.C.-D.; Berghella, L.; Coletta, M.; Lattanzi, L.; Zanchi, M.; Gabriella, M.; Ponzetto, C.; Cossu, G. Skeletal Myogenic Progenitors Originating from Embryonic Dorsal Aorta Coexpress Endothelial and Myogenic Markers and Contribute to Postnatal Muscle Growth and Regeneration. J. Cell Biol. 1999, 147, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Dellavalle, A.; Sampaolesi, M.; Tonlorenzi, R.; Tagliafico, E.; Sacchetti, B.; Perani, L.; Innocenzi, A.; Gálvez, B.G.; Messina, G.; Morosetti, R.; et al. Pericytes of human skeletal muscle are myogenic precursors distinct from satellite cells. Nat. Cell Biol. 2007, 9, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, K.J.; Pannérec, A.; Cadot, B.; Parlakian, A.; Besson, V.; Gomes, E.R.; Marazzi, G.; Sassoon, D.A. Identification and characterization of a non-satellite cell muscle resident progenitor during postnatal development. Nat. Cell Biol. 2010, 12, 257–266. [Google Scholar] [CrossRef]
- Rizzo, G.; Di Maggio, R.; Benedetti, A.; Morroni, J.; Bouche, M.; Lozanoska-Ochser, B. Splenic Ly6Chi monocytes are critical players in dystrophic muscle injury and repair. JCI Insight 2020, 5, e130807. [Google Scholar] [CrossRef] [PubMed]
- Giovannelli, G.; Msc, G.G.; Grosemans, H.; Sampaolesi, M. Morphological and functional analyses of skeletal muscles from an immunodeficient animal model of limb-girdle muscular dystrophy type 2E. Muscle Nerve 2018, 58, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Camps, J.; Grosemans, H.; Gijsbers, R.; Maes, C.; Sampaolesi, M. Growth Factor Screening in Dystrophic Muscles Reveals PDGFB/PDGFRB-Mediated Migration of Interstitial Stem Cells. Int. J. Mol. Sci. 2019, 20, 1118. [Google Scholar] [CrossRef] [PubMed]
- Piñol-Jurado, P.; Gallardo, E.; De Luna, N.; Suárez-Calvet, X.; Sánchez-Riera, C.; Fernández-Simón, E.; Gomis, C.; Illa, I.; Díaz-Manera, J. Platelet-Derived Growth Factor BB Influences Muscle Regeneration in Duchenne Muscle Dystrophy. Am. J. Pathol. 2017, 187, 1814–1827. [Google Scholar] [CrossRef] [PubMed]
- Motohashi, N.; Asakura, A. Muscle satellite cell heterogeneity and self-renewal. Front. Cell Dev. Biol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Relaix, F.; Montarras, D.; Zaffran, S.; Gayraud-Morel, B.; Rocancourt, D.; Tajbakhsh, S.; Mansouri, A.; Cumano, A.; Buckingham, M. Pax3 and Pax7 have distinct and overlapping functions in adult muscle progenitor cells. J. Cell Biol. 2005, 172, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Sambasivan, R.; Gayraud-Morel, B.; Dumas, G.; Cimper, C.; Paisant, S.; Kelly, R.G.; Tajbakhsh, S. Distinct Regulatory Cascades Govern Extraocular and Pharyngeal Arch Muscle Progenitor Cell Fates. Dev. Cell 2009, 16, 810–821. [Google Scholar] [CrossRef]
- Sacco, A.; Doyonnas, R.; Kraft, P.; Vitorovic, S.; Blau, H.M. Self-renewal and expansion of single transplanted muscle stem cells. Nat. Cell Biol. 2008, 456, 502–506. [Google Scholar] [CrossRef] [PubMed]
- Collins, C.A.; Olsen, I.; Zammit, P.S.; Heslop, L.; Petrie, A.; Partridge, T.A.; Morgan, J.E. Stem Cell Function, Self-Renewal, and Behavioral Heterogeneity of Cells from the Adult Muscle Satellite Cell Niche. Cell 2005, 122, 289–301. [Google Scholar] [CrossRef]
- Schultz, E. Satellite Cell Proliferative Compartments in Growing Skeletal Muscles. Dev. Biol. 1996, 175, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Malecova, B.; Gatto, S.; Etxaniz, U.; Passafaro, M.; Cortez, A.; Nicoletti, C.; Giordani, L.; Torcinaro, A.; De Bardi, M.; Bicciato, S.; et al. Dynamics of cellular states of fibro-adipogenic progenitors during myogenesis and muscular dystrophy. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Gatto, S.; Puri, P.L.; Malecova, B. Single Cell Gene Expression Profiling of Skeletal Muscle-Derived Cells. In Methods in Molecular Biology; Springer International Publishing: Cham, Switzerland, 2017; Volume 1556, pp. 191–219. [Google Scholar]
- Giordani, L.; He, G.J.; Negroni, E.; Sakai, H.; Law, J.Y.; Siu, M.M.; Wan, R.; Corneau, A.; Tajbakhsh, S.; Cheung, T.H.; et al. High-Dimensional Single-Cell Cartography Reveals Novel Skeletal Muscle-Resident Cell Populations. Mol. Cell 2019, 74, 609–621.e6. [Google Scholar] [CrossRef] [PubMed]
- Camps, J.; Breuls, N.; Sifrim, A.; Giarratana, N.; Corvelyn, M.; Danti, L.; Grosemans, H.; Vanuytven, S.; Thiry, I.; Belicchi, M.; et al. Interstitial Cell Remodeling Promotes Aberrant Adipogenesis in Dystrophic Muscles. Cell Rep. 2020, 31, 107597. [Google Scholar] [CrossRef] [PubMed]
- Charles, J.P.; Cappellari, O.; Spence, A.J.; Hutchinson, J.R.; Wells, D.J. Musculoskeletal Geometry, Muscle Architecture and Functional Specialisations of the Mouse Hindlimb. PLoS ONE 2016, 11, e0147669. [Google Scholar] [CrossRef]
- Mathewson, M.A.; Chapman, M.A.; Hentzen, E.R.; Fridén, J.; Lieber, R.L. Anatomical, architectural, and biochemical diversity of the murine forelimb muscles. J. Anat. 2012, 221, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Daley, M.A.; Felix, G.; Biewener, A.A. Running stability is enhanced by a proximo-distal gradient in joint neuromechanical control. J. Exp. Biol. 2007, 210, 732. [Google Scholar] [CrossRef][Green Version]
- Turk, R.; Sterrenburg, E.; Van Der Wees, C.G.C.; De Meijer, E.J.; De Menezes, R.X.; Groh, S.; Campbell, K.P.; Noguchi, S.; Van Ommen, G.J.B.; Dunnen, J.T.D.; et al. Common pathological mechanisms in mouse models for muscular dystrophies. FASEB J. 2005, 20, 127–129. [Google Scholar] [CrossRef]
- Van Putten, M.; Lloyd, E.M.; De Greef, J.C.; Raz, V.; Willmann, R.; Grounds, M.D. Mouse models for muscular dystrophies: An overview. Dis. Model. Mech. 2020, 13, dmm043562. [Google Scholar] [CrossRef] [PubMed]
- Kobuke, K.; Piccolo, F.; Garringer, K.W.; Moore, S.A.; Sweezer, E.; Yang, B.; Campbell, K.P. A common disease-associated missense mutation in alpha-sarcoglycan fails to cause muscular dystrophy in mice. Hum. Mol. Genet. 2008, 17, 1201–1213. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.M.; Rader, E.P.; Crawford, R.W.; Iyengar, N.K.; Thedens, D.R.; Faulkner, J.A.; Parikh, S.V.; Weiss, R.M.; Chamberlain, J.S.; Moore, S.A.; et al. Sarcolemma-localized nNOS is required to maintain activity after mild exercise. Nat. Cell Biol. 2008, 456, 511–515. [Google Scholar] [CrossRef]
- Duclos, F.; Straub, V.; Moore, S.A.; Venzke, D.P.; Hrstka, R.F.; Crosbie, R.H.; Durbeej, M.; Lebakken, C.S.; Ettinger, A.J.; Van Der Meulen, J.; et al. Progressive Muscular Dystrophy in α-Sarcoglycan-Deficient Mice. J. Cell Biol. 1998, 142, 1461–1471. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.A.; Engvall, E. Sarcoglycan Isoforms in Skeletal Muscle. J. Biol. Chem. 1999, 274, 38171–38176. [Google Scholar] [CrossRef]
- Mombaerts, P.; Iacomini, J.; Johnson, R.S.; Herrup, K.; Tonegawa, S.; Papaioannou, V.E. RAG-1-deficient mice have no mature B and T lymphocytes. Cell 1992, 68, 869–877. [Google Scholar] [CrossRef]
- Cao, X.; Shores, E.W.; Hu-Li, J.; Anver, M.R.; Kelsail, B.L.; Russell, S.M.; Drago, J.; Noguchi, M.; Grinberg, A.; Bloom, E.T.; et al. Defective lymphoid development in mice lacking expression of the common cytokine receptor γ chain. Immunity 1995, 2, 223–238. [Google Scholar] [CrossRef]
- Durbeej, M.; Cohn, R.D.; Hrstka, R.F.; Moore, S.A.; Allamand, V.; Davidson, B.L.; Williamson, R.A.; Campbell, K.P. Disruption of the β-Sarcoglycan Gene Reveals Pathogenetic Complexity of Limb-Girdle Muscular Dystrophy Type 2E. Mol. Cell 2000, 5, 141–151. [Google Scholar] [CrossRef]
- Araishi, K.; Sasaoka, T.; Imamura, M.; Noguchi, S.; Hama, H.; Wakabayashi, E.; Yoshida, M.; Hori, T.; Ozawa, E. Loss of the Sarcoglycan Complex and Sarcospan Leads to Muscular Dystrophy in -Sarcoglycan-Deficient Mice. Hum. Mol. Genet. 1999, 8, 1589–1598. [Google Scholar] [CrossRef]
- Henriques, S.F.; Patissier, C.; Bourg, N.; Fecchio, C.; Sandonà, D.; Marsolier, J.; Richard, I. Different outcome of sarcoglycan missense mutation between human and mouse. PLoS ONE 2018, 13, e0191274. [Google Scholar] [CrossRef]
- Hack, A.A.; Ly, C.T.; Jiang, F.; Clendenin, C.J.; Sigrist, K.S.; Wollmann, R.L.; McNally, E.M. γ-Sarcoglycan Deficiency Leads to Muscle Membrane Defects and Apoptosis Independent of Dystrophin. J. Cell Biol. 1998, 142, 1279–1287. [Google Scholar] [CrossRef] [PubMed]
- Demonbreun, A.R.; Wyatt, E.J.; Fallon, K.S.; Oosterbaan, C.C.; Page, P.G.; Hadhazy, M.; Quattrocelli, M.; Barefield, D.Y.; McNally, E.M. A gene-edited mouse model of limb-girdle muscular dystrophy 2C for testing exon skipping. Dis. Model. Mech. 2020, 13, dmm040832. [Google Scholar] [CrossRef] [PubMed]
- Straub, V.; Rafael, J.A.; Chamberlain, J.S.; Campbell, K.P. Animal Models for Muscular Dystrophy Show Different Patterns of Sarcolemmal Disruption. J. Cell Biol. 1997, 139, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Sasaoka, T.; Imamura, M.; Araishi, K.; Noguchi, S.; Mizuno, Y.; Takagoshi, N.; Hama, H.; Wakabayashi-Takai, E.; Yoshimoto-Matsuda, Y.; Nonaka, I.; et al. Pathological analysis of muscle hypertrophy and degeneration in muscular dystrophy in γ-sarcoglycan-deficient mice. Neuromuscul. Disord. 2003, 13, 193–206. [Google Scholar] [CrossRef]
- Semplicini, C.; Vissing, J.; Dahlqvist, J.R.; Stojkovic, T.; Bello, L.; Witting, N.; Duno, M.; Leturcq, F.; Bertolin, C.; D’Ambrosio, P.; et al. Clinical and genetic spectrum in limb-girdle muscular dystrophy type 2E. Neurology 2015, 84, 1772–1781. [Google Scholar] [CrossRef]
- Pratt, S.J.; Xu, S.; Mullins, R.J.; Lovering, R.M. Temporal changes in magnetic resonance imaging in the mdx mouse. BMC Res. Notes 2013, 6, 262. [Google Scholar] [CrossRef] [PubMed]
- Heemskerk, A.M.; Strijkers, G.J.; Vilanova, A.; Drost, M.R.; Nicolay, K. Determination of mouse skeletal muscle architecture using three-dimensional diffusion tensor imaging. Magn. Reson. Med. 2005, 53, 1333–1340. [Google Scholar] [CrossRef] [PubMed]
- Chemello, F.; Bean, C.; Cancellara, P.; Laveder, P.; Reggiani, C.; Lanfranchi, G. Microgenomic Analysis in Skeletal Muscle: Expression Signatures of Individual Fast and Slow Myofibers. PLoS ONE 2011, 6, e16807. [Google Scholar] [CrossRef] [PubMed]
- Spangenburg, E.E.; Booth, F.W. Molecular regulation of individual skeletal muscle fibre types. Acta Physiol. Scand. 2003, 178, 413–424. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez Riera, C.; Lozanoska-Ochser, B.; Testa, S.; Fornetti, E.; Bouché, M.; Madaro, L. Muscle Diversity, Heterogeneity, and Gradients: Learning from Sarcoglycanopathies. Int. J. Mol. Sci. 2021, 22, 2502. https://doi.org/10.3390/ijms22052502
Sánchez Riera C, Lozanoska-Ochser B, Testa S, Fornetti E, Bouché M, Madaro L. Muscle Diversity, Heterogeneity, and Gradients: Learning from Sarcoglycanopathies. International Journal of Molecular Sciences. 2021; 22(5):2502. https://doi.org/10.3390/ijms22052502
Chicago/Turabian StyleSánchez Riera, Carles, Biliana Lozanoska-Ochser, Stefano Testa, Ersilia Fornetti, Marina Bouché, and Luca Madaro. 2021. "Muscle Diversity, Heterogeneity, and Gradients: Learning from Sarcoglycanopathies" International Journal of Molecular Sciences 22, no. 5: 2502. https://doi.org/10.3390/ijms22052502
APA StyleSánchez Riera, C., Lozanoska-Ochser, B., Testa, S., Fornetti, E., Bouché, M., & Madaro, L. (2021). Muscle Diversity, Heterogeneity, and Gradients: Learning from Sarcoglycanopathies. International Journal of Molecular Sciences, 22(5), 2502. https://doi.org/10.3390/ijms22052502