
Integrin Regulation in Immunological and Cancerous Cells and Exosomes



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
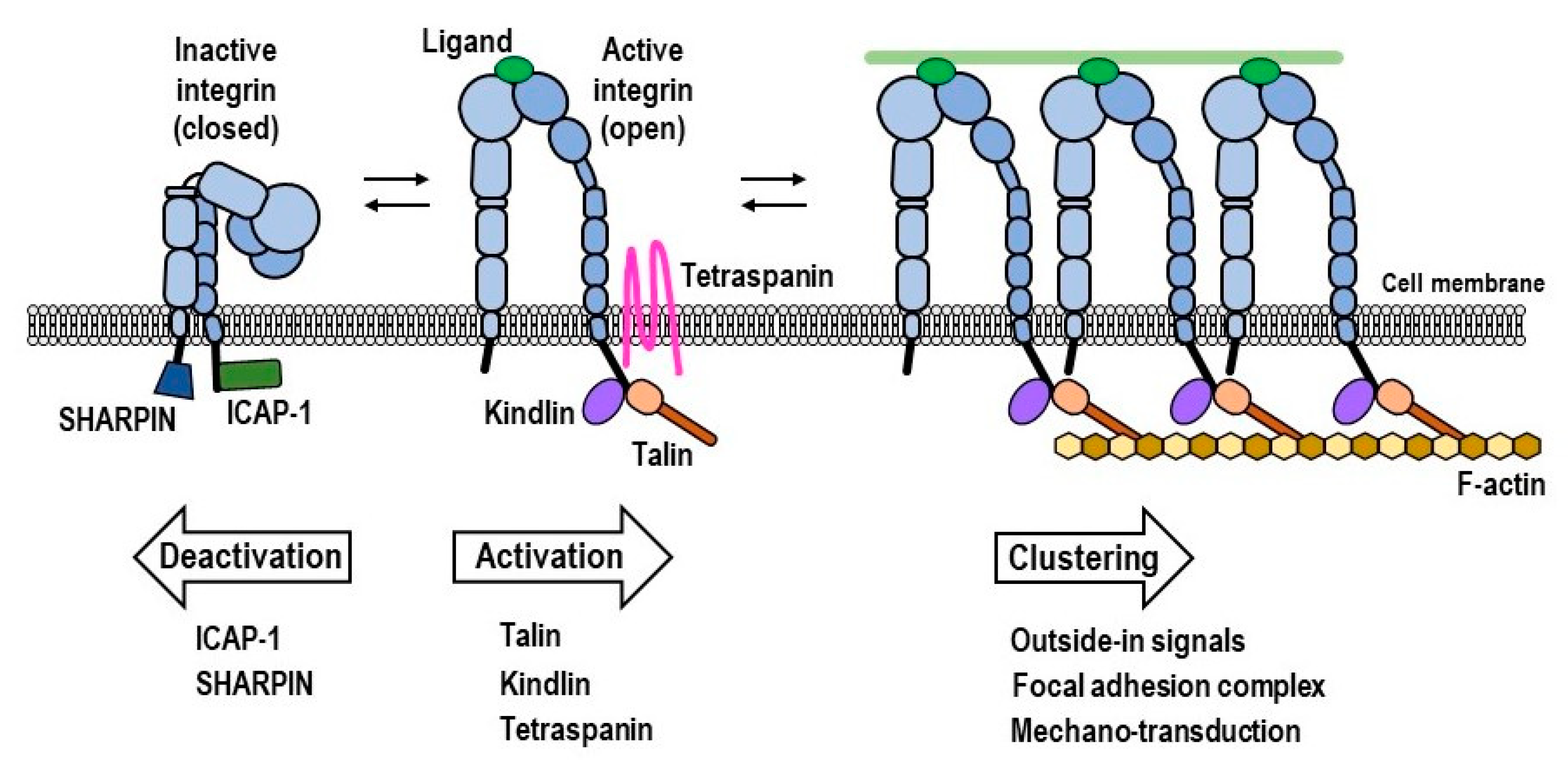
1. Integrin Regulation in Cells
1.1. Structures of Integrins
1.2. Intracellular Integrin Activators: Talin and Kindlin
1.3. Intracellular Integrin Inactivators: SHARPIN and ICAP-1
1.4. Force-Driven Integrin Activation
1.5. Heterotypic Lateral Association of Integrins with Tetraspanins and Other Membrane Proteins
1.6. Homotypic Lateral Association of Integrins, Leading to Integrin Clustering
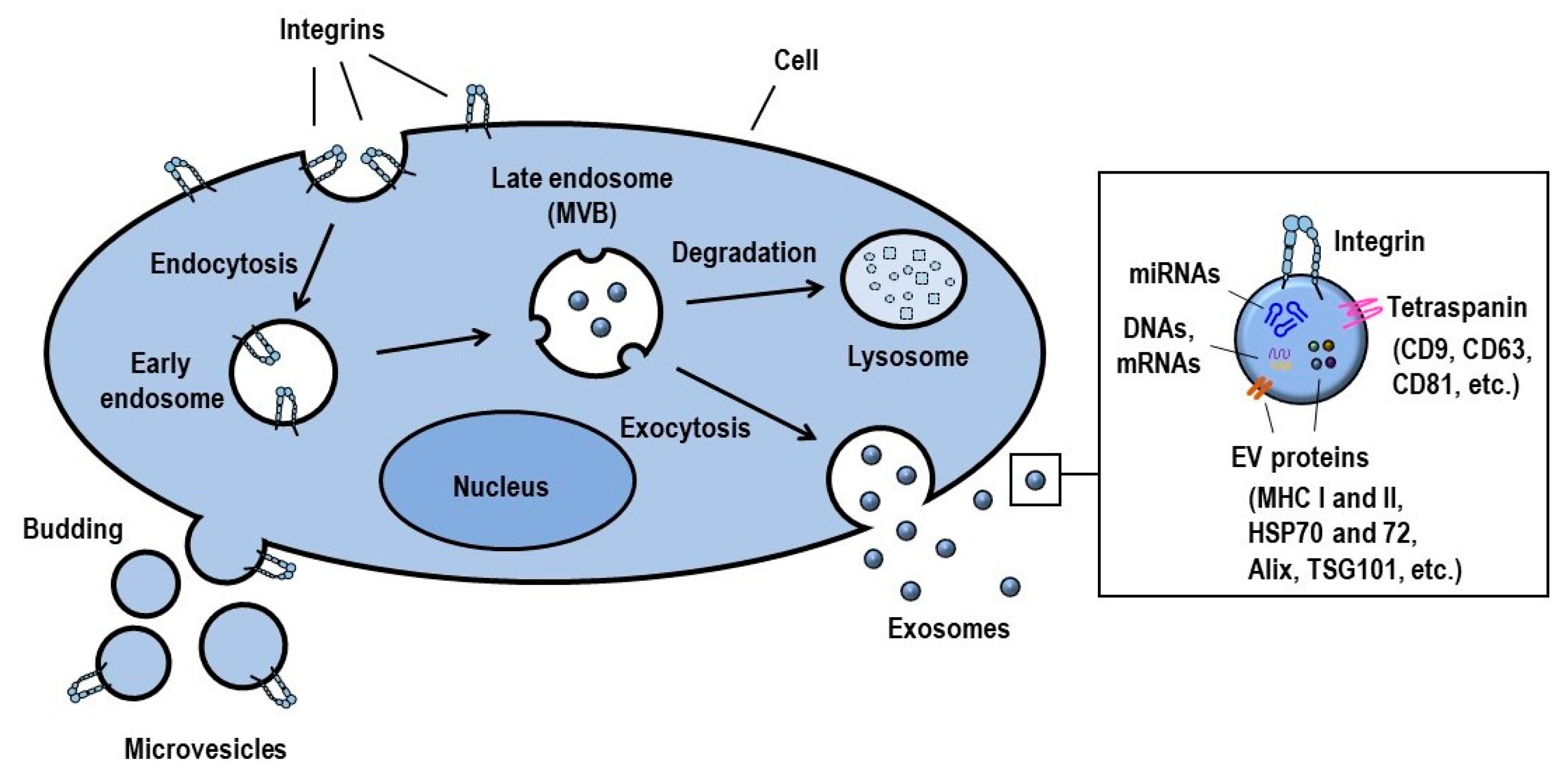
2. Integrin Regulation in Exosomes
2.1. Definition, Biogenesis and Function of Exosomes
2.2. Exosomal Integrin Functions
2.3. Regulation of Integrin Function on Exosomes
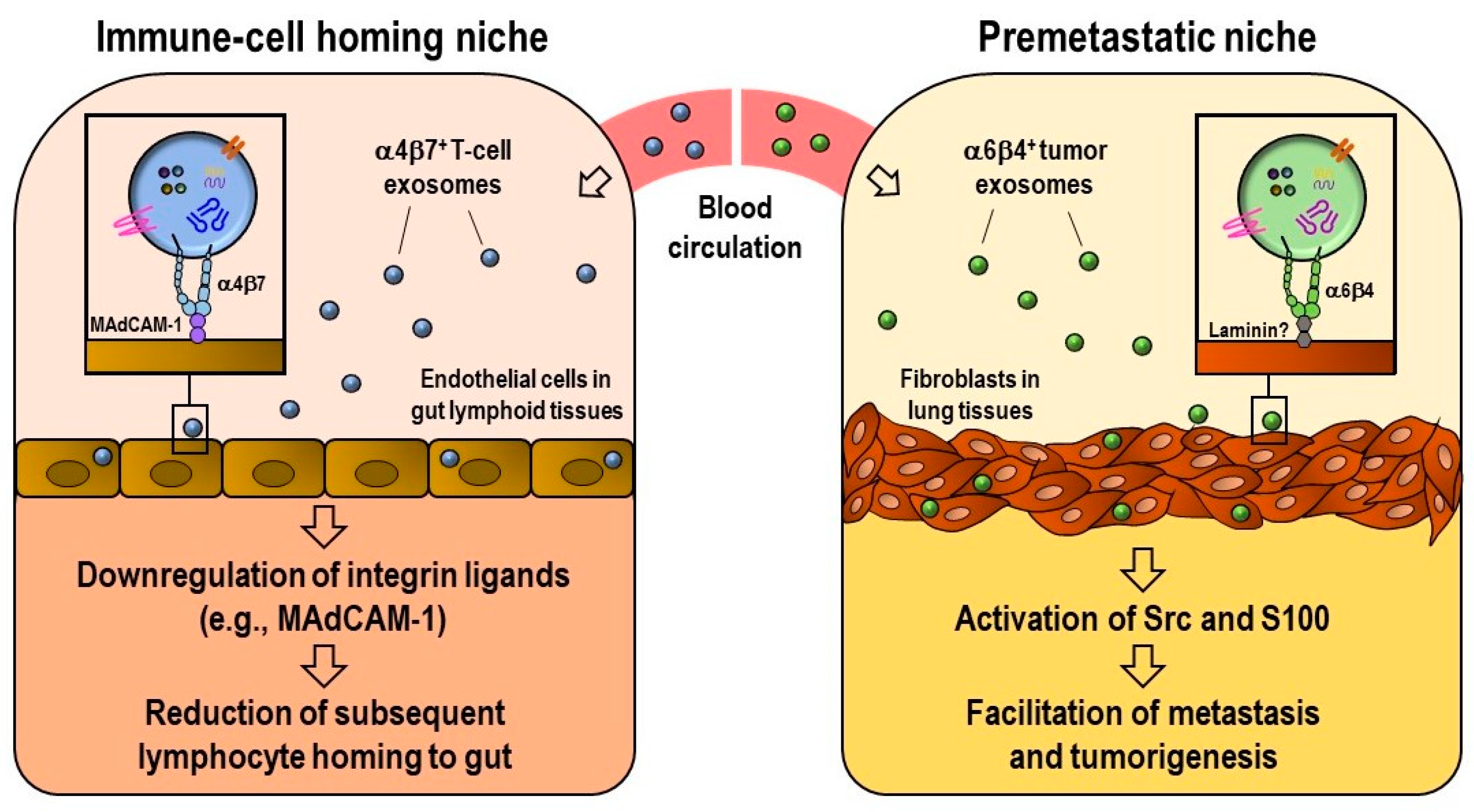
2.4. Integrin-Mediated Determination of the Biodistribution of Exosomes
2.5. Integrin-Mediated Internalization of Exosomes
2.6. Exosomal Transfer of Integrin Proteins and Associated Kinases
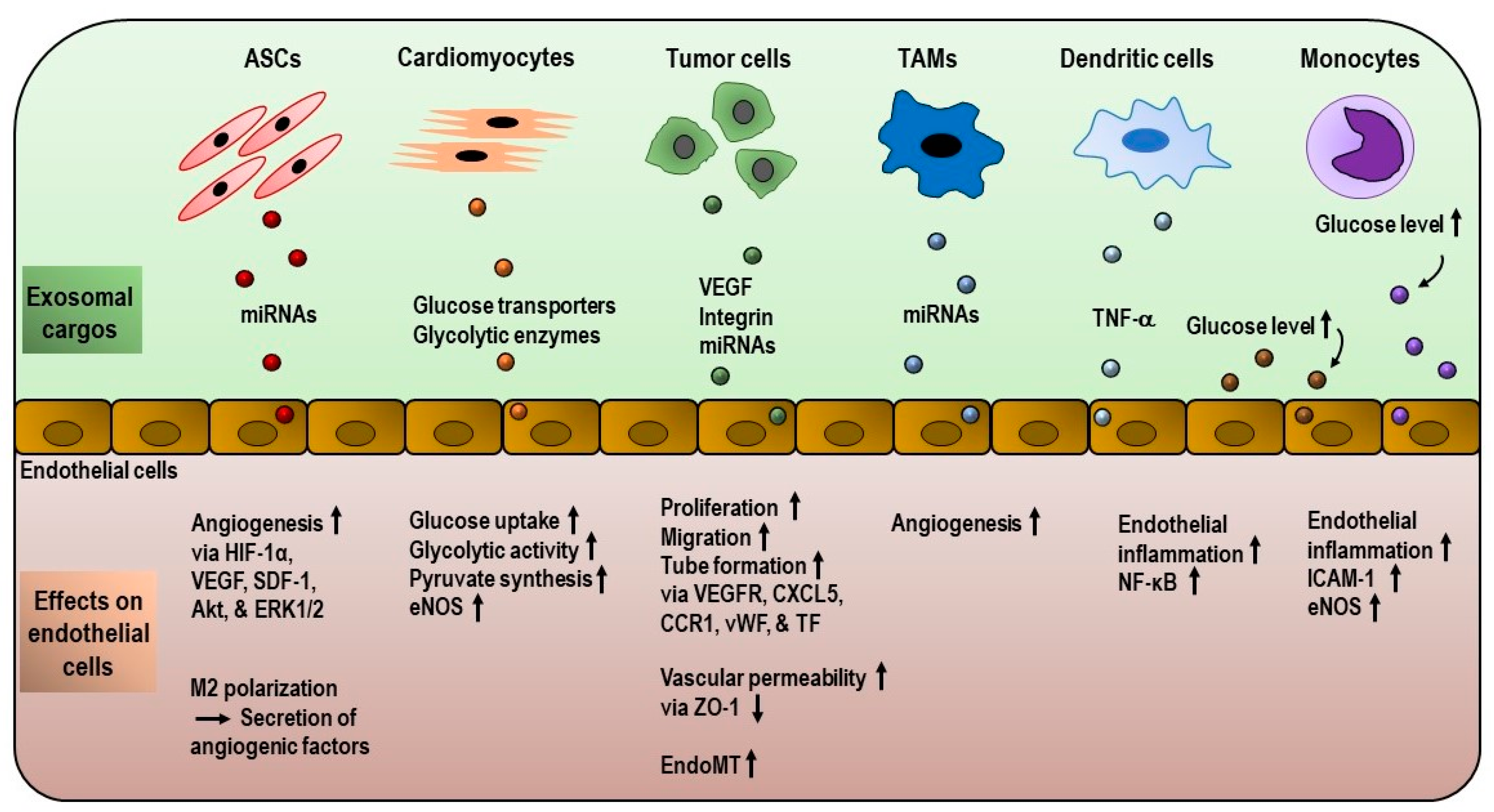
3. Exosome-Mediated Remodeling of Endothelial Gene Expression and Metabolism
3.1. Role of Exosomes in Endothelial Metabolism
3.2. Role of Exosomes in Angiogenesis and Tumor Microenvironment
4. Summary
Author Contributions
Funding
Conflicts of Interest
References
- Shimaoka, M.; Takagi, J.; Springer, T.A. Conformational Regulation of Integrin Structure and Function. Annu. Rev. Biophys. Biomol. Struct. 2002, 31, 485–516. [Google Scholar] [CrossRef] [PubMed]
- Anthis, N.J.; Campbell, I.D. The tail of integrin activation. Trends Biochem. Sci. 2011, 36, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Ye, F.; Ginsberg, M.H. Regulation of Integrin Activation. Annu. Rev. Cell Dev. Biol. 2011, 27, 321–345. [Google Scholar] [CrossRef]
- Park, E.J.; Yuki, Y.; Kiyono, H.; Shimaoka, M. Structural basis of blocking integrin activation and deactivation for anti-inflammation. J. Biomed. Sci. 2015, 22, 51. [Google Scholar] [CrossRef] [PubMed]
- Lafoya, B.; Munroe, J.A.; Miyamoto, A.; Detweiler, M.A.; Crow, J.J.; Gazdik, T.; Albig, A.R. Beyond the matrix: The many non-ECM ligands for integrins. Int. J. Mol. Sci. 2018, 19, 449. [Google Scholar] [CrossRef]
- Ginsberg, M.H. Integrin activation. BMB Rep. 2014, 47, 655–659. [Google Scholar] [CrossRef]
- Shattil, S.J.; Kim, C.; Ginsberg, M.H. The final steps of integrin activation: The end game. Nat. Rev. Mol. Cell Biol. 2010, 11, 288–300. [Google Scholar] [CrossRef]
- Tadokoro, S.; Shattil, S.J.; Eto, K.; Tai, V.; Liddington, R.C.; De Pereda, J.M.; Ginsberg, M.H.; Calderwood, D.A. Talin binding to integrin β tails: A final common step in integrin activation. Science 2003, 302, 103–106. [Google Scholar] [CrossRef]
- Gough, R.E.; Goult, B.T. The tale of two talins—Two isoforms to fine-tune integrin signalling. FEBS Lett. 2018, 592, 2108–2125. [Google Scholar] [CrossRef]
- Calderwood, D.A.; Zent, R.; Grant, R.; Rees, D.J.G.; Hynes, R.O.; Ginsberg, M.H. The talin head domain binds to integrin β subunit cytoplasmic tails and regulates integrin activation. J. Biol. Chem. 1999, 274, 28071–28074. [Google Scholar] [CrossRef] [PubMed]
- Calderwood, D.A.; Yan, B.; de Pereda, J.M.; Alvarez, B.G.; Fujioka, Y.; Liddington, R.C.; Ginsberg, M.H. The Phosphotyrosine Binding-like Domain of Talin Activates Integrins. J. Biol. Chem. 2002, 277, 21749–21758. [Google Scholar] [CrossRef]
- Vinogradova, O.; Velyvis, A.; Velyviene, A.; Hu, B.; Haas, T.A.; Plow, E.F.; Qin, J. A structural mechanism of integrin $α$IIb$β$3 “inside-out” activation as regulated by its cytoplasmic face. Cell 2002, 110, 587–597. [Google Scholar] [CrossRef]
- Calderwood, D.A. The Rap1-RIAM pathway prefers β2 integrins. Blood 2015, 126, 2658–2659. [Google Scholar] [CrossRef]
- Goksoy, E.; Ma, Y.; Wang, X.; Kong, X.; Perera, D.; Plow, E.F.; Qin, J. Structural Basis for the Autoinhibition of Talin in Regulating Integrin Activation. Mol. Cell 2008, 31, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Wang, J. Pull and push: Talin activation for integrin signaling. Cell Res. 2012, 22, 1512–1514. [Google Scholar] [CrossRef]
- Schiemer, J.; Bohm, A.; Lin, L.; Merrill-Skoloff, G.; Flaumenhaft, R.; Huang, J.S.; Le Breton, G.C.; Chishti, A.H. Gα13 switch region 2 relieves talin autoinhibition to activate αiIbβ3 integrin. J. Biol. Chem. 2016, 291, 26598–26612. [Google Scholar] [CrossRef] [PubMed]
- Rognoni, E.; Ruppert, R.; Fässler, R. The kindlin family: Functions, signaling properties and implications for human disease. J. Cell Sci. 2016, 129, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Plow, E.F.; Qin, J.; Byzova, T. Kindling the flame of integrin activation and function with kindlins. Curr. Opin. Hematol. 2009, 16, 323–328. [Google Scholar] [CrossRef]
- Moretti, F.A.; Moser, M.; Lyck, R.; Abadier, M.; Ruppert, R.; Engelhardt, B.; Fassler, R. Kindlin-3 regulates integrin activation and adhesion reinforcement of effector T cells. Proc. Natl. Acad. Sci. USA 2013, 110, 17005–17010. [Google Scholar] [CrossRef] [PubMed]
- Lefort, C.T.; Rossaint, J.; Moser, M.; Petrich, B.G.; Zarbock, A.; Monkley, S.J.; Critchley, D.R.; Ginsberg, M.H.; Fässler, R.; Ley, K. Distinct roles for talin-1 and kindlin-3 in LFA-1 extension and affinity regulation. Blood 2012, 119, 4275–4282. [Google Scholar] [CrossRef]
- Wen, L.; Marki, A.; Roy, P.; McArdle, S.; Sun, H.; Fan, Z.; Gingras, A.R.; Ginsberg, M.H.; Ley, K. Kindlin-3 recruitment to the plasma membrane precedes high affinity β2 integrin and neutrophil arrest from rolling. Blood 2020. [Google Scholar] [CrossRef]
- Margraf, A.; Germena, G.; Drexler, H.C.A.; Rossaint, J.; Ludwig, N.; Prystaj, B.; Mersmann, S.; Thomas, K.; Block, H.; Gottschlich, W.; et al. The integrin-linked kinase is required for chemokine-triggered high-affinity conformation of the neutrophil β2-integrin LFA-1. Blood 2020, 136, 2200–2205. [Google Scholar] [CrossRef]
- Marcovecchio, P.M.; Zhu, Y.P.; Hanna, R.N.; Dinh, H.Q.; Tacke, R.; Wu, R.; McArdle, S.; Reynolds, S.; Araujo, D.J.; Ley, K.; et al. Frontline Science: Kindlin-3 is essential for patrolling and phagocytosis functions of nonclassical monocytes during metastatic cancer surveillance. J. Leukoc. Biol. 2020, 107, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Huang, M.; Lai, J.; Mao, K.; Sun, P.; Cao, Z.; Hu, Y.; Zhang, Y.; Schulte, M.L.; Jin, C.; et al. Kindlin supports platelet integrin αIIbβ3 activation by interacting with paxillin. J. Cell Sci. 2017, 130, 3764–3775. [Google Scholar] [CrossRef] [PubMed]
- Haydari, Z.; Shams, H.; Jahed, Z.; Mofrad, M.R.K. Kindlin Assists Talin to Promote Integrin Activation. Biophys. J. 2020, 118, 1977–1991. [Google Scholar] [CrossRef]
- Pouwels, J.; Nevo, J.; Pellinen, T.; Ylänne, J.; Ivaska, J. Negative regulators of integrin activity. J. Cell Sci. 2012, 125, 3271–3280. [Google Scholar] [CrossRef]
- Rantala, J.K.; Pouwels, J.; Pellinen, T.; Veltel, S.; Laasola, P.; Mattila, E.; Potter, C.S.; Duffy, T.; Sundberg, J.P.; Kallioniemi, O.; et al. SHARPIN is an endogenous inhibitor of β1-integrin activation. Nat. Cell Biol. 2011, 13, 1315–1324. [Google Scholar] [CrossRef]
- Zeng, C.; Xiong, D.; Zhang, K.; Yao, J. Shank-associated RH domain interactor signaling in tumorigenesis (review). Oncol. Lett. 2020, 20, 2579–2586. [Google Scholar] [CrossRef]
- Gao, J.; Bao, Y.; Ge, S.; Sun, P.; Sun, J.; Liu, J.; Chen, F.; Han, L.; Cao, Z.; Qin, J.; et al. Sharpin suppresses β1-integrin activation by complexing with the β1 tail and kindlin-1. Cell Commun. Signal. 2019, 17, 101. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.A.; Hemler, M.E. Interaction of the integrin β1 cytoplasmic domain with ICAP-1 protein. J. Biol. Chem. 1999, 274, 11–19. [Google Scholar] [CrossRef]
- Bouvard, D.; Vignoud, L.; Dupé-Manet, S.; Abed, N.; Fournier, H.N.; Vincent-Monegat, C.; Francesco Retta, S.; Fässler, R.; Block, M.R. Disruption of focal adhesions by integrin cytoplasmic domain-associated protein-1α. J. Biol. Chem. 2003, 278, 6567–6574. [Google Scholar] [CrossRef] [PubMed]
- Faurobert, E.; Rome, C.; Lisowska, J.; Manet-Dupé, S.; Boulday, G.; Malbouyres, M.; Balland, M.; Bouin, A.P.; Kéramidas, M.; Bouvard, D.; et al. CCM1-ICAP-1 complex controls β1 integrin-dependent endothelial contractility and fibronectin remodeling. J. Cell Biol. 2013, 202, 545–561. [Google Scholar] [CrossRef]
- Kiema, T.; Lad, Y.; Jiang, P.; Oxley, C.L.; Baldassarre, M.; Wegener, K.L.; Campbell, I.D.; Ylänne, J.; Calderwood, D.A. The molecular basis of filamin binding to integrins and competition with talin. Mol. Cell 2006, 21, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Lad, Y.; Jiang, P.; Ruskamo, S.; Harburger, D.S.; Ylänne, J.; Campbell, I.D.; Calderwood, D.A. Structural basis of the migfilin-filamin interaction and competition with integrin β tails. J. Biol. Chem. 2008, 283, 35154–35163. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Guo, S.S.; Fässler, R. Integrin-mediated mechanotransduction. J. Cell Biol. 2016, 215, 445–456. [Google Scholar] [CrossRef]
- Yakovenko, O.; Sharma, S.; Forero, M.; Tchesnokova, V.; Aprikian, P.; Kidd, B.; Mach, A.; Vogel, V.; Sokurenko, E.; Thomas, W.E. FimH Forms Catch Bonds That Are Enhanced by Mechanical Force Due to Allosteric Regulation. J. Biol. Chem. 2008, 283, 11596–11605. [Google Scholar] [CrossRef]
- Ross, T.D.; Coon, B.G.; Yun, S.; Baeyens, N.; Tanaka, K.; Ouyang, M.; Schwartz, M.A. Integrins in mechanotransduction. Curr. Opin. Cell Biol. 2013, 25, 613–618. [Google Scholar] [CrossRef] [PubMed]
- Comrie, W.A.; Babich, A.; Burkhardt, J.K. F-actin flow drives affinity maturation and spatial organization of LFA-1 at the immunological synapse. J. Cell Biol. 2015, 208, 475–491. [Google Scholar] [CrossRef]
- Nordenfelt, P.; Elliott, H.L.; Springer, T.A. Coordinated integrin activation by actin-dependent force during T-cell migration. Nat. Commun. 2016, 7, 13119. [Google Scholar] [CrossRef]
- Li, Z.; Lee, H.; Zhu, C. Molecular mechanisms of mechanotransduction in integrin-mediated cell-matrix adhesion. Exp. Cell Res. 2016, 349, 85–94. [Google Scholar] [CrossRef]
- Zuidema, A.; Wang, W.; Sonnenberg, A. Crosstalk between Cell Adhesion Complexes in Regulation of Mechanotransduction. BioEssays 2020, 42, 2000119. [Google Scholar] [CrossRef]
- Driscoll, T.P.; Ahn, S.J.; Huang, B.; Kumar, A.; Schwartz, M.A. Actin flow-dependent and -independent force transmission through integrins. Proc. Natl. Acad. Sci. USA 2020, 117, 32413–32422. [Google Scholar] [CrossRef]
- Lin, G.L.; Cohen, D.M.; Desai, R.A.; Breckenridge, M.T.; Gao, L.; Humphries, M.J.; Chen, C.S. Activation of beta 1 but not beta 3 integrin increases cell traction forces. FEBS Lett. 2013, 587, 763–769. [Google Scholar] [CrossRef] [PubMed]
- Milloud, R.; Destaing, O.; de Mets, R.; Bourrin-Reynard, I.; Oddou, C.; Delon, A.; Wang, I.; Albigès-Rizo, C.; Balland, M. αvβ3 integrins negatively regulate cellular forces by phosphorylation of its distal NPXY site. Biol. Cell 2017, 109, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Berditchevski, F. Complexes of tetraspanins with integrins: More than meets the eye. J. Cell Sci. 2001, 114, 4143–4151. [Google Scholar] [PubMed]
- Berditchevski, F.; Zutter, M.M.; Hemler, M.E. Characterization of novel complexes on the cell surface between integrins and proteins with 4 transmembrane domains (TM4 proteins). Mol. Biol. Cell 1996, 7, 193–207. [Google Scholar] [CrossRef] [PubMed]
- Kotha, J.; Longhurst, C.; Appling, W.; Jennings, L.K. Tetraspanin CD9 regulates beta 1 integrin activation and enhances cell motility to fibronectin via a PI-3 kinase-dependent pathway. Exp. Cell Res. 2008, 314, 1811–1822. [Google Scholar] [CrossRef]
- Nishiuchi, R.; Sanzen, N.; Nada, S.; Sumida, Y.; Wada, Y.; Okada, M.; Takagi, J.; Hasegawa, H.; Sekiguchi, K. Potentiation of the ligand-binding activity of integrin $α$3$β$1 via association with tetraspanin CD151. Proc. Natl. Acad. Sci. USA 2005, 102, 1939–1944. [Google Scholar] [CrossRef] [PubMed]
- Reyes, R.; Monjas, A.; Yánez-Mó, M.; Cardeñes, B.; Morlino, G.; Gilsanz, A.; Machado-Pineda, Y.; Lafuente, E.; Monk, P.; Sánchez-Madrid, F.; et al. Different states of integrin LFA-1 aggregation are controlled through its association with tetraspanin CD9. Biochim. Biophys. Acta Mol. Cell Res. 2015, 1853, 2464–2480. [Google Scholar] [CrossRef]
- Shibagaki, N.; Hanada, K.I.; Yamashita, H.; Shimada, S.; Hamada, H. Overexpression of CD82 on human T cells enhances LFA-1/ICAM-1-mediated cell-cell adhesion: Functional association between CD82 and LFA-1 in T cell activation. Eur. J. Immunol. 1999, 29, 4081–4091. [Google Scholar] [CrossRef]
- Vancompernolle, S.E.; Levy, S.; Todd, S.C. Anti-CD81 activates LFA-1 on T cells and promotes T cell-B cell collaboration. Eur. J. Immunol. 2001, 31, 823–831. [Google Scholar] [CrossRef]
- Arokiasamy, S.; Balderstone, M.J.M.; De Rossi, G.; Whiteford, J.R. Syndecan-3 in Inflammation and Angiogenesis. Front. Immunol. 2020, 10, 3031. [Google Scholar] [CrossRef]
- Soares, M.A.; Teixeira, F.C.O.B.; Fontes, M.; Arêas, A.L.; Leal, M.G.; Pavão, M.S.G.; Stelling, M.P. Heparan Sulfate Proteoglycans May Promote or Inhibit Cancer Progression by Interacting with Integrins and Affecting Cell Migration. BioMed Res. Int. 2015, 2015. [Google Scholar] [CrossRef]
- Morgan, M.R.; Humphries, M.J.; Bass, M.D. Synergistic control of cell adhesion by integrins and syndecans. Nat. Rev. Mol. Cell Biol. 2007, 8, 957–969. [Google Scholar] [CrossRef] [PubMed]
- Rovira-Clavé, X.; Angulo-Ibáñez, M.; Reina, M.; Espel, E. The PDZ-binding domain of syndecan-2 inhibits LFA-1 high-affinity conformation. Cell. Signal. 2014, 26, 1489–1499. [Google Scholar] [CrossRef] [PubMed]
- Beauvais, D.L.M.; Burbach, B.J.; Rapraeger, A.C. The syndecan-1 ectodomain regulates αvβ3 integrin activily in human mammary carcinoma cells. J. Cell Biol. 2004, 167, 171–181. [Google Scholar] [CrossRef]
- Afratis, N.A.; Nikitovic, D.; Multhaupt, H.A.B.; Theocharis, A.D.; Couchman, J.R.; Karamanos, N.K. Syndecans—Key regulators of cell signaling and biological functions. FEBS J. 2017, 284, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.R.; Hamidi, H.; Bass, M.D.; Warwood, S.; Ballestrem, C.; Humphries, M.J. Syndecan-4 Phosphorylation Is a Control Point for Integrin Recycling. Dev. Cell 2013, 24, 472–485. [Google Scholar] [CrossRef] [PubMed]
- De Rossi, G.; Evans, A.R.; Kay, E.; Woodfin, A.; McKay, T.R.; Nourshargh, S.; Whiteford, J.R. Shed syndecan-2 inhibits angiogenesis. J. Cell Sci. 2014, 127, 4788–4799. [Google Scholar] [CrossRef]
- Beauvais, D.M.; Ell, B.J.; McWhorter, A.R.; Rapraeger, A.C. Syndecan-1 regulates α vβ 3 and α vβ 5 integrin activation during angiogenesis and is blocked by synstatin, a novel peptide inhibitor. J. Exp. Med. 2009, 206, 691–705. [Google Scholar] [CrossRef]
- Beauvais, D.M.; Rapraeger, A.C. Syndecan-1 couples the insulin-like growth factor-1 receptor to inside-out integrin activation. J. Cell Sci. 2010, 123, 3796–3807. [Google Scholar] [CrossRef]
- Wang, H.; Jin, H.; Beauvais, D.M.; Rapraeger, A.C. Cytoplasmic Domain Interactions of Syndecan-1 and Syndecan-4 with α6β4 Integrin Mediate Human Epidermal Growth Factor Receptor (HER1 and HER2)-dependent Motility and Survival. J. Biol. Chem. 2014, 289, 30318–30332. [Google Scholar] [CrossRef]
- Fiore, V.F.; Ju, L.; Chen, Y.; Zhu, C.; Barker, T.H. Dynamic catch of a Thy-1–α5β1+syndecan-4 trimolecular complex. Nat. Commun. 2014, 5, 4886. [Google Scholar] [CrossRef]
- Chronopoulos, A.; Thorpe, S.D.; Cortes, E.; Lachowski, D.; Rice, A.J.; Mykuliak, V.V.; Róg, T.; Lee, D.A.; Hytönen, V.P.; del Río Hernández, A.E. Syndecan-4 tunes cell mechanics by activating the kindlin-integrin-RhoA pathway. Nat. Mater. 2020, 19, 669–678. [Google Scholar] [CrossRef] [PubMed]
- Kukkurainen, S.; Azizi, L.; Zhang, P.; Jacquier, M.-C.; Baikoghli, M.; von Essen, M.; Tuukkanen, A.; Laitaoja, M.; Liu, X.; Rahikainen, R.; et al. The F1 loop of the talin head domain acts as a gatekeeper in integrin activation and clustering. J. Cell Sci. 2020, 133, jcs239202. [Google Scholar] [CrossRef] [PubMed]
- Bakker, G.J.; Eich, C.; Torreno-Pina, J.A.; Diez-Ahedo, R.; Perez-Samper, G.; Van Zanten, T.S.; Figdor, C.G.; Cambi, A.; Garcia-Parajo, M.F. Lateral mobility of individual integrin nanoclusters orchestrates the onset for leukocyte adhesion. Proc. Natl. Acad. Sci. USA 2012, 109, 4869–4874. [Google Scholar] [CrossRef] [PubMed]
- Jin, T.; Li, J. Dynamitin Controls β 2 Integrin Avidity by Modulating Cytoskeletal Constraint on Integrin Molecules. J. Biol. Chem. 2002, 277, 32963–32969. [Google Scholar] [CrossRef]
- Cluzel, C.; Saltel, F.; Lussi, J.; Paulhe, F.; Imhof, B.A.; Wehrle-Haller, B. The mechanisms and dynamics of αvβ3 integrin clustering in living cells. J. Cell Biol. 2005, 171, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Cambi, A.; Joosten, B.; Koopman, M.; de Lange, F.; Beeren, I.; Torensma, R.; Fransen, J.A.; Garcia-Parajó, M.; van Leeuwen, F.N.; Figdor, C.G. Organization of the Integrin LFA-1 in Nanoclusters Regulates Its Activity. Mol. Biol. Cell 2006, 17, 4270–4281. [Google Scholar] [CrossRef]
- Yáñez-Mó, M.; Siljander, P.R.-M.; Andreu, Z.; Bedina Zavec, A.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef]
- Zaborowski, M.P.; Balaj, L.; Breakefield, X.O.; Lai, C.P. Extracellular Vesicles: Composition, Biological Relevance, and Methods of Study. Bioscience 2015, 65, 783–797. [Google Scholar] [CrossRef] [PubMed]
- Abels, E.R.; Breakefield, X.O. Introduction to Extracellular Vesicles: Biogenesis, RNA Cargo Selection, Content, Release, and Uptake. Cell. Mol. Neurobiol. 2016, 36, 301–312. [Google Scholar] [CrossRef]
- Mashouri, L.; Yousefi, H.; Aref, A.R.; Ahadi, A.M.; Molaei, F.; Alahari, S.K. Exosomes: Composition, biogenesis, and mechanisms in cancer metastasis and drug resistance. Mol. Cancer 2019, 18, 75. [Google Scholar] [CrossRef]
- Hessvik, N.P.; Llorente, A. Current knowledge on exosome biogenesis and release. Cell. Mol. Life Sci. 2018, 75, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Meldolesi, J. Exosomes and Ectosomes in Intercellular Communication. Curr. Biol. 2018, 28, R435–R444. [Google Scholar] [CrossRef] [PubMed]
- Lőrincz, Á.M.; Bartos, B.; Szombath, D.; Szeifert, V.; Timár, C.I.; Turiák, L.; Drahos, L.; Kittel, Á.; Veres, D.S.; Kolonics, F.; et al. Role of Mac-1 integrin in generation of extracellular vesicles with antibacterial capacity from neutrophilic granulocytes. J. Extracell. Vesicles 2020, 9, 1698889. [Google Scholar] [CrossRef] [PubMed]
- Vicencio, J.M.; Yellon, D.M.; Sivaraman, V.; Das, D.; Boi-Doku, C.; Arjun, S.; Zheng, Y.; Riquelme, J.A.; Kearney, J.; Sharma, V.; et al. Plasma Exosomes Protect the Myocardium From Ischemia-Reperfusion Injury. J. Am. Coll. Cardiol. 2015, 65, 1525–1536. [Google Scholar] [CrossRef]
- Frühbeis, C.; Fröhlich, D.; Kuo, W.P.; Amphornrat, J.; Thilemann, S.; Saab, A.S.; Kirchhoff, F.; Möbius, W.; Goebbels, S.; Nave, K.-A.; et al. Neurotransmitter-Triggered Transfer of Exosomes Mediates Oligodendrocyte–Neuron Communication. PLoS Biol. 2013, 11, e1001604. [Google Scholar] [CrossRef]
- Hur, Y.H.; Feng, S.; Wilson, K.F.; Cerione, R.A.; Antonyak, M.A. Embryonic Stem Cell-Derived Extracellular Vesicles Maintain ESC Stemness by Activating FAK. Dev. Cell 2020. [Google Scholar] [CrossRef]
- Segura, E.; Nicco, C.; Lombard, B.; Véron, P.; Raposo, G.; Batteux, F.; Amigorena, S.; Théry, C. ICAM-1 on exosomes from mature dendritic cells is critical for efficient naive T-cell priming. Blood 2005, 106, 216–223. [Google Scholar] [CrossRef]
- Segura, E.; Guérin, C.; Hogg, N.; Amigorena, S.; Théry, C. CD8 + Dendritic Cells Use LFA-1 to Capture MHC-Peptide Complexes from Exosomes In Vivo. J. Immunol. 2007, 179, 1489–1496. [Google Scholar] [CrossRef] [PubMed]
- Wahlgren, J.; Karlson, T.D.L.; Glader, P.; Telemo, E.; Valadi, H. Activated Human T Cells Secrete Exosomes That Participate in IL-2 Mediated Immune Response Signaling. PLoS ONE 2012, 7, e49723. [Google Scholar] [CrossRef] [PubMed]
- Torralba, D.; Baixauli, F.; Villarroya-Beltri, C.; Fernández-Delgado, I.; Latorre-Pellicer, A.; Acín-Pérez, R.; Martín-Cófreces, N.B.; Jaso-Tamame, Á.L.; Iborra, S.; Jorge, I.; et al. Priming of dendritic cells by DNA-containing extracellular vesicles from activated T cells through antigen-driven contacts. Nat. Commun. 2018, 9, 2658. [Google Scholar] [CrossRef] [PubMed]
- Park, E.J.; Prajuabjinda, O.; Soe, Z.Y.; Darkwah, S.; Appiah, M.G.; Kawamoto, E.; Momose, F.; Shiku, H.; Shimaoka, M. Exosomal regulation of lymphocyte homing to the gut. Blood Adv. 2019, 3, 1–11. [Google Scholar] [CrossRef]
- Raposo, G.; Nijman, H.W.; Stoorvogel, W.; Liejendekker, R.; Harding, C.V.; Melief, C.J.; Geuze, H.J. B lymphocytes secrete antigen-presenting vesicles. J. Exp. Med. 1996, 183, 1161–1172. [Google Scholar] [CrossRef]
- Jong, A.Y.; Wu, C.-H.; Li, J.; Sun, J.; Fabbri, M.; Wayne, A.S.; Seeger, R.C. Large-scale isolation and cytotoxicity of extracellular vesicles derived from activated human natural killer cells. J. Extracell. Vesicles 2017, 6, 1294368. [Google Scholar] [CrossRef]
- Rajendran, L.; Honsho, M.; Zahn, T.R.; Keller, P.; Geiger, K.D.; Verkade, P.; Simons, K. Alzheimer’s disease beta-amyloid peptides are released in association with exosomes. Proc. Natl. Acad. Sci. USA 2006, 103, 11172–11177. [Google Scholar] [CrossRef]
- Grey, M.; Dunning, C.J.; Gaspar, R.; Grey, C.; Brundin, P.; Sparr, E.; Linse, S. Acceleration of α-Synuclein Aggregation by Exosomes. J. Biol. Chem. 2015, 290, 2969–2982. [Google Scholar] [CrossRef]
- Singh, A.; Fedele, C.; Lu, H.; Nevalainen, M.T.; Keen, J.H.; Languino, L.R. Exosome-mediated Transfer of avb3 Integrin from Tumorigenic to Nontumorigenic Cells Promotes a Migratory Phenotype. Mol. Cancer Res. 2016, 14, 1136–1146. [Google Scholar] [CrossRef]
- Tang, M.K.S.; Yue, P.Y.K.; Ip, P.P.; Huang, R.-L.; Lai, H.-C.; Cheung, A.N.Y.; Tse, K.Y.; Ngan, H.Y.S.; Wong, A.S.T. Soluble E-cadherin promotes tumor angiogenesis and localizes to exosome surface. Nat. Commun. 2018, 9, 2270. [Google Scholar] [CrossRef]
- Yu, S.; Liu, C.; Su, K.; Wang, J.; Liu, Y.; Zhang, L.; Li, C.; Cong, Y.; Kimberly, R.; Grizzle, W.E.; et al. Tumor Exosomes Inhibit Differentiation of Bone Marrow Dendritic Cells. J. Immunol. 2007, 178, 6867–6875. [Google Scholar] [CrossRef]
- Gaballa, R.; Ali, H.E.A.; Mahmoud, M.O.; Rhim, J.S.; Ali, H.I.; Salem, H.F.; Saleem, M.; Kandeil, M.A.; Ambs, S.; Abd Elmageed, Z.Y. Exosomes-Mediated Transfer of Itga2 Promotes Migration and Invasion of Prostate Cancer Cells by Inducing Epithelial-Mesenchymal Transition. Cancers 2020, 12, 2300. [Google Scholar] [CrossRef]
- Krishn, S.R.; Singh, A.; Bowler, N.; Duffy, A.N.; Friedman, A.; Fedele, C.; Kurtoglu, S.; Tripathi, S.K.; Wang, K.; Hawkins, A.; et al. Prostate cancer sheds the αvβ3 integrin in vivo through exosomes. Matrix Biol. 2019, 77, 41–57. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Bowler, N.; Harshyne, L.A.; Craig Hooper, D.; Krishn, S.R.; Kurtoglu, S.; Fedele, C.; Liu, Q.; Tang, H.-Y.; Kossenkov, A.V.; et al. Exosomal αvβ6 integrin is required for monocyte M2 polarization in prostate cancer. Matrix Biol. 2018, 70, 20–35. [Google Scholar] [CrossRef]
- Zhao, J.; Schlößer, H.A.; Wang, Z.; Qin, J.; Li, J.; Popp, F.; Popp, M.C.; Alakus, H.; Chon, S.-H.; Hansen, H.P.; et al. Tumor-Derived Extracellular Vesicles Inhibit Natural Killer Cell Function in Pancreatic Cancer. Cancers 2019, 11, 874. [Google Scholar] [CrossRef] [PubMed]
- Klibi, J.; Niki, T.; Riedel, A.; Pioche-Durieu, C.; Souquere, S.; Rubinstein, E.; Le Moulec, S.; Guigay, J.; Hirashima, M.; Guemira, F.; et al. Blood diffusion and Th1-suppressive effects of galectin-9–containing exosomes released by Epstein-Barr virus–infected nasopharyngeal carcinoma cells. Blood 2009, 113, 1957–1966. [Google Scholar] [CrossRef]
- Costa-Silva, B.; Aiello, N.M.; Ocean, A.J.; Singh, S.; Zhang, H.; Thakur, B.K.; Becker, A.; Hoshino, A.; Mark, M.T.; Molina, H.; et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat. Cell Biol. 2015, 17, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Melo, S.A.; Luecke, L.B.; Kahlert, C.; Fernandez, A.F.; Gammon, S.T.; Kaye, J.; LeBleu, V.S.; Mittendorf, E.A.; Weitz, J.; Rahbari, N.; et al. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature 2015, 523, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Hornick, N.I.; Huan, J.; Doron, B.; Goloviznina, N.A.; Lapidus, J.; Chang, B.H.; Kurre, P. Serum Exosome MicroRNA as a Minimally-Invasive Early Biomarker of AML. Sci. Rep. 2015, 5, 11295. [Google Scholar] [CrossRef]
- Whiteside, T.L. Tumor-Derived Exosomes and Their Role in Cancer Progression. Adv. Clin. Chem. 2016, 74, 103–141. [Google Scholar] [CrossRef]
- Hoshino, A.; Costa-Silva, B.; Shen, T.-L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Krishn, S.R.; Salem, I.; Quaglia, F.; Naranjo, N.M.; Agarwal, E.; Liu, Q.; Sarker, S.; Kopenhaver, J.; McCue, P.A.; Weinreb, P.H.; et al. The αvβ6 integrin in cancer cell-derived small extracellular vesicles enhances angiogenesis. J. Extracell. Vesicles 2020, 9, 1763594. [Google Scholar] [CrossRef]
- Soe, Z.Y.; Prajuabjinda, O.; Myint, P.K.; Gaowa, A.; Kawamoto, E.; Park, E.J.; Shimaoka, M. Talin-2 regulates integrin functions in exosomes. Biochem. Biophys. Res. Commun. 2019, 512, 429–434. [Google Scholar] [CrossRef]
- Fedele, C.; Singh, A.; Zerlanko, B.J.; Iozzo, R.V.; Languino, L.R. The αvβ6 Integrin Is Transferred Intercellularly via Exosomes. J. Biol. Chem. 2015, 290, 4545–4551. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, B.C.; Melis, N.; Chen, D.; Wang, W.; Gallardo, D.; Weigert, R.; Parent, C.A. The LTB4–BLT1 axis regulates actomyosin and β2-integrin dynamics during neutrophil extravasation. J. Cell Biol. 2020, 219. [Google Scholar] [CrossRef]
- Genschmer, K.R.; Russell, D.W.; Lal, C.; Szul, T.; Bratcher, P.E.; Noerager, B.D.; Abdul Roda, M.; Xu, X.; Rezonzew, G.; Viera, L.; et al. Activated PMN Exosomes: Pathogenic Entities Causing Matrix Destruction and Disease in the Lung. Cell 2019, 176, 113–126.e15. [Google Scholar] [CrossRef] [PubMed]
- Kawamoto, E.; Masui-Ito, A.; Eguchi, A.; Soe, Z.Y.; Prajuabjinda, O.; Darkwah, S.; Park, E.J.; Imai, H.; Shimaoka, M. Integrin and PD-1 Ligand Expression on Circulating Extracellular Vesicles in Systemic Inflammatory Response Syndrome and Sepsis. SHOCK 2019, 52, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Domenis, R.; Marino, M.; Cifù, A.; Scardino, G.; Curcio, F.; Fabris, M. Circulating exosomes express α4β7 integrin and compete with CD4+ T cells for the binding to Vedolizumab. PLoS ONE 2020, 15, e0242342. [Google Scholar] [CrossRef] [PubMed]
- Kowal, J.; Arras, G.; Colombo, M.; Jouve, M.; Morath, J.P.; Primdal-Bengtson, B.; Dingli, F.; Loew, D.; Tkach, M.; Théry, C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA 2016, 113, E968–E977. [Google Scholar] [CrossRef] [PubMed]
- Nazarenko, I.; Rana, S.; Baumann, A.; McAlear, J.; Hellwig, A.; Trendelenburg, M.; Lochnit, G.; Preissner, K.T.; Zoller, M. Cell Surface Tetraspanin Tspan8 Contributes to Molecular Pathways of Exosome-Induced Endothelial Cell Activation. Cancer Res. 2010, 70, 1668–1678. [Google Scholar] [CrossRef]
- Wiklander, O.P.B.; Nordin, J.Z.; O’Loughlin, A.; Gustafsson, Y.; Corso, G.; Mäger, I.; Vader, P.; Lee, Y.; Sork, H.; Seow, Y.; et al. Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting. J. Extracell. Vesicles 2015, 4, 26316. [Google Scholar] [CrossRef]
- Mulcahy, L.A.; Pink, R.C.; Carter, D.R.F. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 2014, 3, 24641. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Brigstock, D.R. Integrins and heparan sulfate proteoglycans on hepatic stellate cells (HSC) are novel receptors for HSC-derived exosomes. FEBS Lett. 2016, 590, 4263–4274. [Google Scholar] [CrossRef] [PubMed]
- Clayton, A.; Turkes, A.; Dewitt, S.; Steadman, R.; Mason, M.D.; Hallett, M.B. Adhesion and signaling by B cell-derived exosomes: The role of integrins. FASEB J. 2004, 18, 977–979. [Google Scholar] [CrossRef]
- Fuentes, P.; Sesé, M.; Guijarro, P.J.; Emperador, M.; Sánchez-Redondo, S.; Peinado, H.; Hümmer, S.; Ramón y Cajal, S. ITGB3-mediated uptake of small extracellular vesicles facilitates intercellular communication in breast cancer cells. Nat. Commun. 2020, 11, 4261. [Google Scholar] [CrossRef]
- Yuan, D.; Zhao, Y.; Banks, W.A.; Bullock, K.M.; Haney, M.; Batrakova, E.; Kabanov, A.V. Macrophage exosomes as natural nanocarriers for protein delivery to inflamed brain. Biomaterials 2017, 142, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wubbolts, R.; Leckie, R.S.; Veenhuizen, P.T.M.; Schwarzmann, G.; Möbius, W.; Hoernschemeyer, J.; Slot, J.-W.; Geuze, H.J.; Stoorvogel, W. Proteomic and Biochemical Analyses of Human B Cell-derived Exosomes. J. Biol. Chem. 2003, 278, 10963–10972. [Google Scholar] [CrossRef]
- Altei, W.F.; Pachane, B.C.; dos Santos, P.K.; Ribeiro, L.N.M.; Sung, B.H.; Weaver, A.M.; Selistre-de-Araújo, H.S. Inhibition of αvβ3 integrin impairs adhesion and uptake of tumor-derived small extracellular vesicles. Cell Commun. Signal. 2020, 18, 158. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.S.; Kang, C.W.; Kang, S.; Jang, Y.; Chae, Y.C.; Kim, B.G.; Cho, N.H. ITGB4-mediated metabolic reprogramming of cancer-associated fibroblasts. Oncogene 2020, 39, 664–676. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Gao, W.; Tang, Q.; Yu, Y.; You, W.; Wu, Z.; Fan, Y.; Zhang, L.; Wu, C.; Han, G.; et al. M2 macrophage-derived exosomes facilitate hepatocarcinoma metastasis by transferring αMβ2 integrin to tumor cells. Hepatology 2020, hep.31432. [Google Scholar] [CrossRef]
- Guo, Q.; Furuta, K.; Lucien, F.; Gutierrez Sanchez, L.H.; Hirsova, P.; Krishnan, A.; Kabashima, A.; Pavelko, K.D.; Madden, B.; Alhuwaish, H.; et al. Integrin β1-enriched extracellular vesicles mediate monocyte adhesion and promote liver inflammation in murine NASH. J. Hepatol. 2019, 71, 1193–1205. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.; Wang, C.; Benedict, C.; Huang, G.; Truongcao, M.; Roy, R.; Cimini, M.; Garikipati, V.N.S.; Cheng, Z.; Koch, W.J.; et al. Interleukin-10 Deficiency Alters Endothelial Progenitor Cell–Derived Exosome Reparative Effect on Myocardial Repair via Integrin-Linked Kinase Enrichment. Circ. Res. 2020, 126, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Bertolini, I.; Ghosh, J.C.; Kossenkov, A.V.; Mulugu, S.; Krishn, S.R.; Vaira, V.; Qin, J.; Plow, E.F.; Languino, L.R.; Altieri, D.C. Small Extracellular Vesicle Regulation of Mitochondrial Dynamics Reprograms a Hypoxic Tumor Microenvironment. Dev. Cell 2020, 55, 163–177.e6. [Google Scholar] [CrossRef] [PubMed]
- DeRita, R.M.; Sayeed, A.; Garcia, V.; Krishn, S.R.; Shields, C.D.; Sarker, S.; Friedman, A.; McCue, P.; Molugu, S.K.; Rodeck, U.; et al. Tumor-Derived Extracellular Vesicles Require β1 Integrins to Promote Anchorage-Independent Growth. iScience 2019, 14, 199–209. [Google Scholar] [CrossRef]
- Song, X.; Ding, Y.; Liu, G.; Yang, X.; Zhao, R.; Zhang, Y.; Zhao, X.; Anderson, G.J.; Nie, G. Cancer Cell-derived Exosomes Induce Mitogen-activated Protein Kinase-dependent Monocyte Survival by Transport of Functional Receptor Tyrosine Kinases. J. Biol. Chem. 2016, 291, 8453–8464. [Google Scholar] [CrossRef]
- Itkin, T.; Gur-Cohen, S.; Spencer, J.A.; Schajnovitz, A.; Ramasamy, S.K.; Kusumbe, A.P.; Ledergor, G.; Jung, Y.; Milo, I.; Poulos, M.G.; et al. Distinct bone marrow blood vessels differentially regulate haematopoiesis. Nature 2016, 532, 323–328. [Google Scholar] [CrossRef]
- Li, X.; Kumar, A.; Carmeliet, P. Metabolic Pathways Fueling the Endothelial Cell Drive. Annu. Rev. Physiol. 2019, 81, 483–503. [Google Scholar] [CrossRef] [PubMed]
- Rohlenova, K.; Veys, K.; Miranda-Santos, I.; De Bock, K.; Carmeliet, P. Endothelial Cell Metabolism in Health and Disease. Trends Cell Biol. 2018, 28, 224–236. [Google Scholar] [CrossRef]
- Bibli, S.-I.; Hu, J.; Looso, M.; Weigert, A.; Ratiu, C.; Wittig, J.; Drekolia, M.K.; Tombor, L.; Randriamboavonjy, V.; Leisegang, M.S.; et al. Mapping the Endothelial Cell S -Sulfhydrome Highlights the Crucial Role of Integrin Sulfhydration in Vascular Function. Circulation 2020, CIRCULATIONAHA.120.051877. [Google Scholar] [CrossRef]
- Li, X.; Sun, X.; Carmeliet, P. Hallmarks of Endothelial Cell Metabolism in Health and Disease. Cell Metab. 2019, 30, 414–433. [Google Scholar] [CrossRef]
- Sáez, T.; de Vos, P.; Kuipers, J.; Sobrevia, L.; Faas, M.M. Fetoplacental endothelial exosomes modulate high d-glucose-induced endothelial dysfunction. Placenta 2018, 66, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Garcia, N.A.; Moncayo-Arlandi, J.; Sepulveda, P.; Diez-Juan, A. Cardiomyocyte exosomes regulate glycolytic flux in endothelium by direct transfer of GLUT transporters and glycolytic enzymes. Cardiovasc. Res. 2016, 109, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Xu, C.; Gillette, T.G.; Huang, T.; Huang, P.; Li, Q.; Li, X.; Li, Q.; Ning, Y.; Tang, R.; et al. Cardiomyocyte-derived small extracellular vesicles can signal eNOS activation in cardiac microvascular endothelial cells to protect against Ischemia/Reperfusion injury. Theranostics 2020, 10, 11754–11774. [Google Scholar] [CrossRef]
- Sáez, T.; de Vos, P.; Kuipers, J.; Sobrevia, L.; Faas, M.M. Exosomes derived from monocytes and from endothelial cells mediate monocyte and endothelial cell activation under high d-glucose conditions. Immunobiology 2019, 224, 325–333. [Google Scholar] [CrossRef]
- Gao, W.; Liu, H.; Yuan, J.; Wu, C.; Huang, D.; Ma, Y.; Zhu, J.; Ma, L.; Guo, J.; Shi, H.; et al. Exosomes derived from mature dendritic cells increase endothelial inflammation and atherosclerosis via membrane TNF-α mediated NF-κB pathway. J. Cell. Mol. Med. 2016, 20, 2318–2327. [Google Scholar] [CrossRef]
- Huang, C.; Huang, Y.; Zhou, Y.; Nie, W.; Pu, X.; Xu, X.; Zhu, J. Exosomes derived from oxidized LDL-stimulated macrophages attenuate the growth and tube formation of endothelial cells. Mol. Med. Rep. 2018. [Google Scholar] [CrossRef]
- Dumas, S.J.; García-Caballero, M.; Carmeliet, P. Metabolic Signatures of Distinct Endothelial Phenotypes. Trends Endocrinol. Metab. 2020, 31, 580–595. [Google Scholar] [CrossRef]
- Zhu, D.; Johnson, T.K.; Wang, Y.; Thomas, M.; Huynh, K.; Yang, Q.; Bond, V.C.; Chen, Y.E.; Liu, D. Macrophage M2 polarization induced by exosomes from adipose-derived stem cells contributes to the exosomal proangiogenic effect on mouse ischemic hindlimb. Stem Cell Res. Ther. 2020, 11, 162. [Google Scholar] [CrossRef]
- An, Y.; Zhao, J.; Nie, F.; Qin, Z.; Xue, H.; Wang, G.; Li, D. Exosomes from Adipose-Derived Stem Cells (ADSCs) Overexpressing miR-21 Promote Vascularization of Endothelial Cells. Sci. Rep. 2019, 9, 12861. [Google Scholar] [CrossRef]
- Yang, E.; Wang, X.; Gong, Z.; Yu, M.; Wu, H.; Zhang, D. Exosome-mediated metabolic reprogramming: The emerging role in tumor microenvironment remodeling and its influence on cancer progression. Signal Transduct. Target. Ther. 2020, 5, 242. [Google Scholar] [CrossRef] [PubMed]
- Fong, M.Y.; Zhou, W.; Liu, L.; Alontaga, A.Y.; Chandra, M.; Ashby, J.; Chow, A.; O’Connor, S.T.F.; Li, S.; Chin, A.R.; et al. Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat. Cell Biol. 2015, 17, 183–194. [Google Scholar] [CrossRef]
- Zhao, H.; Yang, L.; Baddour, J.; Achreja, A.; Bernard, V.; Moss, T.; Marini, J.C.; Tudawe, T.; Seviour, E.G.; San Lucas, F.A.; et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. Elife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Harjes, U.; Bensaad, K.; Harris, A.L. Endothelial cell metabolism and implications for cancer therapy. Br. J. Cancer 2012, 107, 1207–1212. [Google Scholar] [CrossRef] [PubMed]
- Cantelmo, A.R.; Conradi, L.-C.; Brajic, A.; Goveia, J.; Kalucka, J.; Pircher, A.; Chaturvedi, P.; Hol, J.; Thienpont, B.; Teuwen, L.-A.; et al. Inhibition of the Glycolytic Activator PFKFB3 in Endothelium Induces Tumor Vessel Normalization, Impairs Metastasis, and Improves Chemotherapy. Cancer Cell 2016, 30, 968–985. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wang, X.; Hou, D.; Huang, Q.; Zhan, W.; Chen, C.; Liu, J.; You, R.; Xie, J.; Chen, P.; et al. Exosomes derived from acute myeloid leukemia cells promote chemoresistance by enhancing glycolysis-mediated vascular remodeling. J. Cell. Physiol. 2019, 234, 10602–10614. [Google Scholar] [CrossRef]
- Ko, S.Y.; Lee, W.; Kenny, H.A.; Dang, L.H.; Ellis, L.M.; Jonasch, E.; Lengyel, E.; Naora, H. Cancer-derived small extracellular vesicles promote angiogenesis by heparin-bound, bevacizumab-insensitive VEGF, independent of vesicle uptake. Commun. Biol. 2019, 2, 386. [Google Scholar] [CrossRef]
- Zeng, Z.; Li, Y.; Pan, Y.; Lan, X.; Song, F.; Sun, J.; Zhou, K.; Liu, X.; Ren, X.; Wang, F.; et al. Cancer-derived exosomal miR-25-3p promotes pre-metastatic niche formation by inducing vascular permeability and angiogenesis. Nat. Commun. 2018, 9, 5395. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Fong, M.Y.; Min, Y.; Somlo, G.; Liu, L.; Palomares, M.R.; Yu, Y.; Chow, A.; O’Connor, S.T.F.; Chin, A.R.; et al. Cancer-Secreted miR-105 Destroys Vascular Endothelial Barriers to Promote Metastasis. Cancer Cell 2014, 25, 501–515. [Google Scholar] [CrossRef]
- Hsu, Y.-L.; Hung, J.-Y.; Chang, W.-A.; Lin, Y.-S.; Pan, Y.-C.; Tsai, P.-H.; Wu, C.-Y.; Kuo, P.-L. Hypoxic lung cancer-secreted exosomal miR-23a increased angiogenesis and vascular permeability by targeting prolyl hydroxylase and tight junction protein ZO-1. Oncogene 2017, 36, 4929–4942. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Sohn, Y.J.; Lee, R.; Yoo, H.J.; Kang, J.Y.; Choi, N.; Na, D.; Yeon, J.H. Cancer-Associated Fibroblasts Differentiated by Exosomes Isolated from Cancer Cells Promote Cancer Cell Invasion. Int. J. Mol. Sci. 2020, 21, 8153. [Google Scholar] [CrossRef] [PubMed]
- Yeon, J.H.; Jeong, H.E.; Seo, H.; Cho, S.; Kim, K.; Na, D.; Chung, S.; Park, J.; Choi, N.; Kang, J.Y. Cancer-derived exosomes trigger endothelial to mesenchymal transition followed by the induction of cancer-associated fibroblasts. Acta Biomater. 2018, 76, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Guo, Z.; Chen, W.; Wang, X.; Cao, M.; Han, X.; Zhang, K.; Teng, B.; Cao, J.; Wu, W.; et al. M2 Macrophage-Derived Exosomes Promote Angiogenesis and Growth of Pancreatic Ductal Adenocarcinoma by Targeting E2F2. Mol. Ther. 2020. [Google Scholar] [CrossRef] [PubMed]
- Nakamori, Y.; Park, E.J.; Shimaoka, M. Immune Deregulation in Sepsis and Septic Shock: Reversing Immune Paralysis by Targeting PD-1/PD-L1 Pathway. Front. Immunol. 2021, 11, 624279. [Google Scholar] [CrossRef]
- Poggio, M.; Hu, T.; Pai, C.-C.; Chu, B.; Belair, C.D.; Chang, A.; Montabana, E.; Lang, U.E.; Fu, Q.; Fong, L.; et al. Suppression of Exosomal PD-L1 Induces Systemic Anti-tumor Immunity and Memory. Cell 2019, 177, 414–427.e13. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H.; et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018, 560, 382–386. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soe, Z.Y.; Park, E.J.; Shimaoka, M. Integrin Regulation in Immunological and Cancerous Cells and Exosomes. Int. J. Mol. Sci. 2021, 22, 2193. https://doi.org/10.3390/ijms22042193
Soe ZY, Park EJ, Shimaoka M. Integrin Regulation in Immunological and Cancerous Cells and Exosomes. International Journal of Molecular Sciences. 2021; 22(4):2193. https://doi.org/10.3390/ijms22042193
Chicago/Turabian StyleSoe, Zay Yar, Eun Jeong Park, and Motomu Shimaoka. 2021. "Integrin Regulation in Immunological and Cancerous Cells and Exosomes" International Journal of Molecular Sciences 22, no. 4: 2193. https://doi.org/10.3390/ijms22042193
APA StyleSoe, Z. Y., Park, E. J., & Shimaoka, M. (2021). Integrin Regulation in Immunological and Cancerous Cells and Exosomes. International Journal of Molecular Sciences, 22(4), 2193. https://doi.org/10.3390/ijms22042193