Novel lncRNAs Co-Expression Networks Identifies LINC00504 with Oncogenic Role in Luminal A Breast Cancer Cells

 ,
,  and
and 
Abstract
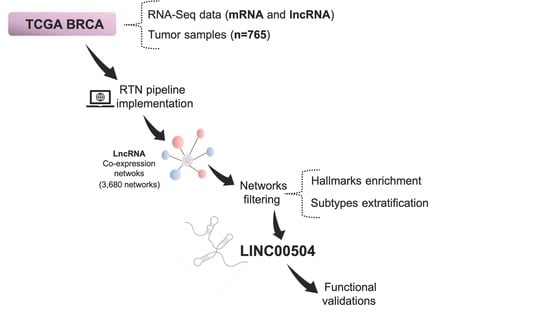
1. Introduction
2. Results
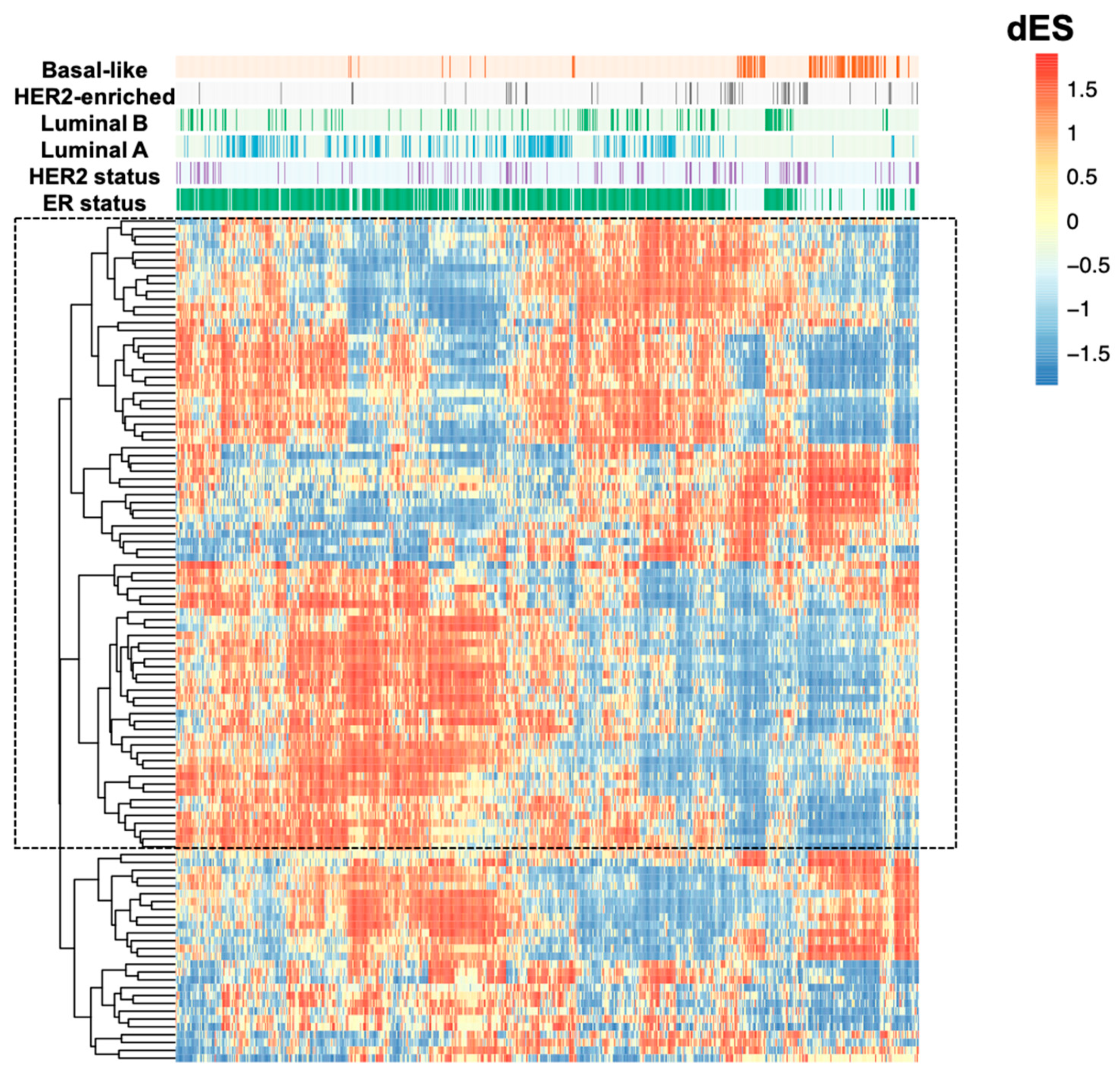
2.1. LncRNAs Co-Expression Networks Activity Are Different among Breast Cancer Molecular Subtypes
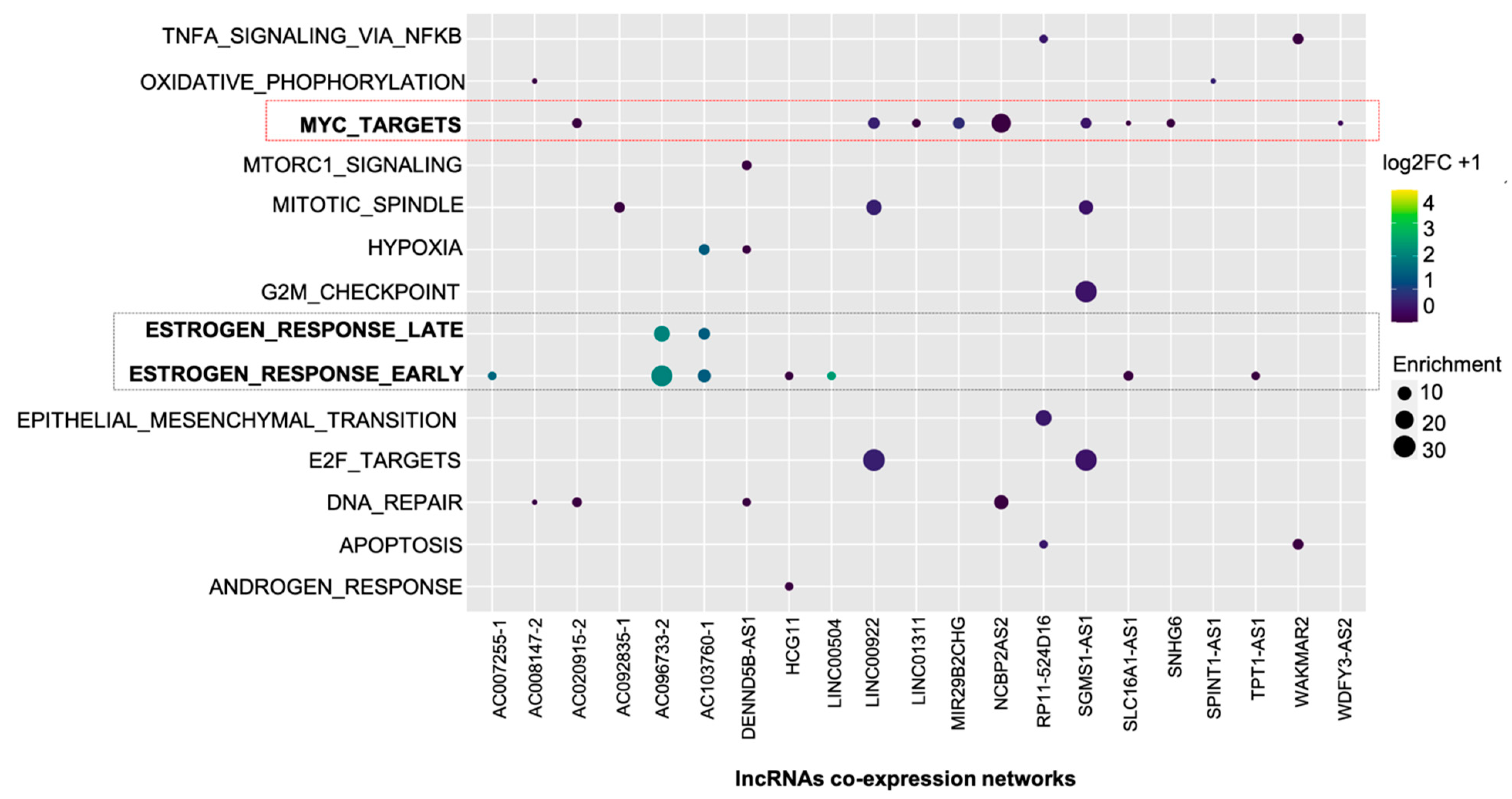
2.2. LncRNAs Co-Expression Networks Are Enriched for Cancer-Related Function
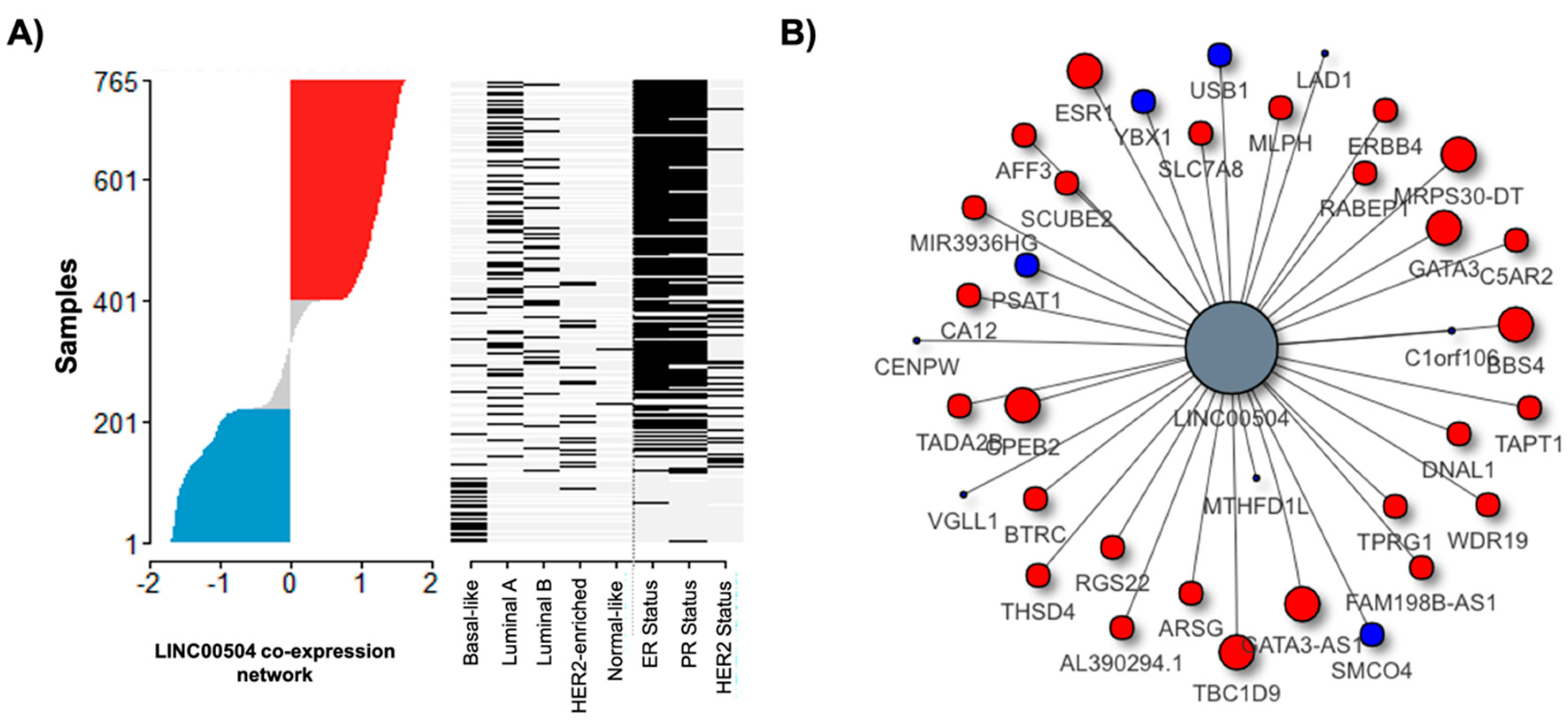
2.3. LINC00504 Co-Expression Network in Breast Cancer Luminal-A and Basal-Like Subtypes
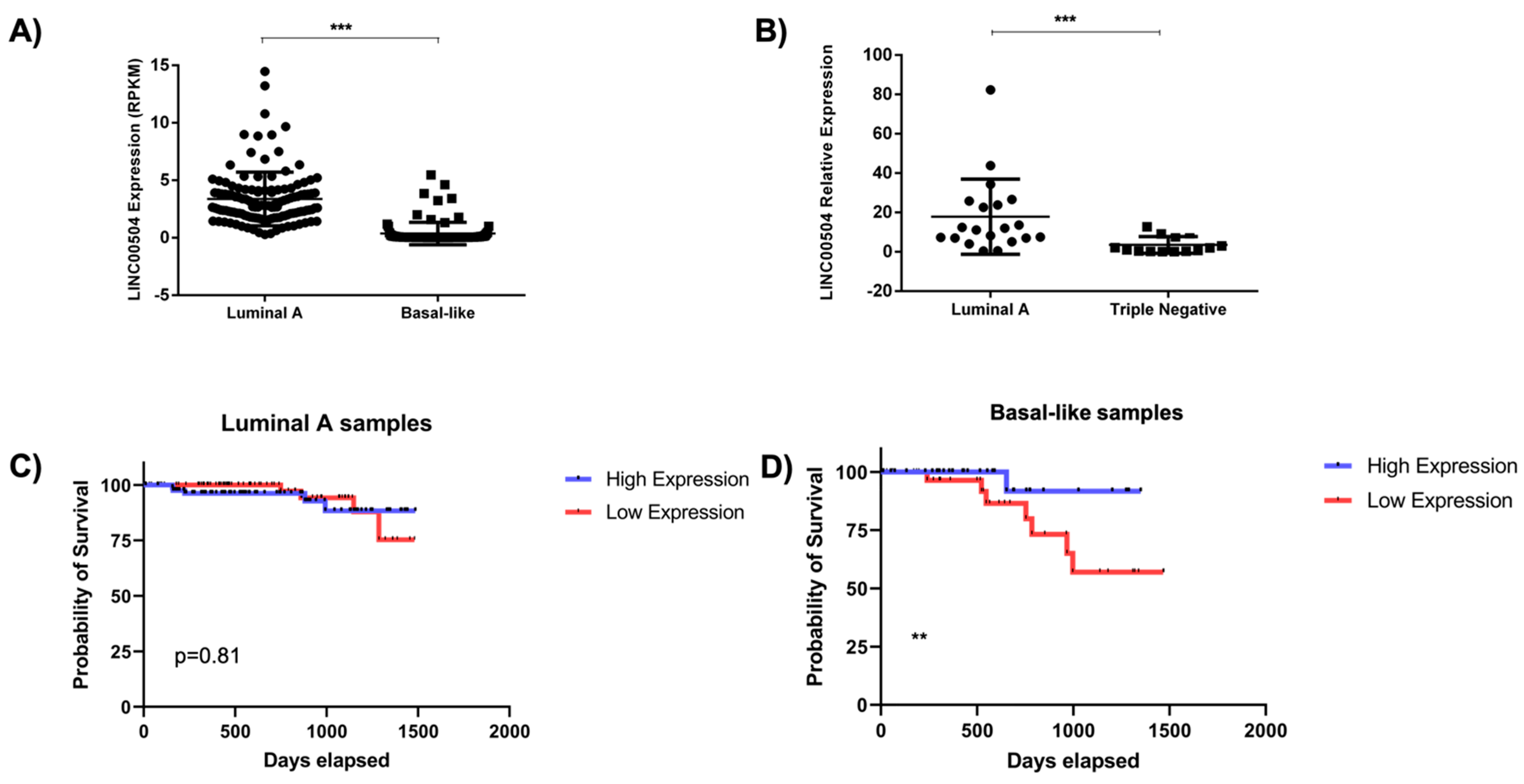
2.4. LINC00504 Is Up-Regulated in Luminal A and Is Related to Better Prognosis in Basal-Like Subtype
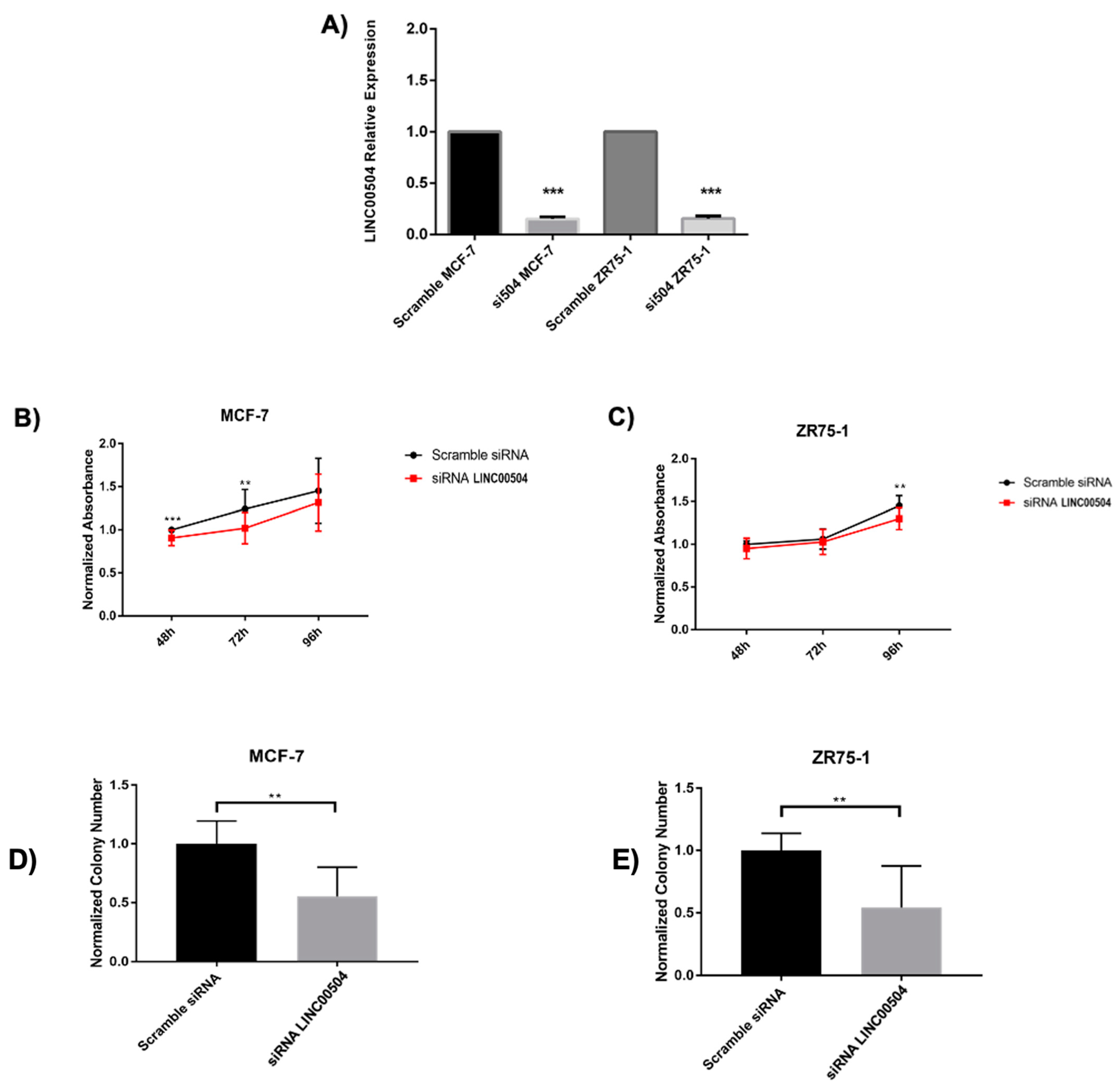
2.5. LINC00504 Is Related to Colony Formation and Cell Viability in Luminal A Cell Lines
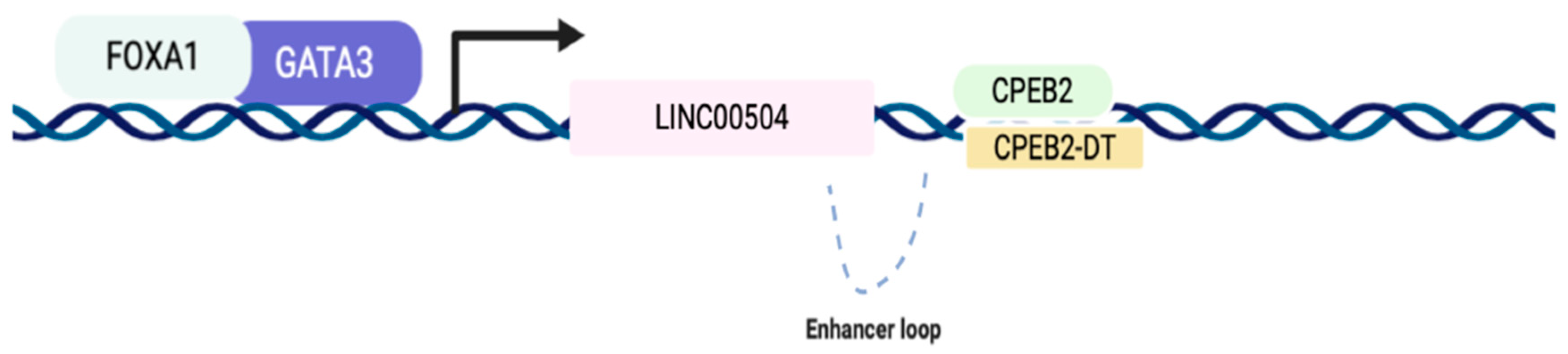
2.6. LINC00504 Co-Expression Network Has Transcriptional Factors Related to Its Regulation
3. Discussion
4. Materials and Methods
4.1. TCGA Data Extraction
4.2. RTN Pipeline Implementation
4.3. Regulon Filtering in Breast Cancer Molecular Subtypes
4.4. Cancer-Related Processes Enrichment Analysis
4.5. Breast Cancer Tissue Sample Characterization
4.6. RNA Isolation and Expression Quantification
4.7. Cell Culture and Growth Conditions
4.8. siRNA Treatment
4.9. Cell Viability
4.10. Colony-Forming Assay
4.11. Apoptosis
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hombach, S.; Kretz, M. Non-coding RNAs: Classification, Biology and Functioning. Adv. Exp. Med. Biol. 2016, 937, 3–17. [Google Scholar] [CrossRef]
- Clark, M.B.; Mercer, T.R.; Bussotti, G.; Leonardi, T.; Haynes, K.R.; Crawford, J.; Brunck, M.E.; Le Cao, K.A.; Thomas, G.P.; Chen, W.Y.; et al. Quantitative gene profiling of long noncoding RNAs with targeted RNA sequencing. Nat. Methods 2015, 12, 339. [Google Scholar] [CrossRef]
- Chan, J.J.; Tay, Y. Noncoding RNA:RNA Regulatory Networks in Cancer. Int. J. Mol. Sci. 2018, 19, 1310. [Google Scholar] [CrossRef]
- Bartonicek, N.; Maag, J.L.; Dinger, M.E. Long noncoding RNAs in cancer: Mechanisms of action and technological advancements. Mol. Cancer 2016, 15, 43. [Google Scholar] [CrossRef] [PubMed]
- Chi, Y.; Wang, D.; Wang, J.; Yu, W.; Yang, J. Long Non-Coding RNA in the Pathogenesis of Cancers. Cells 2019, 8, 1015. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Taheri, M. Maternally expressed gene 3 (MEG3): A tumor suppressor long non coding RNA. Biomed. Pharmacother. 2019, 118, 109129. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Shi, L.; Luo, Z. Long Non-coding RNAs in Cancer: Implications for Diagnosis, Prognosis, and Therapy. Front Med. 2020, 7, 612393. [Google Scholar] [CrossRef]
- Jalali, S.; Kapoor, S.; Sivadas, A.; Bhartiya, D.; Scaria, V. Computational approaches towards understanding human long noncoding RNA biology. Bioinformatics 2015, 31, 2241–2251. [Google Scholar] [CrossRef]
- Liao, Q.; Liu, C.; Yuan, X.; Kang, S.; Miao, R.; Xiao, H.; Zhao, G.; Luo, H.; Bu, D.; Zhao, H.; et al. Large-scale prediction of long noncoding RNA functions in a coding-non-coding gene coexpression network. Nucleic Acids Res. 2011, 39, 3864–3878. [Google Scholar] [CrossRef] [PubMed]
- Necsulea, A.; Soumillon, M.; Warnefors, M.; Liechti, A.; Daish, T.; Zeller, U.; Baker, J.C.; Grützner, F.; Kaessmann, H. The evolution of lncRNA repertoires and expression patterns in tetrapods. Nature 2014, 505, 635–640. [Google Scholar] [CrossRef]
- Fletcher, M.N.; Castro, M.A.; Wang, X.; De Santiago, I.; O’Reilly, M.; Chin, S.F.; Rueda, O.M.; Caldas, C.; Ponder, B.A.; Markowetz, F.; et al. Master Regulators of FGFR2 Signalling and Breast Cancer Risk. Nat. Commun. 2013, 4, 2464. [Google Scholar] [CrossRef]
- Castro, M.A.; de Santiago, I.; Campbell, T.M.; Vaughn, C.; Hickey, T.E.; Ross, E.; Tilley, W.D.; Markowetz, F.; Ponder, B.A.; Meyer, K.B. Regulators of Genetic Risk of Breast Cancer Identified by Integrative Network Analysis. Nat. Genet. 2016, 48, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; Van De Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Yersal, O.; Barutca, S. Biological subtypes of breast cancer: Prognostic and therapeutic implications. World J. Clin. Oncol. 2014, 5, 412–424. [Google Scholar] [CrossRef]
- Beniey, M.; Haque, T.; Hassan, S. Translating the role of PARP inhibitors in triple-negative breast cancer. Oncoscience 2019, 6, 287–288. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries published correction. CA Cancer J. Clin. 2018, 68, 394–424, Erratum in 2020, 70, 313. [Google Scholar] [CrossRef] [PubMed]
- Polyak, K. Breast cancer: Origins and evolution. J. Clin. Investig. 2007, 117, 3155–3163. [Google Scholar] [CrossRef]
- Davis, C.A.; Hitz, B.C.; Sloan, C.A.; Chan, E.T.; Davidson, J.M.; Gabdank, I.; Hilton, J.A.; Jain, K.; Baymuradov, U.K.; Narayanan, A.K.; et al. The Encyclopedia of DNA elements (ENCODE): Data portal update. Nucleic Acids Res. 2018, 46, D794–D801. [Google Scholar] [CrossRef]
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef]
- Giangarrà, V.; Igea, A.; Castellazzi, C.L.; Bava, F.A.; Mendez, R. Global Analysis of CPEBs Reveals Sequential and Non-Redundant Functions in Mitotic Cell Cycle. PLoS ONE 2015, 10, e0138794. [Google Scholar] [CrossRef] [PubMed]
- Grudzien-Nogalska, E.; Reed, B.C.; Rhoads, R.E. CPEB1 promotes differentiation and suppresses EMT in mammary epithelial cells. J. Cell Sci. 2014, 127 Pt 10, 2326–2338. [Google Scholar] [CrossRef]
- Pascual, R.; Martín, J.; Salvador, F.; Reina, O.; Chanes, V.; Millanes-Romero, A.; Suñer, C.; Fernández-Miranda, G.; Bartomeu, A.; Huang, Y.S.; et al. The RNA binding protein CPEB2 regulates hormone sensing in mammary gland development and luminal breast cancer. Sci. Adv. 2020, 6, eaax3868. [Google Scholar] [CrossRef]
- Spicuglia, S.; Vanhille, L. Chromatin signatures of active enhancers. Nucleus 2012, 3, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Al-Thoubaity, F.K. Molecular classification of breast cancer: A retrospective cohort study. Ann. Med. Surg. 2019, 49, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Ung, M.; Schaafsma, E.; Mattox, D.; Wang, G.L.; Cheng, C. Pan-cancer systematic identification of lncRNAs associated with cancer prognosis. PeerJ 2020, 8, e8797. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Zhou, L.; Chi, Y.; Wu, H.; Shi, L. LncRNA profile study reveals a seven-lncRNA signature predicts the prognosis of patients with colorectal cancer. Biomark Res. 2020, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Gao, T.; Sun, Y.; Zheng, X.; Wang, J.; Ma, J.; Hu, X.; Li, J.; Hu, M. LncRNA expression profile reveals the potential role of lncRNAs in gastric carcinogenesis. Cancer Biomark. 2015, 15, 249–258. [Google Scholar] [CrossRef]
- Jiang, C.; Li, Y.; Zhao, Z.; Lu, J.; Chen, H.; Ding, N.; Wang, G.; Xu, J.; Li, X. Identifying and functionally characterizing tissue-specific and ubiquitously expressed human lncRNAs. Oncotarget 2016, 7, 7120–7133. [Google Scholar] [CrossRef]
- Yan, C.; Zhang, Z.; Bao, S.; Hou, P.; Zhou, M.; Xu, C.; Sun, J. Computational Methods and Applications for Identifying Disease-Associated lncRNAs as Potential Biomarkers and Therapeutic Targets. Mol. Ther. Nucleic Acids 2020, 21, 156–171. [Google Scholar] [CrossRef]
- Campbell, T.M.; Castro, M.A.; de Oliveira, K.G.; Ponder, B.A.; Meyer, K.B. ERα Binding by Transcription Factors Nfib and Ybx1 Enables Fgfr2 Signaling to Modulate Estrogen Responsiveness in Breast Cancer. Cancer Res. 2018, 78, 410–421. [Google Scholar] [CrossRef]
- Campbell, T.M.; Castro, M.A.; de Santiago, I.; Fletcher, M.N.; Halim, S.; Prathalingam, R.; Ponder, B.A.; Meyer, K.B. FGFR2 Risk Snps Confer Breast Cancer Risk by Augmenting Oestrogen Responsiveness. Carcinogenesis 2016, 37, 741–750. [Google Scholar] [CrossRef]
- Feng, J.; Ma, J.; Liu, S.; Wang, J.; Chen, Y. A noncoding RNA LINC00504 interacts with c-Myc to regulate tumor metabolism in colon cancer. J. Cell Biochem. 2019, 120, 14725–14734. [Google Scholar] [CrossRef]
- Zhang, Z. Silencing LINC00504 inhibits cell proliferation, invasion as well as migration and promotes cell apoptosis in lung cancer cells via upregulating miR-876-3p. Cytotechnology 2020. [Google Scholar] [CrossRef]
- Ma, H.P.; Wang, L.X.; Li, W.; Guo, H.H.; Wu, Y.; Li, X.Y. Upregulation of LINC00504 is associated with aggressive progression and poor prognosis in non-small cell lung cancer. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 699–703. [Google Scholar] [CrossRef]
- Liu, Y.; He, X.; Chen, Y.; Cao, D. Long noncoding RNA LINC00504 regulates the Warburg effect in ovarian cancer through inhibition of miR-1244. Mol. Cell Biochem. 2020, 464, 39–50. [Google Scholar] [CrossRef]
- Theodorou, V.; Stark, R.; Menon, S.; Carroll, J.S. GATA3 acts upstream of FOXA1 in mediating ESR1 binding by shaping enhancer accessibility. Genome Res. 2013, 23, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Hang, J.; Lei, T.; He, Y.; Kuang, Z.; Wang, L.; Chen, L.; He, J.; Zhang, W.; Liao, Y.; et al. Identification of key genes relevant to the prognosis of ER-positive and ER-negative breast cancer based on a prognostic prediction system. Mol. Biol. Rep. 2019, 46, 2111–2119. [Google Scholar] [CrossRef] [PubMed]
- Shehata, M.; Teschendorff, A.; Sharp, G.; Novcic, N.; Russell, I.A.; Avril, S.; Prater, M.; Eirew, P.; Caldas, C.; Watson, C.J.; et al. Phenotypic and functional characterisation of the luminal cell hierarchy of the mammary gland. Breast Cancer Res. 2012, 14, R134. [Google Scholar] [CrossRef] [PubMed]
- Carro, M.S.; Lim, W.K.; Alvarez, M.J.; Bollo, R.J.; Zhao, X.; Snyder, E.Y.; Sulman, E.P.; Anne, S.L.; Doetsch, F.; Colman, H.; et al. The Transcriptional Network for Mesenchymal Transformation of Brain Tumours. Nature 2010, 463, 318–325. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2015, 102, 15545–15550. [Google Scholar] [CrossRef]
- Groeneveld, C.; Chagas, V.; Jones, S.; Robertson, G.; Ponder, B.; Meyer, K.; Castro, M. RTNsurvival: An R/Bioconductor package for regulatory network survival analysis. Bioinformatics 2019, btz229. [Google Scholar] [CrossRef] [PubMed]
- Goldhirsch, A.; Winer, E.P.; Coates, A.S.; Gelber, R.D.; Piccart-Gebhart, M.; Thürlimann, B.; Senn, H.J.; Panel Members. Personalizing the treatment of women with early breast cancer: Highlights of the St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2013. Ann. Oncol. 2013, 24, 2206–2223. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Network Element | Mutual Information | logFC |
|---|---|---|
| CPEB2 | 0.40 | 1.85 |
| CPEB2-DT | −0.11 | −0.05 |
| ESR1 | 0.23 | 6.96 |
| FOXA1 | 0.20 | 6.21 |
| GATA3 | 0.24 | 4.33 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mathias, C.; Groeneveld, C.S.; Trefflich, S.; Zambalde, E.P.; Lima, R.S.; Urban, C.A.; Prado, K.B.; Ribeiro, E.M.S.F.; Castro, M.A.A.; Gradia, D.F.; et al. Novel lncRNAs Co-Expression Networks Identifies LINC00504 with Oncogenic Role in Luminal A Breast Cancer Cells. Int. J. Mol. Sci. 2021, 22, 2420. https://doi.org/10.3390/ijms22052420
Mathias C, Groeneveld CS, Trefflich S, Zambalde EP, Lima RS, Urban CA, Prado KB, Ribeiro EMSF, Castro MAA, Gradia DF, et al. Novel lncRNAs Co-Expression Networks Identifies LINC00504 with Oncogenic Role in Luminal A Breast Cancer Cells. International Journal of Molecular Sciences. 2021; 22(5):2420. https://doi.org/10.3390/ijms22052420
Chicago/Turabian StyleMathias, Carolina, Clarice S. Groeneveld, Sheyla Trefflich, Erika P. Zambalde, Rubens S. Lima, Cícero A. Urban, Karin B. Prado, Enilze M. S. F. Ribeiro, Mauro A. A. Castro, Daniela F. Gradia, and et al. 2021. "Novel lncRNAs Co-Expression Networks Identifies LINC00504 with Oncogenic Role in Luminal A Breast Cancer Cells" International Journal of Molecular Sciences 22, no. 5: 2420. https://doi.org/10.3390/ijms22052420
APA StyleMathias, C., Groeneveld, C. S., Trefflich, S., Zambalde, E. P., Lima, R. S., Urban, C. A., Prado, K. B., Ribeiro, E. M. S. F., Castro, M. A. A., Gradia, D. F., & de Oliveira, J. C. (2021). Novel lncRNAs Co-Expression Networks Identifies LINC00504 with Oncogenic Role in Luminal A Breast Cancer Cells. International Journal of Molecular Sciences, 22(5), 2420. https://doi.org/10.3390/ijms22052420