
The ALS-Associated FUS (P525L) Variant Does Not Directly Interfere with Microtubule-Dependent Kinesin-1 Motility

 , and
, and 
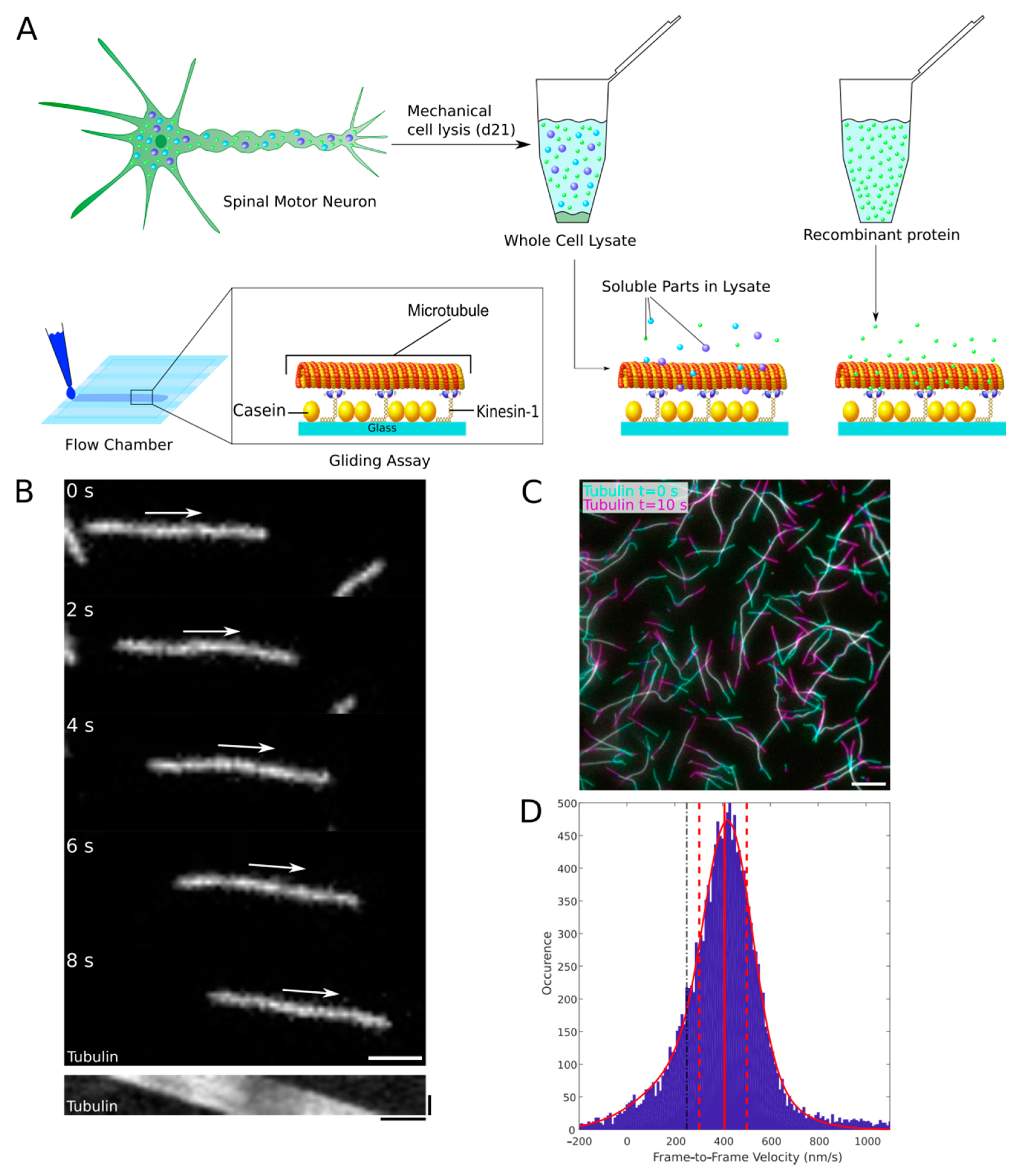{kind=link}
{kind=link}
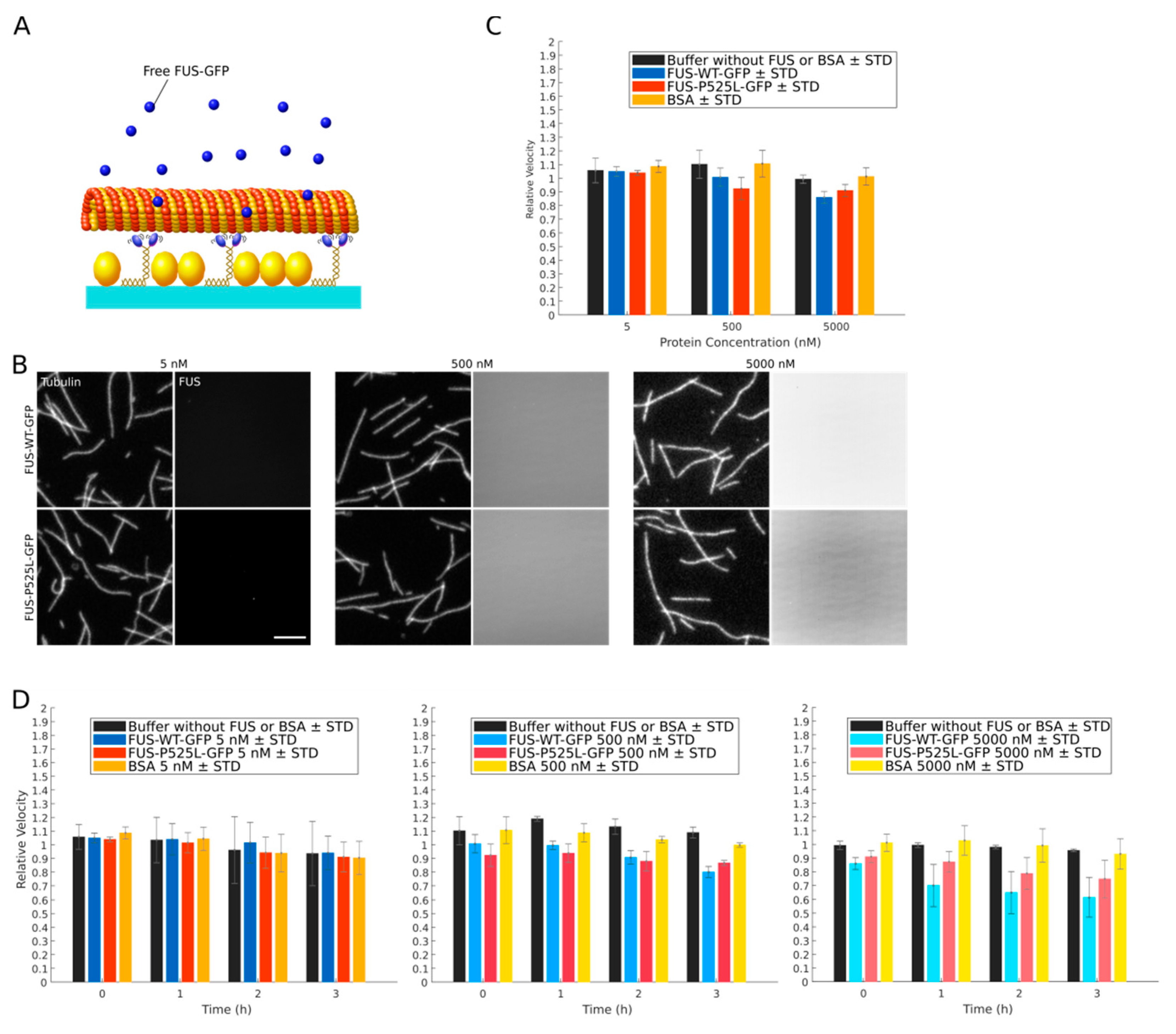{kind=link}
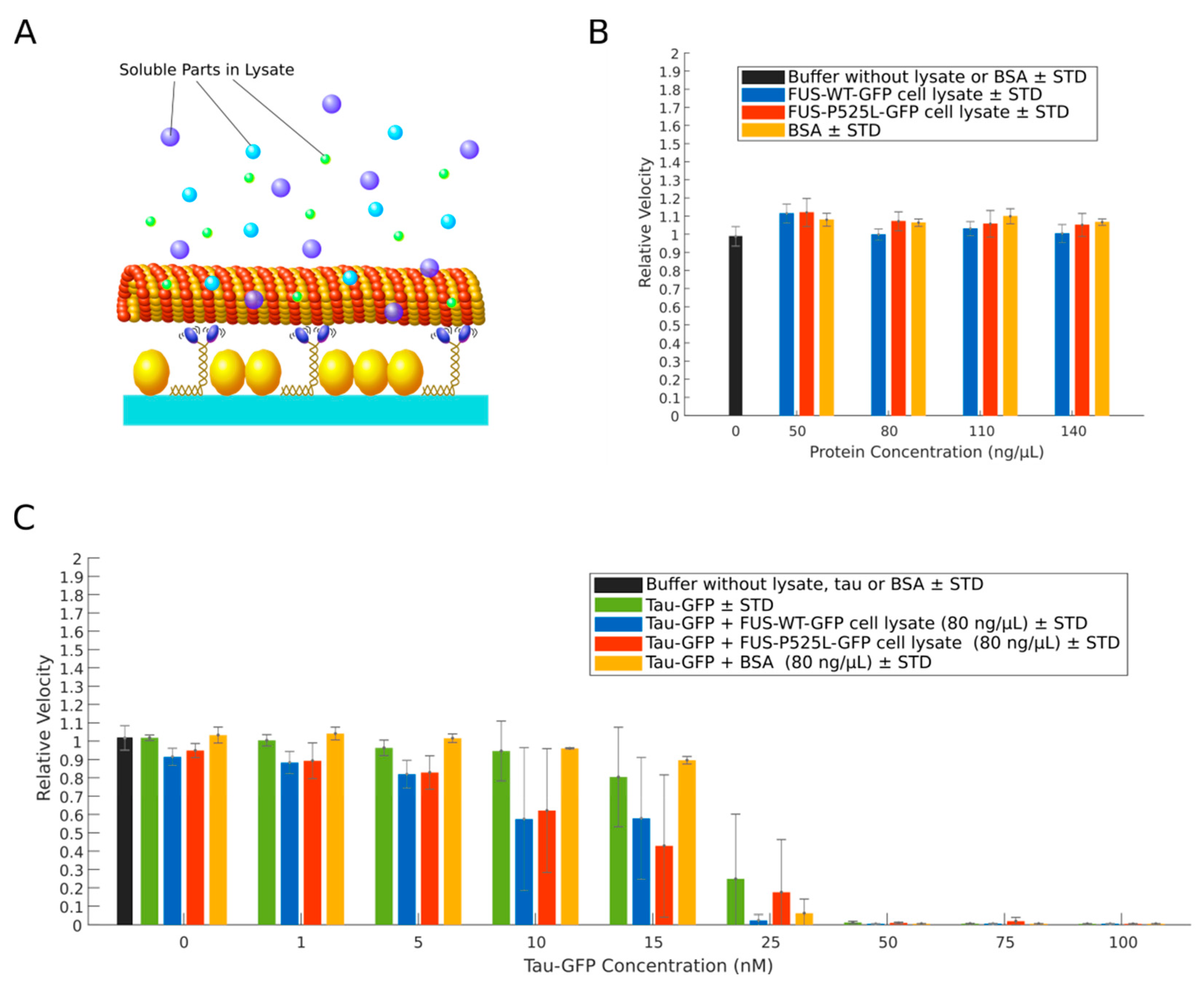{kind=link}
Abstract
1. Introduction
2. Results
2.1. The Optimized Microtubule Gliding Motility Assay Detects Recombinant Road-Block Proteins in the Low Nanomolar Range
2.2. Recombinant FUS Does Neither Directly Bind to Microtubules nor Directly Inhibit Kinesin-1 Motility
2.3. Cell Lysates from Neurons Expressing FUS-P525L-GFP Do Not Inhibit Kinesin-1 Motility
3. Discussion
4. Materials and Methods
4.1. NPC Line Generation, Cell Culture and Motor Neuron Differentiation
4.2. Preparation of Cell Lysates and Protein Concentration Determination
4.3. Expression and Purification of Tubulin and Kinesin-1
4.4. Protein-Enriched Microtubule Gliding Motility Assay
4.5. Microtubule Pull-Down Assay
4.6. Western Blot
4.7. Data Analysis and Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Grafstein, B.; Forman, D.S. Intracellular transport in neurons. Physiol. Rev. 1980, 60, 1167–1283. [Google Scholar] [CrossRef]
- A Fletcher, D.; A Theriot, J. An introduction to cell motility for the physical scientist. Phys. Biol. 2004, 1, T1–T10. [Google Scholar] [CrossRef]
- Holzbaur, E.L.; Scherer, S.S. Microtubules, Axonal Transport, and Neuropathy. N. Engl. J. Med. 2011, 365, 2330–2332. [Google Scholar] [CrossRef] [PubMed]
- Gindhart, J.; Weber, K. Lysosome and Endosome Organization and Transport in Neurons. In Encyclopedia of Neuroscience; Elsevier: Amsterdam, The Netherlands, 2009; pp. 581–587. ISBN 978-0-08-045046-9. [Google Scholar]
- Barra, H.S.; Arce, C.A.; Argaraña, C.E. Posttranslational tyrosination/detyrosination of tubulin. Mol. Neurobiol. 1988, 2, 133–153. [Google Scholar] [CrossRef]
- Ludueña, R.F. Multiple Forms of Tubulin: Different Gene Products and Covalent Modifications. Int. Rev. Cytol. 1997, 178, 207–275. [Google Scholar] [CrossRef]
- Wloga, R.; Gaertig, J. Post-translational modifications of microtubules. J. Cell Sci. 2010, 123, 3447–3455. [Google Scholar] [CrossRef] [PubMed]
- Tarhan, M.C.; Orazov, Y.; Yokokawa, R.; Karsten, S.L.; Fujita, H. Biosensing MAPs as “roadblocks”: Kinesin-based functional analysis of tau protein isoforms and mutants using suspended microtubules (sMTs). Lab Chip 2013, 13, 3217. [Google Scholar] [CrossRef]
- Semenova, I.; Ikeda, K.; Resaul, K.; Kraikivski, P.; Aguiar, M.; Gygi, S.; Zaliapin, I.; Cowan, A.; Rodionov, V. Regulation of microtubule-based transport by MAP4. Mol. Biol. Cell 2014, 25, 3119–3132. [Google Scholar] [CrossRef]
- Monroy, B.Y.; Sawyer, D.L.; Ackermann, B.E.; Borden, M.M.; Tan, T.C.; Ori-McKenney, K.M. Competition between microtubule-associated proteins directs motor transport. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Chaudhary, A.R.; Lu, H.; Krementsova, E.B.; Bookwalter, C.S.; Trybus, K.M.; Hendricks, A.G. MAP7 regulates organelle transport by recruiting kinesin-1 to microtubules. J. Biol. Chem. 2019, 294, 10160–10171. [Google Scholar] [CrossRef]
- Rodríguez-Martín, T.; Cuchillo-Ibáñez, I.; Noble, W.; Nyenya, F.; Anderton, B.H.; Hanger, D.P. Tau phosphorylation affects its axonal transport and degradation. Neurobiol. Aging 2013, 34, 2146–2157. [Google Scholar] [CrossRef]
- Kanaan, N.M.; Morfini, G.A.; Lapointe, N.E.; Pigino, G.F.; Patterson, K.R.; Song, Y.; Andreadis, A.; Fu, Y.; Brady, S.T.; Binder, L.I. Pathogenic Forms of Tau Inhibit Kinesin-Dependent Axonal Transport through a Mechanism Involving Activation of Axonal Phosphotransferases. J. Neurosci. 2011, 31, 9858–9868. [Google Scholar] [CrossRef]
- Kim, J.; Choi, I.-Y.; Michaelis, M.L.; Lee, P. Quantitative in vivo measurement of early axonal transport deficits in a triple transgenic mouse model of Alzheimer’s disease using manganese-enhanced MRI. NeuroImage 2011, 56, 1286–1292. [Google Scholar] [CrossRef]
- Bilsland, L.G.; Sahai, E.; Kelly, G.; Golding, M.; Greensmith, L.; Schiavo, G. Deficits in axonal transport precede ALS symptoms in vivo. Proc. Natl. Acad. Sci. USA 2010, 107, 20523–20528. [Google Scholar] [CrossRef]
- De Vos, K.J.; Hafezparast, M. Neurobiology of axonal transport defects in motor neuron diseases: Opportunities for translational research? Neurobiol. Dis. 2017, 105, 283–299. [Google Scholar] [CrossRef]
- Van Es, M.A.; Hardiman, O.; Chio, A.; Al-Chalabi, A.; Pasterkamp, R.J.; Veldink, J.H.; Van den Berg, L.H. Amyotrophic lateral sclerosis. Lancet 2017, 390, 2084–2098. [Google Scholar] [CrossRef]
- Chen, S.; Sayana, P.; Zhang, X.; Le, W. Genetics of amyotrophic lateral sclerosis: An update. Mol. Neurodegener. 2013, 8, 28. [Google Scholar] [CrossRef]
- E Renton, A.; Chiò, A.; Traynor, B.J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 2014, 17, 17–23. [Google Scholar] [CrossRef]
- Naumann, M.; Peikert, K.; Günther, R.; Van Der Kooi, A.J.; Aronica, E.; Hübers, A.; Danel, V.; Corcia, P.; Pan-Montojo, F.; Cirak, S.; et al. Phenotypes and malignancy risk of different FUS mutations in genetic amyotrophic lateral sclerosis. Ann. Clin. Transl. Neurol. 2019, 6, 2384–2394. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, I.R.; Neumann, M. Fused in Sarcoma Neuropathology in Neurodegenerative Disease. Cold Spring Harb. Perspect. Med. 2017, 7, a024299. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Lee, H.O.; Jawerth, L.; Maharana, S.; Jahnel, M.; Hein, M.Y.; Stoynov, S.; Mahamid, J.; Saha, S.; Franzmann, T.M.; et al. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell 2015, 162, 1066–1077. [Google Scholar] [CrossRef]
- Chen, C.; Ding, X.; Akram, N.; Xue, S.; Luo, S.-Z. Fused in Sarcoma: Properties, Self-Assembly and Correlation with Neurodegenerative Diseases. Molecules 2019, 24, 1622. [Google Scholar] [CrossRef]
- Shang, Y.; Huang, E.J. Mechanisms of FUS mutations in familial amyotrophic lateral sclerosis. Brain Res. 2016, 1647, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Vance, C.; Scotter, E.L.; Nishimura, A.L.; Troakes, C.; Mitchell, J.C.; Kathe, C.; Urwin, H.; Manser, C.; Miller, C.C.; Hortobágyi, T.; et al. ALS mutant FUS disrupts nuclear localization and sequesters wild-type FUS within cytoplasmic stress granules. Hum. Mol. Genet. 2013, 22, 2676–2688. [Google Scholar] [CrossRef] [PubMed]
- Japtok, J.; Lojewski, X.; Naumann, M.; Klingenstein, M.; Reinhardt, P.; Sterneckert, J.; Putz, S.; Demestre, M.; Boeckers, T.M.; Ludolph, A.C.; et al. Stepwise acquirement of hallmark neuropathology in FUS-ALS iPSC models depends on mutation type and neuronal aging. Neurobiol. Dis. 2015, 82, 420–429. [Google Scholar] [CrossRef]
- Naumann, M.; Pal, A.; Goswami, A.; Lojewski, X.; Japtok, J.; Vehlow, A.; Naujock, M.; Günther, R.; Jin, M.; Stanslowsky, N.; et al. Impaired DNA damage response signaling by FUS-NLS mutations leads to neurodegeneration and FUS aggregate formation. Nat. Commun. 2018, 9, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, K.R.; Godena, V.K.; Hewitt, V.L.; Whitworth, A.J. Axonal transport defects are a common phenotype in Drosophila models of ALS. Hum. Mol. Genet. 2016, 25, 2378–2392. [Google Scholar] [CrossRef]
- Guo, W.; Naujock, M.; Fumagalli, L.; Vandoorne, T.; Baatsen, P.; Boon, R.; Ordovás, L.; Patel, A.; Welters, M.; Vanwelden, T.; et al. HDAC6 inhibition reverses axonal transport defects in motor neurons derived from FUS-ALS patients. Nat. Commun. 2017, 8, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Daigle, J.G.; Lanson, J.N.A.; Smith, R.B.; Casci, I.; Maltare, A.; Monaghan, J.; Nichols, C.D.; Kryndushkin, D.; Shewmaker, F.; Pandey, U.B. RNA-binding ability of FUS regulates neurodegeneration, cytoplasmic mislocalization and incorporation into stress granules associated with FUS carrying ALS-linked mutations. Hum. Mol. Genet. 2012, 22, 1193–1205. [Google Scholar] [CrossRef]
- Sama, R.R.K.; Ward, C.L.; Kaushansky, L.J.; Lemay, N.; Ishigaki, S.; Urano, F.; Bosco, D.A. FUS/TLS assembles into stress granules and is a prosurvival factor during hyperosmolar stress. J. Cell. Physiol. 2013, 228, 2222–2231. [Google Scholar] [CrossRef]
- Shelkovnikova, T.A.; Robinson, H.K.; Southcombe, J.A.; Ninkina, N.; Buchman, V.L. Multistep process of FUS aggregation in the cell cytoplasm involves RNA-dependent and RNA-independent mechanisms. Hum. Mol. Genet. 2014, 23, 5211–5226. [Google Scholar] [CrossRef] [PubMed]
- Marrone, L.; Drexler, H.C.A.; Wang, J.; Tripathi, P.; Distler, T.; Heisterkamp, P.; Anderson, E.N.; Kour, S.; Moraiti, A.; Maharana, S.; et al. FUS pathology in ALS is linked to alterations in multiple ALS-associated proteins and rescued by drugs stimulating autophagy. Acta Neuropathol. 2019, 138, 67–84. [Google Scholar] [CrossRef]
- Aulas, A.; Velde, C.V. Alterations in stress granule dynamics driven by TDP-43 and FUS: A link to pathological inclusions in ALS? Front. Cell. Neurosci. 2015, 9, 423. [Google Scholar] [CrossRef]
- Kanai, Y.; Dohmae, N.; Hirokawa, N. Kinesin Transports RNA: Isolation and Characterization of an RNA-Transporting Granule. Neuron 2004, 43, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, K.; Clatterbuck-Soper, S.F.; Jackrel, M.E.; Shorter, J.; Mili, S. FUS inclusions disrupt RNA localization by sequestering kinesin-1 and inhibiting microtubule detyrosination. J. Cell Biol. 2017, 216, 1015–1034. [Google Scholar] [CrossRef]
- Nitzsche, B.; Bormuth, V.; Bräuer, C.; Howard, J.; Ionov, L.; Kerssemakers, J.; Korten, T.; LeDuc, C.; Ruhnow, F.; Diez, S. Studying Kinesin Motors by Optical 3D-Nanometry in Gliding Motility Assays. In Methods in Cell Biology; Wilson, L., Correia, J.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2010; Volume 95, pp. 247–271. ISBN 978-0-12-374815-7. [Google Scholar]
- Mackall, J.; Meredith, M.; Lane, M.D. A mild procedure for the rapid release of cytoplasmic enzymes from cultured animal cells. Anal. Biochem. 1979, 95, 270–274. [Google Scholar] [CrossRef]
- Harlow, E.; Lane, D. Immunoprecipitation: Lysing Yeast Cells Using Glass Beads. Cold Spring Harb. Protoc. 2006, 2006. [Google Scholar] [CrossRef]
- Kruger, N.J.; Walker, J.M. The Protein Protocols Handbook; Humana Press: Totowa, NJ, USA, 2009; ISBN 978-1-59745-198-7. [Google Scholar]
- A Cohn, S.; Ingold, A.L.; Scholey, J.M. Quantitative analysis of sea urchin egg kinesin-driven microtubule motility. J. Biol. Chem. 1989, 264, 4290–4297. [Google Scholar] [CrossRef]
- Inoue, D.; Mahmot, B.; Kabir, A.M.R.; Farhana, T.I.; Tokuraku, K.; Sada, K.; Konagaya, A.; Kakugo, A. Depletion force induced collective motion of microtubules driven by kinesin. Nanoscale 2015, 7, 18054–18061. [Google Scholar] [CrossRef]
- Saito, A.; Farhana, T.I.; Kabir, A.M.R.; Inoue, D.; Konagaya, A.; Sada, K.; Kakugo, A. Understanding the emergence of collective motion of microtubules driven by kinesins: Role of concentration of microtubules and depletion force. RSC Adv. 2017, 7, 13191–13197. [Google Scholar] [CrossRef]
- Böhm, K.J.; Stracke, R.; Baum, M.; Zieren, M.; Unger, E. Effect of temperature on kinesin-driven microtubule gliding and kinesin ATPase activity. FEBS Lett. 2000, 466, 59–62. [Google Scholar] [CrossRef]
- Ruhnow, F.; Zwicker, D.; Diez, S. Tracking Single Particles and Elongated Filaments with Nanometer Precision. Biophys. J. 2011, 100, 2820–2828. [Google Scholar] [CrossRef] [PubMed]
- Korten, T.; Tavkin, E.; Scharrel, L.; Kushwaha, V.S.; Diez, S. An automated in vitro motility assay for high-throughput studies of molecular motors. Lab Chip 2018, 18, 3196–3206. [Google Scholar] [CrossRef] [PubMed]
- Hoeprich, G.J.; Thompson, A.R.; McVicker, D.P.; Hancock, W.O.; Berger, C.L. Kinesin’s Neck-Linker Determines its Ability to Navigate Obstacles on the Microtubule Surface. Biophys. J. 2014, 106, 1691–1700. [Google Scholar] [CrossRef] [PubMed]
- Fujioka, Y.; Ishigaki, S.; Masuda, A.; Iguchi, Y.; Udagawa, T.; Watanabe, H.; Katsuno, M.; Ohno, K.; Sobue, G. FUS-regulated region- and cell-type-specific transcriptome is associated with cell selectivity in ALS/FTLD. Sci. Rep. 2013, 3, 2388. [Google Scholar] [CrossRef]
- Akiyama, T.; Suzuki, N.; Ishikawa, M.; Fujimori, K.; Sone, T.; Kawada, J.; Funayama, R.; Fujishima, F.; Mitsuzawa, S.; Ikeda, K.; et al. Aberrant axon branching via Fos-B dysregulation in FUS-ALS motor neurons. EBioMedicine 2019, 45, 362–378. [Google Scholar] [CrossRef]
- Pal, A.; Glaß, H.; Naumann, M.; Kreiter, N.; Japtok, J.; Sczech, R.; Hermann, A. High content organelle trafficking enables disease state profiling as powerful tool for disease modelling. Sci. Data 2018, 5, 180241. [Google Scholar] [CrossRef]
- Korten, S.; Albet-Torres, N.; Paderi, F.; Siethoff, L.T.; Diez, S.; Korten, T.; Kronnie, G.T.; Månsson, A. Sample solution constraints on motor-driven diagnostic nanodevices. Lab Chip 2013, 13, 866–876. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.A. Use of vanadate as protein-phosphotyrosine phosphatase inhibitor. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1991; Volume 201, pp. 477–482. [Google Scholar] [CrossRef]
- Wang, W.; Himes, R.; Dentler, W. The binding of a ciliary microtubule plus-end binding protein complex to microtubules is regulated by ciliary protein kinase and phosphatase activities. J. Biol. Chem. 1994, 269, 21460–21466. [Google Scholar] [CrossRef]
- Yun, M.; Zhang, X.; Park, C.; Park, H.-W.; Endow, S.A. A structural pathway for activation of the kinesin motor ATPase. EMBO J. 2001, 20, 2611–2618. [Google Scholar] [CrossRef]
- Okada, Y.; Hirokawa, N. Mechanism of the single-headed processivity: Diffusional anchoring between the K-loop of kinesin and the C terminus of tubulin. Proc. Natl. Acad. Sci. USA 2000, 97, 640–645. [Google Scholar] [CrossRef] [PubMed]
- Farhadi, L.; Rosario, C.F.D.; DeBold, E.P.; Baskaran, A.; Ross, J.L. Active Self-Organization of Actin-Microtubule Composite Self-Propelled Rods. Front. Phys. 2018, 6, 75. [Google Scholar] [CrossRef]
- Hong, W.; Takshak, A.; Osunbayo, O.; Kunwar, A.; Vershinin, M. The Effect of Temperature on Microtubule-Based Transport by Cytoplasmic Dynein and Kinesin-1 Motors. Biophys. J. 2016, 111, 1287–1294. [Google Scholar] [CrossRef]
- Peck, A.; Sargin, M.E.; Lapointe, N.E.; Rose, K.; Manjunath, B.S.; Feinstein, S.C.; Wilson, L. Tau isoform-specific modulation of kinesin-driven microtubule gliding rates and trajectories as determined with tau-stabilized microtubules. Cytoskeleton 2010, 68, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Parimalam, S.S.; Tarhan, M.C.; Karsten, S.L.; Fujita, H.; Shintaku, H.; Kotera, H.; Yokokawa, R. On-chip microtubule gliding assay for parallel measurement of tau protein species. Lab Chip 2016, 16, 1691–1697. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Wang, P.; Chen, X.; Cheng, H.; Liu, J.; Fushimi, K.; Zhu, L.; Wu, J.Y. FUS interacts with ATP synthase beta subunit and induces mitochondrial unfolded protein response in cellular and animal models. Proc. Natl. Acad. Sci. USA 2018, 115, E9678–E9686. [Google Scholar] [CrossRef]
- MacKenzie, I.R.; Neumann, M. FET proteins in frontotemporal dementia and amyotrophic lateral sclerosis. Brain Res. 2012, 1462, 40–43. [Google Scholar] [CrossRef]
- Martinez-Macias, M.I.; Moore, D.A.; Green, R.L.; Gomez-Herreros, F.; Naumann, M.; Hermann, A.; Van Damme, P.; Hafezparast, M.; Caldecott, K.W. FUS (fused in sarcoma) is a component of the cellular response to topoisomerase I–induced DNA breakage and transcriptional stress. Life Sci. Alliance 2019, 2, e201800222. [Google Scholar] [CrossRef]
- Coady, T.H.; Manley, J.L. ALS mutations in TLS/FUS disrupt target gene expression. Genes Dev. 2015, 29, 1696–1706. [Google Scholar] [CrossRef]
- Kim, S.H.; Shanware, N.P.; Bowler, M.J.; Tibbetts, R.S. Amyotrophic Lateral Sclerosis-associated Proteins TDP-43 and FUS/TLS Function in a Common Biochemical Complex to Co-regulate HDAC6 mRNA. J. Biol. Chem. 2010, 285, 34097–34105. [Google Scholar] [CrossRef] [PubMed]
- Tas, R.P.; Chazeau, A.; Cloin, B.M.; Lambers, M.L.; Hoogenraad, C.C.; Kapitein, L.C. Differentiation between Oppositely Oriented Microtubules Controls Polarized Neuronal Transport. Neuron 2017, 96, 1264–1271.e5. [Google Scholar] [CrossRef]
- Murakami, T.; Qamar, S.; Lin, J.Q.; Schierle, G.S.K.; Rees, E.; Miyashita, A.; Costa, A.R.; Dodd, R.B.; Chan, F.T.; Michel, C.H.; et al. ALS/FTD Mutation-Induced Phase Transition of FUS Liquid Droplets and Reversible Hydrogels into Irreversible Hydrogels Impairs RNP Granule Function. Neuron 2015, 88, 678–690. [Google Scholar] [CrossRef]
- Sau, D.; Rusmini, P.; Crippa, V.; Onesto, E.; Bolzoni, E.; Ratti, A.; Poletti, A. Dysregulation of axonal transport and motorneuron diseases. Biol. Cell 2011, 103, 87–107. [Google Scholar] [CrossRef]
- Lattante, S.; Doronzio, P.N.; Marangi, G.; Conte, A.; Bisogni, G.; Bernardo, D.; Russo, T.; Lamberti, D.; Patrizi, S.; Apollo, F.P.; et al. Coexistence of variants in TBK1 and in other ALS-related genes elucidates an oligogenic model of pathogenesis in sporadic ALS. Neurobiol. Aging 2019, 84, 239.e9–239.e14. [Google Scholar] [CrossRef] [PubMed]
- Borghero, G.; Pugliatti, M.; Marrosu, F.; Marrosu, M.G.; Murru, M.R.; Floris, G.; Cannas, A.; Occhineri, P.; Cau, T.B.; Loi, D.; et al. TBK1 is associated with ALS and ALS-FTD in Sardinian patients. Neurobiol. Aging 2016, 43, 180.e1–180.e5. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, P.; Glatza, M.; Hemmer, K.; Tsytsyura, Y.; Thiel, C.S.; Höing, S.; Moritz, S.; Parga, J.A.; Wagner, L.; Bruder, J.M.; et al. Derivation and Expansion Using Only Small Molecules of Human Neural Progenitors for Neurodegenerative Disease Modeling. PLoS ONE 2013, 8, e59252. [Google Scholar] [CrossRef]
- Castoldi, M.; Popov, A.V. Purification of brain tubulin through two cycles of polymerization–depolymerization in a high-molarity buffer. Protein Expr. Purif. 2003, 32, 83–88. [Google Scholar] [CrossRef]
- Korten, T.; Chaudhuri, S.; Tavkin, E.; Braun, M.; Diez, S. Kinesin-1 Expressed in Insect Cells Improves Microtubule in vitro Gliding Performance, Long-Term Stability and Guiding Efficiency in Nanostructures. IEEE Trans. NanoBioscience 2016, 15, 62–69. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seifert, A.; Drechsler, H.; Japtok, J.; Korten, T.; Diez, S.; Hermann, A. The ALS-Associated FUS (P525L) Variant Does Not Directly Interfere with Microtubule-Dependent Kinesin-1 Motility. Int. J. Mol. Sci. 2021, 22, 2422. https://doi.org/10.3390/ijms22052422
Seifert A, Drechsler H, Japtok J, Korten T, Diez S, Hermann A. The ALS-Associated FUS (P525L) Variant Does Not Directly Interfere with Microtubule-Dependent Kinesin-1 Motility. International Journal of Molecular Sciences. 2021; 22(5):2422. https://doi.org/10.3390/ijms22052422
Chicago/Turabian StyleSeifert, Anne, Hauke Drechsler, Julia Japtok, Till Korten, Stefan Diez, and Andreas Hermann. 2021. "The ALS-Associated FUS (P525L) Variant Does Not Directly Interfere with Microtubule-Dependent Kinesin-1 Motility" International Journal of Molecular Sciences 22, no. 5: 2422. https://doi.org/10.3390/ijms22052422
APA StyleSeifert, A., Drechsler, H., Japtok, J., Korten, T., Diez, S., & Hermann, A. (2021). The ALS-Associated FUS (P525L) Variant Does Not Directly Interfere with Microtubule-Dependent Kinesin-1 Motility. International Journal of Molecular Sciences, 22(5), 2422. https://doi.org/10.3390/ijms22052422