The Role of Collagen Triple Helix Repeat-Containing 1 Protein (CTHRC1) in Rheumatoid Arthritis



Abstract
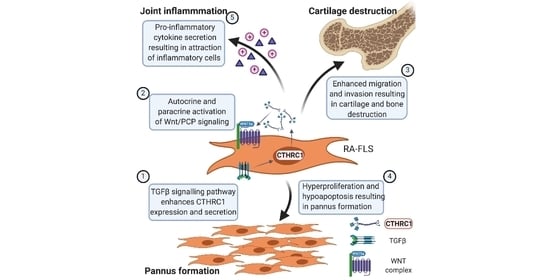
1. Introduction
2. CTHRC1 Domain Structure
3. Identification and Physiological Function of CTHRC1
4. Expression of CTHRC1
5. Signaling Roles of CTHRC1
5.1. Role of CTHRC1 in the TGF-β Pathway
5.2. CTHRC1 Is a Component of Canonical and Non-Canonical Wnt Signaling Pathways
6. CTHRC1 Is Associated with RA Development and Disease Severity
7. Invasive Synoviocytes Are Key Drivers of Joint Destruction in RA
8. RA-FLS Are One Source of CTHRC1
9. CTHRC1 Plays a Central Role in Bone Remodeling
10. Sex Disparity and CTHRC1
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACPA | Anti-citrullinated protein antibody |
| ADAMTs12 | A disintegrin and metallopeptidase with thrombospondin type 1 motif 12 |
| Ang2 | angiopoietin 2 |
| BMP2/4 | Bone morphogenetic protein 2/4 |
| CAIA | Collagen Antibody-Induced Arthritis |
| CCL2 | C-C Motif Chemokine Ligand 2 |
| CD34, CD90 | Cluster of differentiation 34/90 |
| CDH11 | cadherin 11 |
| COMP | cartilage oligomeric matrix protein |
| C1qtnf3 | Complement C1q tumor necrosis factor-related protein 3 |
| CRP | C-reactive protein |
| CSF-1 | Colony stimulating factor 1 |
| CTHRC1 | Collagen triple helix repeat-containing 1 protein |
| CXCL12 | C-X-C Motif Chemokine Ligand 12 |
| Daam2 | Dishevelled associated activator of morphogenesis |
| DPAGT 1 | Dolichyl-Phosphate N-Acetylglucosaminephosphotransferase 1 |
| Dvl | Dishevelled |
| FZD | Frizzled |
| GSK3β | Glycogen synthase kinase 3beta |
| IL-1/6/8/11/15 | Interleukin 1/6/8/11/15 |
| INF-γ | Interferon gamma |
| LRP | Lipoprotein receptor-related protein |
| OA | Osteoarthritis |
| OPG | Osteoprotegerin |
| PCP Pathway | Planar cell polarity pathway |
| Pgia8 | Proteoglycan induced arthritis 8 |
| RA | Rheumatoid arthritis |
| RA-FLS | Rheumatoid arthritis fibroblast like synoviocyte |
| RANKL | Receptor Activator of Nuclear Factor-Kappa B ligand |
| RF | Rheumatoid factor |
| ROR2 | Receptor tyrosine kinase-like orphan receptor 2 |
| RSPO2 | R-spondin 2 |
| Sdc2 | Syndecan 2 |
| SLE | Systemic lupus erythematosus |
| SMAD 2/3/4 | The abbreviation refers to the homologies to the Caenorhabditis elegans “small” worm phenotype and Drosophila MAD (“Mothers Against Decapentaplegic”) family of genes |
| SOST | Sclerostin |
| TPBG | trophoblast glycoprotein |
| TCF | T cell factor |
| TGF-β | Transforming growth factor beta |
| TNF-α | Tumor necrosis factor alpha |
| THY1 | Thy-1 Cell Surface Antigen |
| VANGL2 | VANGL planar cell polarity protein 2 |
| WAIF1 | Wnt-activated inhibitory factor 1 |
| Wnt | Wingless and Int-1 |
References
- Gibofsky, A. Epidemiology, pathophysiology, and diagnosis of rheumatoid arthritis: A Synopsis. Am. J. Manag. Care 2014, 20, S128–S135. [Google Scholar]
- Smolen, J.S.; Aletaha, D.; Barton, A.; Burmester, G.R.; Emery, P.; Firestein, G.S.; Kavanaugh, A.; McInnes, I.B.; Solomon, D.H.; Strand, V.; et al. Rheumatoid arthritis. Nat. Rev. Dis Primers 2018, 4, 18001. [Google Scholar] [CrossRef]
- Feldmann, M. Development of anti-TNF therapy for rheumatoid arthritis. Nat. Rev. Immunol. 2002, 2, 364–371. [Google Scholar] [CrossRef]
- Rein, P.; Mueller, R.B. Treatment with Biologicals in Rheumatoid Arthritis: An Overview. Rheumatol. Ther. 2017, 4, 247–261. [Google Scholar] [CrossRef]
- Brzustewicz, E.; Bryl, E. The role of cytokines in the pathogenesis of rheumatoid arthritis--Practical and potential application of cytokines as biomarkers and targets of personalized therapy. Cytokine 2015, 76, 527–536. [Google Scholar] [CrossRef]
- Van Delft, M.A.M.; Huizinga, T.W.J. An overview of autoantibodies in rheumatoid arthritis. J. Autoimmun. 2020, 110, 102392. [Google Scholar] [CrossRef]
- Myngbay, A.; Bexeitov, Y.; Adilbayeva, A.; Assylbekov, Z.; Yevstratenko, B.P.; Aitzhanova, R.M.; Matkarimov, B.; Adarichev, V.A.; Kunz, J. CTHRC1: A New Candidate Biomarker for Improved Rheumatoid Arthritis Diagnosis. Front. Immunol. 2019, 10, 1353. [Google Scholar] [CrossRef]
- Hu, T.; Liu, Y.; Tan, L.; Huang, J.; Yu, J.; Wu, Y.; Pei, Z.; Zhang, X.; Li, J.; Song, L.; et al. Value of serum collagen triple helix repeat containing-1(CTHRC1) and 14-3-3eta protein compared to anti-CCP antibodies and anti-MCV antibodies in the diagnosis of rheumatoid arthritis. Br. J. Biomed. Sci. 2020, 1–5. [Google Scholar] [CrossRef]
- Mizoguchi, F.; Slowikowski, K.; Wei, K.; Marshall, J.L.; Rao, D.A.; Chang, S.K.; Nguyen, H.N.; Noss, E.H.; Turner, J.D.; Earp, B.E.; et al. Functionally distinct disease-associated fibroblast subsets in rheumatoid arthritis. Nat. Commun. 2018, 9, 789. [Google Scholar] [CrossRef]
- Kimura, H.; Kwan, K.M.; Zhang, Z.; Deng, J.M.; Darnay, B.G.; Behringer, R.R.; Nakamura, T.; de Crombrugghe, B.; Akiyama, H. Cthrc1 is a positive regulator of osteoblastic bone formation. PLoS ONE 2008, 3, e3174. [Google Scholar] [CrossRef]
- Takeshita, S.; Fumoto, T.; Matsuoka, K.; Park, K.A.; Aburatani, H.; Kato, S.; Ito, M.; Ikeda, K. Osteoclast-secreted CTHRC1 in the coupling of bone resorption to formation. J. Clin. Investig. 2013, 123, 3914–3924. [Google Scholar] [CrossRef]
- Pyagay, P.; Heroult, M.; Wang, Q.; Lehnert, W.; Belden, J.; Liaw, L.; Friesel, R.E.; Lindner, V. Collagen triple helix repeat containing 1, a novel secreted protein in injured and diseased arteries, inhibits collagen expression and promotes cell migration. Circ. Res. 2005, 96, 261–268. [Google Scholar] [CrossRef]
- Leclere, L.; Nir, T.S.; Bazarsky, M.; Braitbard, M.; Schneidman-Duhovny, D.; Gat, U. Dynamic Evolution of the Cthrc1 Genes, a Newly Defined Collagen-Like Family. Genome Biol. Evol. 2020, 12, 3957–3970. [Google Scholar] [CrossRef]
- Kishore, U.; Gaboriaud, C.; Waters, P.; Shrive, A.K.; Greenhough, T.J.; Reid, K.B.; Sim, R.B.; Arlaud, G.J. C1q and tumor necrosis factor superfamily: Modularity and versatility. Trends Immunol. 2004, 25, 551–561. [Google Scholar] [CrossRef]
- Yamamoto, S.; Nishimura, O.; Misaki, K.; Nishita, M.; Minami, Y.; Yonemura, S.; Tarui, H.; Sasaki, H. Cthrc1 selectively activates the planar cell polarity pathway of Wnt signaling by stabilizing the Wnt-receptor complex. Dev. Cell 2008, 15, 23–36. [Google Scholar] [CrossRef]
- LeClair, R.; Lindner, V. The role of collagen triple helix repeat containing 1 in injured arteries, collagen expression, and transforming growth factor beta signaling. Trends Cardiovasc. Med. 2007, 17, 202–205. [Google Scholar] [CrossRef]
- Jin, J.; Togo, S.; Kadoya, K.; Tulafu, M.; Namba, Y.; Iwai, M.; Watanabe, J.; Nagahama, K.; Okabe, T.; Hidayat, M.; et al. Pirfenidone attenuates lung fibrotic fibroblast responses to transforming growth factor-beta1. Respir. Res. 2019, 20, 119. [Google Scholar] [CrossRef]
- Bauer, Y.; Tedrow, J.; de Bernard, S.; Birker-Robaczewska, M.; Gibson, K.F.; Guardela, B.J.; Hess, P.; Klenk, A.; Lindell, K.O.; Poirey, S.; et al. A novel genomic signature with translational significance for human idiopathic pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2015, 52, 217–231. [Google Scholar] [CrossRef]
- Li, J.; Wang, Y.; Ma, M.; Jiang, S.; Zhang, X.; Zhang, Y.; Yang, X.; Xu, C.; Tian, G.; Li, Q.; et al. Autocrine CTHRC1 activates hepatic stellate cells and promotes liver fibrosis by activating TGF-beta signaling. EBioMedicine 2019, 40, 43–55. [Google Scholar] [CrossRef]
- Li, Y.K.; Li, Y.M.; Li, Y.; Wei, Y.R.; Zhang, J.; Li, B.; You, Z.R.; Chen, Y.; Huang, B.Y.; Miao, Q.; et al. CTHRC1 expression in primary biliary cholangitis. J. Dig. Dis. 2019, 20, 371–376. [Google Scholar] [CrossRef]
- Bian, Z.; Miao, Q.; Zhong, W.; Zhang, H.; Wang, Q.; Peng, Y.; Chen, X.; Guo, C.; Shen, L.; Yang, F.; et al. Treatment of cholestatic fibrosis by altering gene expression of Cthrc1: Implications for autoimmune and non-autoimmune liver disease. J. Autoimmun. 2015, 63, 76–87. [Google Scholar] [CrossRef]
- Binks, A.P.; Beyer, M.; Miller, R.; LeClair, R.J. Cthrc1 lowers pulmonary collagen associated with bleomycin-induced fibrosis and protects lung function. Physiol. Rep. 2017, 5, 1–9. [Google Scholar] [CrossRef]
- Tsukui, T.; Sun, K.H.; Wetter, J.B.; Wilson-Kanamori, J.R.; Hazelwood, L.A.; Henderson, N.C.; Adams, T.S.; Schupp, J.C.; Poli, S.D.; Rosas, I.O.; et al. Collagen-producing lung cell atlas identifies multiple subsets with distinct localization and relevance to fibrosis. Nat. Commun. 2020, 11, 1920. [Google Scholar] [CrossRef]
- Ruiz-Villalba, A.; Romero, J.P.; Hernandez, S.C.; Vilas-Zornoza, A.; Fortelny, N.; Castro-Labrador, L.; San Martin-Uriz, P.; Lorenzo-Vivas, E.; Garcia-Olloqui, P.; Palacio, M.; et al. Single-Cell RNA Sequencing Analysis Reveals a Crucial Role for CTHRC1 (Collagen Triple Helix Repeat Containing 1) Cardiac Fibroblasts After Myocardial Infarction. Circulation 2020, 142, 1831–1847. [Google Scholar] [CrossRef]
- Bai, L.; Zhang, W.; Tan, L.; Yang, H.; Ge, M.; Zhu, C.; Zhang, R.; Cao, Y.; Chen, J.; Luo, Z.; et al. Hepatitis B virus hijacks CTHRC1 to evade host immunity and maintain replication. J. Mol. Cell Biol. 2015, 7, 543–556. [Google Scholar] [CrossRef]
- Zhang, R.; Cao, Y.; Bai, L.; Zhu, C.; Li, R.; He, H.; Liu, Y.; Wu, K.; Liu, F.; Wu, J. The collagen triple helix repeat containing 1 facilitates hepatitis B virus-associated hepatocellular carcinoma progression by regulating multiple cellular factors and signal cascades. Mol. Carcinog. 2015, 54, 1554–1566. [Google Scholar] [CrossRef]
- Wu, Q.; Yang, Q.; Sun, H. Role of collagen triple helix repeat containing-1 in tumor and inflammatory diseases. J. Cancer Res. Ther. 2017, 13, 621–624. [Google Scholar] [CrossRef]
- Jiang, N.; Cui, Y.; Liu, J.; Zhu, X.; Wu, H.; Yang, Z.; Ke, Z. Multidimensional Roles of Collagen Triple Helix Repeat Containing 1 (CTHRC1) in Malignant Cancers. J. Cancer 2016, 7, 2213–2220. [Google Scholar] [CrossRef]
- Durmus, T.; LeClair, R.J.; Park, K.S.; Terzic, A.; Yoon, J.K.; Lindner, V. Expression analysis of the novel gene collagen triple helix repeat containing-1 (Cthrc1). Gene Expr. Patterns 2006, 6, 935–940. [Google Scholar] [CrossRef]
- Leclair, R.J.; Wang, Q.; Benson, M.A.; Prudovsky, I.; Lindner, V. Intracellular localization of Cthrc1 characterizes differentiated smooth muscle. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1332–1338. [Google Scholar] [CrossRef]
- Duarte, C.W.; Stohn, J.P.; Wang, Q.; Emery, I.F.; Prueser, A.; Lindner, V. Elevated plasma levels of the pituitary hormone Cthrc1 in individuals with red hair but not in patients with solid tumors. PLoS ONE 2014, 9, e100449. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Budi, E.H. Specificity, versatility, and control of TGF-beta family signaling. Sci. Signal. 2019, 12, eaav5183. [Google Scholar] [CrossRef] [PubMed]
- Tzavlaki, K.; Moustakas, A. TGF-beta Signaling. Biomolecules 2020, 10, 487. [Google Scholar] [CrossRef] [PubMed]
- Vander Ark, A.; Cao, J.; Li, X. TGF-beta receptors: In and beyond TGF-beta signaling. Cell Signal. 2018, 52, 112–120. [Google Scholar] [CrossRef]
- Wang, P.; Wang, Y.C.; Chen, X.Y.; Shen, Z.Y.; Cao, H.; Zhang, Y.J.; Yu, J.; Zhu, J.D.; Lu, Y.Y.; Fang, J.Y. CTHRC1 is upregulated by promoter demethylation and transforming growth factor-beta1 and may be associated with metastasis in human gastric cancer. Cancer Sci. 2012, 103, 1327–1333. [Google Scholar] [CrossRef]
- LeClair, R.J.; Durmus, T.; Wang, Q.; Pyagay, P.; Terzic, A.; Lindner, V. Cthrc1 is a novel inhibitor of transforming growth factor-beta signaling and neointimal lesion formation. Circ. Res. 2007, 100, 826–833. [Google Scholar] [CrossRef]
- Kikuchi, K.; Kubo, M.; Sato, S.; Fujimoto, M.; Tamaki, K. Serum tissue inhibitor of metalloproteinases in patients with systemic sclerosis. J. Am. Acad. Dermatol. 1995, 33, 973–978. [Google Scholar] [CrossRef]
- Clevers, H.; Nusse, R. Wnt/beta-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef]
- Miao, C.G.; Yang, Y.Y.; He, X.; Li, X.F.; Huang, C.; Huang, Y.; Zhang, L.; Lv, X.W.; Jin, Y.; Li, J. Wnt signaling pathway in rheumatoid arthritis, with special emphasis on the different roles in synovial inflammation and bone remodeling. Cell Signal. 2013, 25, 2069–2078. [Google Scholar] [CrossRef]
- Liu, G.; Sengupta, P.K.; Jamal, B.; Yang, H.Y.; Bouchie, M.P.; Lindner, V.; Varelas, X.; Kukuruzinska, M.A. N-glycosylation induces the CTHRC1 protein and drives oral cancer cell migration. J. Biol. Chem. 2013, 288, 20217–20227. [Google Scholar] [CrossRef]
- Sengupta, P.K.; Bouchie, M.P.; Kukuruzinska, M.A. N-glycosylation gene DPAGT1 is a target of the Wnt/beta-catenin signaling pathway. J. Biol. Chem. 2010, 285, 31164–31173. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.Y.; Pan, Y.F.; Guo, X.H.; Wu, Y.Q.; Gu, J.R.; Cai, D.Z. Expression of beta-catenin in rheumatoid arthritis fibroblast-like synoviocytes. Scand. J. Rheumatol. 2011, 40, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Dong, B.; Vold, S.; Olvera-Jaramillo, C.; Chang, H. Functional redundancy of frizzled 3 and frizzled 6 in planar cell polarity control of mouse hair follicles. Development 2018, 145, dev168468. [Google Scholar] [CrossRef] [PubMed]
- Kagermeier-Schenk, B.; Wehner, D.; Ozhan-Kizil, G.; Yamamoto, H.; Li, J.; Kirchner, K.; Hoffmann, C.; Stern, P.; Kikuchi, A.; Schambony, A.; et al. Waif1/5T4 inhibits Wnt/beta-catenin signaling and activates noncanonical Wnt pathways by modifying LRP6 subcellular localization. Dev. Cell 2011, 21, 1129–1143. [Google Scholar] [CrossRef]
- Adarichev, V.A.; Nesterovitch, A.B.; Bardos, T.; Biesczat, D.; Chandrasekaran, R.; Vermes, C.; Mikecz, K.; Finnegan, A.; Glant, T.T. Sex effect on clinical and immunologic quantitative trait loci in a murine model of rheumatoid arthritis. Arthritis Rheum. 2003, 48, 1708–1720. [Google Scholar] [CrossRef]
- Kudryavtseva, E.; Forde, T.S.; Pucker, A.D.; Adarichev, V.A. Wnt signaling genes of murine chromosome 15 are involved in sex-affected pathways of inflammatory arthritis. Arthritis Rheum. 2012, 64, 1057–1068. [Google Scholar] [CrossRef]
- Adarichev, V.A.; Vegvari, A.; Szabo, Z.; Kis-Toth, K.; Mikecz, K.; Glant, T.T. Congenic strains displaying similar clinical phenotype of arthritis represent different immunologic models of inflammation. Genes Immun. 2008, 9, 591–601. [Google Scholar] [CrossRef]
- Glant, T.T.; Szanto, S.; Vegvari, A.; Szabo, Z.; Kis-Toth, K.; Mikecz, K.; Adarichev, V.A. Two loci on chromosome 15 control experimentally induced arthritis through the differential regulation of IL-6 and lymphocyte proliferation. J. Immunol. 2008, 181, 1307–1314. [Google Scholar] [CrossRef]
- Libioulle, C.; Louis, E.; Hansoul, S.; Sandor, C.; Farnir, F.; Franchimont, D.; Vermeire, S.; Dewit, O.; de Vos, M.; Dixon, A.; et al. Novel Crohn disease locus identified by genome-wide association maps to a gene desert on 5p13.1 and modulates expression of PTGER4. PLoS Genet. 2007, 3, e58. [Google Scholar] [CrossRef]
- Kurz, T.; Hoffjan, S.; Hayes, M.G.; Schneider, D.; Nicolae, R.; Heinzmann, A.; Jerkic, S.P.; Parry, R.; Cox, N.J.; Deichmann, K.A.; et al. Fine mapping and positional candidate studies on chromosome 5p13 identify multiple asthma susceptibility loci. J. Allergy Clin. Immunol. 2006, 118, 396–402. [Google Scholar] [CrossRef]
- Liu, C.J.; Kong, W.; Xu, K.; Luan, Y.; Ilalov, K.; Sehgal, B.; Yu, S.; Howell, R.D.; Di Cesare, P.E. ADAMTS-12 associates with and degrades cartilage oligomeric matrix protein. J. Biol. Chem. 2006, 281, 15800–15808. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wright, G.L.; Peterson, J.M. C1q/TNF-Related Protein 3 (CTRP3) Function and Regulation. Compr. Physiol. 2017, 7, 863–878. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Min, J.Y.; Baek, J.M.; Ahn, S.J.; Jun, H.Y.; Yoon, K.H.; Choi, M.K.; Lee, M.S.; Oh, J. CTRP3 acts as a negative regulator of osteoclastogenesis through AMPK-c-Fos-NFATc1 signaling in vitro and RANKL-induced calvarial bone destruction in vivo. Bone 2015, 79, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Murayama, M.A.; Kakuta, S.; Maruhashi, T.; Shimizu, K.; Seno, A.; Kubo, S.; Sato, N.; Saijo, S.; Hattori, M.; Iwakura, Y. CTRP3 plays an important role in the development of collagen-induced arthritis in mice. Biochem. Biophys. Res. Commun. 2014, 443, 42–48. [Google Scholar] [CrossRef]
- Wei, Z.; Li, M. Genome-wide linkage and association analysis of rheumatoid arthritis in a Canadian population. BMC Proc. 2007, 1 (Suppl. 1), S19. [Google Scholar] [CrossRef]
- Jawaheer, D.; Seldin, M.F.; Amos, C.I.; Chen, W.V.; Shigeta, R.; Etzel, C.; Damle, A.; Xiao, X.; Chen, D.; Lum, R.F.; et al. Screening the genome for rheumatoid arthritis susceptibility genes: A replication study and combined analysis of 512 multicase families. Arthritis Rheum. 2003, 48, 906–916. [Google Scholar] [CrossRef]
- Mukhopadhyay, N.; Halder, I.; Bhattacharjee, S.; Weeks, D.E. Two-dimensional linkage analyses of rheumatoid arthritis. BMC Proc. 2007, 1 (Suppl. 1), S68. [Google Scholar] [CrossRef]
- Plant, D.; Bowes, J.; Potter, C.; Hyrich, K.L.; Morgan, A.W.; Wilson, A.G.; Isaacs, J.D.; Wellcome Trust Case Control Consortium; British Society for Rheumatology Biologics Register; Barton, A. Genome-wide association study of genetic predictors of anti-tumor necrosis factor treatment efficacy in rheumatoid arthritis identifies associations with polymorphisms at seven loci. Arthritis Rheum. 2011, 63, 645–653. [Google Scholar] [CrossRef]
- Wu, Q.; Yang, Q.; Sun, H. Collagen triple helix repeat containing-1: A novel biomarker associated with disease activity in Systemic lupus erythematosus. Lupus 2018, 27, 2076–2085. [Google Scholar] [CrossRef]
- Zvaifler, N.J.; Firestein, G.S. Pannus and pannocytes. Alternative models of joint destruction in rheumatoid arthritis. Arthritis Rheum. 1994, 37, 783–789. [Google Scholar] [CrossRef]
- Gravallese, E.M. Bone destruction in arthritis. Ann. Rheum. Dis. 2002, 61 (Suppl. 2), ii84–ii86. [Google Scholar] [CrossRef] [PubMed]
- Schett, G. Synovitis--an inflammation of joints destroying the bone. Swiss Med. Wkly. 2012, 142, w13692. [Google Scholar] [CrossRef] [PubMed]
- Goldring, S.R.; Gravallese, E.M. Pathogenesis of bone lesions in rheumatoid arthritis. Curr. Rheumatol. Rep. 2002, 4, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Bartok, B.; Firestein, G.S. Fibroblast-like synoviocytes: Key effector cells in rheumatoid arthritis. Immunol. Rev. 2010, 233, 233–255. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, S.; Mima, T.; Sasai, M.; Nishioka, K.; Shimizu, M.; Murata, N.; Yoshikawa, H.; Nakanishi, K.; Suemura, M.; McCloskey, R.V.; et al. Tumour necrosis factor alpha (TNF-alpha) interferes with Fas-mediated apoptotic cell death on rheumatoid arthritis (RA) synovial cells: A possible mechanism of rheumatoid synovial hyperplasia and a clinical benefit of anti-TNF-alpha therapy for RA. Cytokine 2000, 12, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Shekhani, M.T.; Forde, T.S.; Adilbayeva, A.; Ramez, M.; Myngbay, A.; Bexeitov, Y.; Lindner, V.; Adarichev, V.A. Collagen triple helix repeat containing 1 is a new promigratory marker of arthritic pannus. Arthritis Res. Ther. 2016, 18, 171. [Google Scholar] [CrossRef] [PubMed]
- Lindqvist, E.; Jonsson, K.; Saxne, T.; Eberhardt, K. Course of radiographic damage over 10 years in a cohort with early rheumatoid arthritis. Ann. Rheum. Dis. 2003, 62, 611–616. [Google Scholar] [CrossRef] [PubMed]
- Machold, K.P.; Stamm, T.A.; Nell, V.P.; Pflugbeil, S.; Aletaha, D.; Steiner, G.; Uffmann, M.; Smolen, J.S. Very recent onset rheumatoid arthritis: Clinical and serological patient characteristics associated with radiographic progression over the first years of disease. Rheumatology 2007, 46, 342–349. [Google Scholar] [CrossRef]
- Abuwarwar, M.H.; Knoblich, K.; Fletcher, A.L. A pathogenic hierarchy for synovial fibroblasts in rheumatoid arthritis. Ann. Transl. Med. 2018, 6, S75. [Google Scholar] [CrossRef]
- Cheon, H.; Boyle, D.L.; Firestein, G.S. Wnt1 inducible signaling pathway protein-3 regulation and microsatellite structure in arthritis. J. Rheumatol. 2004, 31, 2106–2114. [Google Scholar]
- Nakamura, Y.; Nawata, M.; Wakitani, S. Expression profiles and functional analyses of Wnt-related genes in human joint disorders. Am. J. Pathol. 2005, 167, 97–105. [Google Scholar] [CrossRef]
- Sen, M.; Lauterbach, K.; El-Gabalawy, H.; Firestein, G.S.; Corr, M.; Carson, D.A. Expression and function of wingless and frizzled homologs in rheumatoid arthritis. Proc. Natl. Acad. Sci. USA 2000, 97, 2791–2796. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, D.W.; Ha, Y.; Ihm, M.H.; Kim, H.; Song, K.; Lee, I. Wnt5a induces endothelial inflammation via beta-catenin-independent signaling. J. Immunol. 2010, 185, 1274–1282. [Google Scholar] [CrossRef] [PubMed]
- Rauner, M.; Stein, N.; Winzer, M.; Goettsch, C.; Zwerina, J.; Schett, G.; Distler, J.H.; Albers, J.; Schulze, J.; Schinke, T.; et al. WNT5A is induced by inflammatory mediators in bone marrow stromal cells and regulates cytokine and chemokine production. J. Bone Miner. Res. 2012, 27, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Sen, M.; Chamorro, M.; Reifert, J.; Corr, M.; Carson, D.A. Blockade of Wnt-5A/frizzled 5 signaling inhibits rheumatoid synoviocyte activation. Arthritis Rheum. 2001, 44, 772–781. [Google Scholar] [CrossRef]
- Larson, C.; Oronsky, B.; Carter, C.A.; Oronsky, A.; Knox, S.J.; Sher, D.; Reid, T.R. TGF-beta: A master immune regulator. Expert Opin. Ther. Targets 2020, 24, 427–438. [Google Scholar] [CrossRef]
- Cheon, H.; Yu, S.J.; Yoo, D.H.; Chae, I.J.; Song, G.G.; Sohn, J. Increased expression of pro-inflammatory cytokines and metalloproteinase-1 by TGF-beta1 in synovial fibroblasts from rheumatoid arthritis and normal individuals. Clin. Exp. Immunol. 2002, 127, 547–552. [Google Scholar] [CrossRef]
- Schett, G.; Gravallese, E. Bone erosion in rheumatoid arthritis: Mechanisms, diagnosis and treatment. Nat. Rev. Rheumatol. 2012, 8, 656–664. [Google Scholar] [CrossRef]
- Kong, Y.Y.; Yoshida, H.; Sarosi, I.; Tan, H.L.; Timms, E.; Capparelli, C.; Morony, S.; Oliveira-dos-Santos, A.J.; Van, G.; Itie, A.; et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature 1999, 397, 315–323. [Google Scholar] [CrossRef]
- Geusens, P. The role of RANK ligand/osteoprotegerin in rheumatoid arthritis. Ther. Adv. Musculoskelet. Dis. 2012, 4, 225–233. [Google Scholar] [CrossRef]
- Jann, J.; Gascon, S.; Roux, S.; Faucheux, N. Influence of the TGF-beta Superfamily on Osteoclasts/Osteoblasts Balance in Physiological and Pathological Bone Conditions. Int. J. Mol. Sci. 2020, 21, 7597. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Gu, W.; Sun, B.; Zhang, Y.; Ji, Y.; Xu, X.; Wen, Y. CTHRC1 promotes osteogenic differentiation of periodontal ligament stem cells by regulating TAZ. J. Mol. Histol. 2017, 48, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, K.; Kohara, Y.; Naoe, Y.; Watanabe, A.; Ito, M.; Ikeda, K.; Takeshita, S. WAIF1 Is a Cell-Surface CTHRC1 Binding Protein Coupling Bone Resorption and Formation. J. Bone Miner. Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.R.; Stohn, J.P.; Wang, Q.; Nagano, K.; Baron, R.; Bouxsein, M.L.; Rosen, C.J.; Adarichev, V.A.; Lindner, V. Inhibition of osteoclast differentiation and collagen antibody-induced arthritis by CTHRC1. Bone 2017, 97, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Ohira, T. Mechanisms and therapeutic targets for bone damage in rheumatoid arthritis, in particular the RANK-RANKL system. Curr. Opin. Pharmacol. 2018, 40, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Calle, J.; Sato, A.Y.; Bellido, T. Role and mechanism of action of sclerostin in bone. Bone 2017, 96, 29–37. [Google Scholar] [CrossRef]
- Goldring, S.R.; Purdue, P.E.; Crotti, T.N.; Shen, Z.; Flannery, M.R.; Binder, N.B.; Ross, F.P.; McHugh, K.P. Bone remodelling in inflammatory arthritis. Ann. Rheum. Dis. 2013, 72 (Suppl. 2), ii52–ii55. [Google Scholar] [CrossRef]
- Alpizar-Rodriguez, D.; Pluchino, N.; Canny, G.; Gabay, C.; Finckh, A. The role of female hormonal factors in the development of rheumatoid arthritis. Rheumatology 2017, 56, 1254–1263. [Google Scholar] [CrossRef]
- Taneja, V. Sex Hormones Determine Immune Response. Front. Immunol. 2018, 9, 1931. [Google Scholar] [CrossRef]
- Islander, U.; Jochems, C.; Lagerquist, M.K.; Forsblad-d’Elia, H.; Carlsten, H. Estrogens in rheumatoid arthritis; the immune system and bone. Mol. Cell. Endocrinol. 2011, 335, 14–29. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type | Potential Effect on Cells | Reference |
|---|---|---|
| Rheumatoid arthritis fibroblast-like synoviocytes (RA-FLS) | Proliferation, migration, initiation of cartilage destruction | [66] |
| Osteoclasts | Inhibition of osteoblast differentiation and osteoblast driven bone formation; | [10,11,82,83] |
| activates WAIF1/PKCδ/ERK pathway necessary for RANKL expression leading to reduced bone resorption and formation | [83] | |
| Osteoblasts | Inhibition of monocyte-osteoclast differentiation and osteoclast-driven bone resorption in trabecular bone, inhibition of NFκB-dependent signaling; CTHRC1 may additionally suppress RANKL expression | [84] |
| Osteocytes | Inhibition of monocyte-osteoclast differentiation and osteoclast driven bone resorption in trabecular bone, inhibition of NFκB-dependent signaling; CTHRC1 may additionally suppress RANKL expression | [84] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Myngbay, A.; Manarbek, L.; Ludbrook, S.; Kunz, J. The Role of Collagen Triple Helix Repeat-Containing 1 Protein (CTHRC1) in Rheumatoid Arthritis. Int. J. Mol. Sci. 2021, 22, 2426. https://doi.org/10.3390/ijms22052426
Myngbay A, Manarbek L, Ludbrook S, Kunz J. The Role of Collagen Triple Helix Repeat-Containing 1 Protein (CTHRC1) in Rheumatoid Arthritis. International Journal of Molecular Sciences. 2021; 22(5):2426. https://doi.org/10.3390/ijms22052426
Chicago/Turabian StyleMyngbay, Askhat, Limara Manarbek, Steve Ludbrook, and Jeannette Kunz. 2021. "The Role of Collagen Triple Helix Repeat-Containing 1 Protein (CTHRC1) in Rheumatoid Arthritis" International Journal of Molecular Sciences 22, no. 5: 2426. https://doi.org/10.3390/ijms22052426
APA StyleMyngbay, A., Manarbek, L., Ludbrook, S., & Kunz, J. (2021). The Role of Collagen Triple Helix Repeat-Containing 1 Protein (CTHRC1) in Rheumatoid Arthritis. International Journal of Molecular Sciences, 22(5), 2426. https://doi.org/10.3390/ijms22052426