Development of a Genome-Scale Metabolic Model and Phenome Analysis of the Probiotic Escherichia coli Strain Nissle 1917


Abstract
1. Introduction
2. Results
2.1. Reconstruction of the Draft Metabolic Model
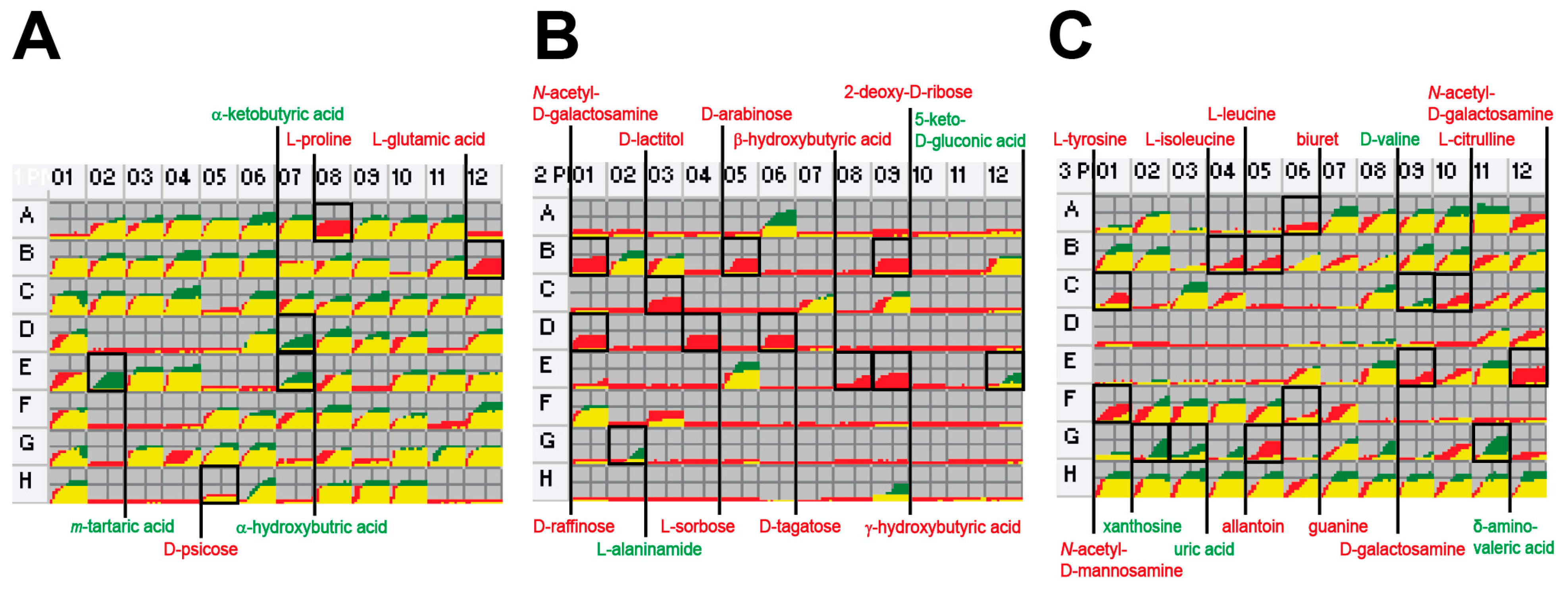
2.2. Phenome Analysis
2.3. Gene Clusters Responsible for the Phenotypic Differences
2.4. Revision and Validation of the Metabolic Model
2.5. Gene Essentiality Analysis
2.6. Simulation of Utilization of Carbon Sources under Anaerobic Intestinal Environments
3. Discussion
4. Materials and Methods
4.1. Identification of Metabolic Genes and Reactions
4.2. PM Test
4.3. FBA
4.4. DNA Sequencing
4.5. Availability of Data and Materials (Nucleotide Sequence Accession Numbers)
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AKG | α-ketoglutarate |
| EcN | Escherichia coli Nissle 1917 |
| ED | Entner–Doudoroff |
| EMP | Embden–Meyerhof–Parnas |
| FBA | Flux balance analysis |
| L-LDH | L-lactate dehydrogenase |
| LPS | Lipopolysaccharide |
| MSP | Methionine salvage pathway |
| PM | Phenotype microarray |
| PTS | Phosphotransferase system |
References
- Wassenaar, T.M. Insights from 100 years of research with probiotic E. coli. Eur. J. Microbiol. Immunol. 2016, 6, 147–161. [Google Scholar] [CrossRef]
- Sonnenborn, U. Escherichia coli strain Nissle 1917–from bench to bedside and back: History of a special Escherichia coli strain with probiotic properties. FEMS Microbiol. Lett. 2016, 363, fnw212. [Google Scholar] [CrossRef]
- Aggarwal, N.; Breedon, A.M.E.; Davis, C.M.; Hwang, I.Y.; Chang, M.W. Engineering probiotics for therapeutic applications: Recent examples and translational outlook. Curr. Opin. Biotechnol. 2020, 65, 171–179. [Google Scholar] [CrossRef]
- Choi, H.J.; Ahn, J.H.; Park, S.H.; Do, K.H.; Kim, J.; Moon, Y. Enhanced wound healing by recombinant Escherichia coli Nissle 1917 via human epidermal growth factor receptor in human intestinal epithelial cells: Therapeutic implication using recombinant probiotics. Infect. Immun. 2012, 80, 1079–1087. [Google Scholar] [CrossRef]
- Hwang, I.Y.; Koh, E.; Wong, A.; March, J.C.; Bentley, W.E.; Lee, Y.S.; Chang, M.W. Engineered probiotic Escherichia coli can eliminate and prevent Pseudomonas aeruginosa gut infection in animal models. Nat. Commun. 2017, 8, 15028. [Google Scholar] [CrossRef]
- Gu, C.; Kim, G.B.; Kim, W.J.; Kim, H.U.; Lee, S.Y. Current status and applications of genome-scale metabolic models. Genome Biol. 2019, 20, 121. [Google Scholar] [CrossRef]
- Li, R.; Helbig, L.; Fu, J.; Bian, X.; Herrmann, J.; Baumann, M.; Stewart, A.F.; Müller, R.; Li, A.; Zips, D.; et al. Expressing cytotoxic compounds in Escherichia coli Nissle 1917 for tumor-targeting therapy. Res. Microbiol. 2019, 170, 74–79. [Google Scholar] [CrossRef]
- Ho, C.L.; Tan, H.Q.; Chua, K.J.; Kang, A.; Lim, K.H.; Ling, K.L.; Yew, W.S.; Lee, Y.S.; Thiery, J.P.; Chang, M.W. Engineered commensal microbes for diet-mediated colorectal-cancer chemoprevention. Nat. Biomed. Eng. 2018, 2, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Isabella, V.M.; Ha, B.N.; Castillo, M.J.; Lubkowicz, D.J.; Rowe, S.E.; Millet, Y.A.; Anderson, C.L.; Li, N.; Fisher, A.B.; West, K.A.; et al. Development of a synthetic live bacterial therapeutic for the human metabolic disease phenylketonuria. Nat. Biotechnol. 2018, 36, 857–864. [Google Scholar] [CrossRef]
- Kurtz, C.B.; Millet, Y.A.; Puurunen, M.K.; Perreault, M.; Charbonneau, M.R.; Isabella, V.M.; Kotula, J.W.; Antipov, E.; Dagon, Y.; Denney, W.S.; et al. An engineered E. coli Nissle improves hyperammonemia and survival in mice and shows dose-dependent exposure in healthy humans. Sci. Transl. Med. 2019, 11, eaau7975. [Google Scholar] [CrossRef]
- Orth, J.D.; Thiele, I.; Palsson, B.O. What is flux balance analysis? Nat. Biotechnol. 2010, 28, 245–248. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, E.J.; Monk, J.M.; Palsson, B.O. Using genome-scale models to predict biological capabilities. Cell 2015, 161, 971–987. [Google Scholar] [CrossRef]
- van der Ark, K.C.H.; van Heck, R.G.A.; Martins Dos Santos, V.A.P.; Belzer, C.; de Vos, W.M. More than just a gut feeling: Constraint-based genome-scale metabolic models for predicting functions of human intestinal microbes. Microbiome 2017, 5, 78. [Google Scholar] [CrossRef]
- Monk, J.M.; Charusanti, P.; Aziz, R.K.; Lerman, J.A.; Premyodhin, N.; Orth, J.D.; Feist, A.M.; Palsson, B.O. Genome-scale metabolic reconstructions of multiple Escherichia coli strains highlight strain-specific adaptations to nutritional environments. Proc. Natl. Acad. Sci. USA 2013, 110, 20338–20343. [Google Scholar] [CrossRef] [PubMed]
- Monk, J.M.; Lloyd, C.J.; Brunk, E.; Mih, N.; Sastry, A.; King, Z.; Takeuchi, R.; Nomura, W.; Zhang, Z.; Mori, H.; et al. iML1515, a knowledgebase that computes Escherichia coli traits. Nat. Biotechnol. 2017, 35, 904–908. [Google Scholar] [CrossRef]
- Kim, H.; Kim, S.; Yoon, S.H. Metabolic network reconstruction and phenome analysis of the industrial microbe, Escherichia coli BL21(DE3). PLoS ONE 2018, 13, e0204375. [Google Scholar] [CrossRef] [PubMed]
- Magnúsdóttir, S.; Heinken, A.; Kutt, L.; Ravcheev, D.A.; Bauer, E.; Noronha, A.; Greenhalgh, K.; Jäger, C.; Baginska, J.; Wilmes, P.; et al. Generation of genome-scale metabolic reconstructions for 773 members of the human gut microbiota. Nat. Biotechnol. 2017, 35, 81–89. [Google Scholar] [CrossRef]
- Reister, M.; Hoffmeier, K.; Krezdorn, N.; Rotter, B.; Liang, C.; Rund, S.; Dandekar, T.; Sonnenborn, U.; Oelschlaeger, T.A. Complete genome sequence of the Gram-negative probiotic Escherichia coli strain Nissle 1917. J. Biotechnol. 2014, 187, 106–107. [Google Scholar] [CrossRef]
- Hayashi, K.; Morooka, N.; Yamamoto, Y.; Fujita, K.; Isono, K.; Choi, S.; Ohtsubo, E.; Baba, T.; Wanner, B.L.; Mori, H.; et al. Highly accurate genome sequences of Escherichia coli K-12 strains MG1655 and W3110. Mol. Syst. Biol. 2006, 2, 2006.0007. [Google Scholar] [CrossRef]
- Welch, R.A.; Burland, V.; Plunkett, G., III; Redford, P.; Roesch, P.; Rasko, D.; Buckles, E.L.; Liou, S.R.; Boutin, A.; Hackett, J.; et al. Extensive mosaic structure revealed by the complete genome sequence of uropathogenic Escherichia coli. Proc. Natl. Acad. Sci. USA 2002, 99, 17020–17024. [Google Scholar] [CrossRef]
- Zdziarski, J.; Brzuszkiewicz, E.; Wullt, B.; Liesegang, H.; Biran, D.; Voigt, B.; Grönberg-Hernandez, J.; Ragnarsdottir, B.; Hecker, M.; Ron, E.Z.; et al. Host imprints on bacterial genomes–rapid, divergent evolution in individual patients. PLoS Pathog. 2010, 6, e1001078. [Google Scholar] [CrossRef] [PubMed]
- Caspi, R.; Billington, R.; Ferrer, L.; Foerster, H.; Fulcher, C.A.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Mueller, L.A.; et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2016, 44, D471–D480. [Google Scholar] [CrossRef] [PubMed]
- Bochner, B.R.; Gadzinski, P.; Panomitros, E. Phenotype microarrays for high-throughput phenotypic testing and assay of gene function. Genome Res. 2001, 11, 1246–1255. [Google Scholar] [CrossRef]
- Yoon, S.H.; Han, M.J.; Jeong, H.; Lee, C.H.; Xia, X.X.; Lee, D.H.; Shim, J.H.; Lee, S.Y.; Oh, T.K.; Kim, J.F. Comparative multi-omics systems analysis of Escherichia coli strains B and K-12. Genome Biol. 2012, 13, R37. [Google Scholar] [CrossRef] [PubMed]
- Lawther, R.P.; Calhoun, D.H.; Adams, C.W.; Hauser, C.A.; Gray, J.; Hatfield, G.W. Molecular basis of valine resistance in Escherichia coli K-12. Proc. Natl. Acad. Sci. USA 1981, 78, 922–925. [Google Scholar] [CrossRef]
- Brinkkötter, A.; Klöss, H.; Alpert, C.; Lengeler, J.W. Pathways for the utilization of N-acetyl-galactosamine and galactosamine in Escherichia coli. Mol. Microbiol. 2000, 37, 125–135. [Google Scholar] [CrossRef]
- Christensen, M.; Borza, T.; Dandanell, G.; Gilles, A.-M.; Barzu, O.; Kelln, R.A.; Neuhard, J. Regulation of expression of the 2-deoxy-d-ribose utilization regulon, deoQKPX, from Salmonella enterica serovar typhimurium. J. Bacteriol. 2003, 185, 6042–6050. [Google Scholar] [CrossRef]
- Bryant, W.A.; Krabben, P.; Baganz, F.; Zhou, Y.; Ward, J.M. The analysis of multiple genome comparisons in genus Escherichia and its application to the discovery of uncharacterised metabolic genes in uropathogenic Escherichia coli CFT073. Comp. Funct. Genom. 2009, 2009, 782924. [Google Scholar] [CrossRef] [PubMed]
- Bausch, C.; Ramsey, M.; Conway, T. Transcriptional organization and regulation of the L-idonic acid pathway (GntII system) in Escherichia coli. J. Bacteriol. 2004, 186, 1388–1397. [Google Scholar] [CrossRef] [PubMed]
- Grozdanov, L.; Zähringer, U.; Blum-Oehler, G.; Brade, L.; Henne, A.; Knirel, Y.A.; Schombel, U.; Schulze, J.; Sonnenborn, U.; Gottschalk, G.; et al. A single nucleotide exchange in the wzy gene is responsible for the semirough O6 lipopolysaccharide phenotype and serum sensitivity of Escherichia coli strain Nissle 1917. J. Bacteriol. 2002, 184, 5912–5925. [Google Scholar] [CrossRef]
- Brzuszkiewicz, E.; Brüggemann, H.; Liesegang, H.; Emmerth, M.; Olschläger, T.; Nagy, G.; Albermann, K.; Wagner, C.; Buchrieser, C.; Emody, L.; et al. How to become a uropathogen: Comparative genomic analysis of extraintestinal pathogenic Escherichia coli strains. Proc. Natl. Acad. Sci. USA 2006, 103, 12879–12884. [Google Scholar] [CrossRef]
- Liu, D.; Reeves, P.R. Escherichia coli K12 regains its O antigen. Microbiology 1994, 140, 49–57. [Google Scholar] [CrossRef]
- Sekowska, A.; Ashida, H.; Danchin, A. Revisiting the methionine salvage pathway and its paralogues. Microb. Biotechnol. 2019, 12, 77–97. [Google Scholar] [CrossRef] [PubMed]
- Simoni, R.D.; Roseman, S.; Saier, M.H., Jr. Sugar transport. Properties of mutant bacteria defective in proteins of the phosphoenolpyruvate: Sugar phosphotransferase system. J. Biol. Chem. 1976, 251, 6584–6597. [Google Scholar] [CrossRef]
- Park, M.S.; Hill, C.M.; Li, Y.; Hardy, R.K.; Khanna, H.; Khang, Y.H.; Raushel, F.M. Catalytic properties of the PepQ prolidase from Escherichia coli. Arch. Biochem. Biophys. 2004, 429, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 2021, 49, D545–D551. [Google Scholar] [CrossRef]
- Chen, Y.M.; Lin, E.C.; Ros, J.; Aguilar, J. Use of operon fusions to examine the regulation of the L-1,2-propanediol oxidoreductase gene of the fucose system in Escherichia coli K12. J. Gen. Microbiol. 1983, 129, 3355–3362. [Google Scholar] [CrossRef]
- Kim, O.B.; Lux, S.; Unden, G. Anaerobic growth of Escherichia coli on D-tartrate depends on the fumarate carrier DcuB and fumarase, rather than the L-tartrate carrier TtdT and L-tartrate dehydratase. Arch. Microbiol. 2007, 188, 583–589. [Google Scholar] [CrossRef]
- Kim, O.B.; Reimann, J.; Lukas, H.; Schumacher, U.; Grimpo, J.; Dünnwald, P.; Unden, G. Regulation of tartrate metabolism by TtdR and relation to the DcuS-DcuR-regulated C4-dicarboxylate metabolism of Escherichia coli. Microbiology 2009, 155, 3632–3640. [Google Scholar] [CrossRef]
- Fang, Y.; Kolmakova-Partensky, L.; Miller, C. A bacterial arginine-agmatine exchange transporter involved in extreme acid resistance. J. Biol. Chem. 2007, 282, 176–182. [Google Scholar] [CrossRef]
- Vinuselvi, P.; Lee, S.K. Engineering Escherichia coli for efficient cellobiose utilization. Appl. Microbiol. Biotechnol. 2011, 92, 125–132. [Google Scholar] [CrossRef]
- Kashiwagi, K.; Shibuya, S.; Tomitori, H.; Kuraishi, A.; Igarashi, K. Excretion and uptake of putrescine by the PotE protein in Escherichia coli. J. Biol. Chem. 1997, 272, 6318–6323. [Google Scholar] [CrossRef]
- Lieven, C.; Beber, M.E.; Olivier, B.G.; Bergmann, F.T.; Ataman, M.; Babaei, P.; Bartell, J.A.; Blank, L.M.; Chauhan, S.; Correia, K.; et al. MEMOTE for standardized genome-scale metabolic model testing. Nat. Biotechnol. 2020, 38, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Schellenberger, J.; Que, R.; Fleming, R.M.; Thiele, I.; Orth, J.D.; Feist, A.M.; Zielinski, D.C.; Bordbar, A.; Lewis, N.E.; Rahmanian, S.; et al. Quantitative prediction of cellular metabolism with constraint-based models: The COBRA Toolbox v2.0. Nat. Protoc. 2011, 6, 1290–1307. [Google Scholar] [CrossRef] [PubMed]
- Ebrahim, A.; Lerman, J.A.; Palsson, B.O.; Hyduke, D.R. COBRApy: COnstraints-Based Reconstruction and Analysis for Python. BMC Syst. Biol. 2013, 7, 74. [Google Scholar] [CrossRef] [PubMed]
- Legrain, C.; Halleux, P.; Stalon, V.; Glansdorff, N. The dual genetic control of ornithine carbamolytransferase in Escherichia coli. A case of bacterial hybrid enzymes. Eur. J. Biochem. 1972, 27, 93–102. [Google Scholar] [CrossRef]
- Merlin, C.; Masters, M.; McAteer, S.; Coulson, A. Why is carbonic anhydrase essential to Escherichia coli? J. Bacteriol. 2003, 185, 6415–6424. [Google Scholar] [CrossRef] [PubMed]
- Vorobieva, A.A.; Khan, M.S.; Soumillion, P. Escherichia coli D-malate dehydrogenase, a generalist enzyme active in the leucine biosynthesis pathway. J. Biol. Chem. 2014, 289, 29086–29096. [Google Scholar] [CrossRef]
- Smith, J.M.; Daum, H.A., III. Identification and nucleotide sequence of a gene encoding 5′-phosphoribosylglycinamide transformylase in Escherichia coli K12. J. Biol. Chem. 1987, 262, 10565–10569. [Google Scholar] [CrossRef]
- Nygaard, P.; Smith, J.M. Evidence for a novel glycinamide ribonucleotide transformylase in Escherichia coli. J. Bacteriol. 1993, 175, 3591–3597. [Google Scholar] [CrossRef] [PubMed]
- Allen, A.; Cunliffe, W.J.; Pearson, J.P.; Sellers, L.A.; Ward, R. Studies on gastrointestinal mucus. Scand. J. Gastroenterol. Suppl. 1984, 93, 101–113. [Google Scholar] [PubMed]
- Fabich, A.J.; Jones, S.A.; Chowdhury, F.Z.; Cernosek, A.; Anderson, A.; Smalley, D.; McHargue, J.W.; Hightower, G.A.; Smith, J.T.; Autieri, S.M.; et al. Comparison of carbon nutrition for pathogenic and commensal Escherichia coli strains in the mouse intestine. Infect. Immun. 2008, 76, 1143–1152. [Google Scholar] [CrossRef]
- Chang, D.E.; Smalley, D.J.; Tucker, D.L.; Leatham, M.P.; Norris, W.E.; Stevenson, S.J.; Anderson, A.B.; Grissom, J.E.; Laux, D.C.; Cohen, P.S.; et al. Carbon nutrition of Escherichia coli in the mouse intestine. Proc. Natl. Acad. Sci. USA 2004, 101, 7427–7432. [Google Scholar] [CrossRef] [PubMed]
- Grubmüller, S.; Schauer, K.; Goebel, W.; Fuchs, T.M.; Eisenreich, W. Analysis of carbon substrates used by Listeria monocytogenes during growth in J774A.1 macrophages suggests a bipartite intracellular metabolism. Front. Cell. Infect. Microbiol. 2014, 4, 156. [Google Scholar] [PubMed]
- Maltby, R.; Leatham-Jensen, M.P.; Gibson, T.; Cohen, P.S.; Conway, T. Nutritional basis for colonization resistance by human commensal Escherichia coli strains HS and Nissle 1917 against E. coli O157:H7 in the mouse intestine. PLoS ONE 2013, 8, e53957. [Google Scholar] [CrossRef] [PubMed]
- McDowall, J.S.; Hjersing, M.C.; Palmer, T.; Sargent, F. Dissection and engineering of the Escherichia coli formate hydrogenlyase complex. FEBS Lett. 2015, 589, 3141–3147. [Google Scholar] [CrossRef]
- Quentmeier, A.; Holzenburg, A.; Mayer, F.; Antranikian, G. Reevaluation of citrate lyase from Escherichia coli. Biochim. Biophys. Acta 1987, 913, 60–65. [Google Scholar] [CrossRef]
- Knappe, J.; Sawers, G. A radical-chemical route to acetyl-CoA: The anaerobically induced pyruvate formate-lyase system of Escherichia coli. FEMS Microbiol. Rev. 1990, 6, 383–398. [Google Scholar]
- Freddolino, P.L.; Goodarzi, H.; Tavazoie, S. Revealing the genetic basis of natural bacterial phenotypic divergence. J. Bacteriol. 2014, 196, 825–839. [Google Scholar] [CrossRef] [PubMed]
- Miranda, R.L.; Conway, T.; Leatham, M.P.; Chang, D.E.; Norris, W.E.; Allen, J.H.; Stevenson, S.J.; Laux, D.C.; Cohen, P.S. Glycolytic and gluconeogenic growth of Escherichia coli O157:H7 (EDL933) and E. coli K-12 (MG1655) in the mouse intestine. Infect. Immun. 2004, 72, 1666–1676. [Google Scholar] [CrossRef]
- Conway, T.; Cohen, P.S. Commensal and pathogenic Escherichia coli metabolism in the gut. Microbiol. Spectr. 2015, 3. [Google Scholar] [CrossRef]
- Hoskins, L.C.; Agustines, M.; McKee, W.B.; Boulding, E.T.; Kriaris, M.; Niedermeyer, G. Mucin degradation in human colon ecosystems. Isolation and properties of fecal strains that degrade ABH blood group antigens and oligosaccharides from mucin glycoproteins. J. Clin. Investig. 1985, 75, 944–953. [Google Scholar] [CrossRef]
- Martinez-Jéhanne, V.; du Merle, L.; Bernier-Fébreau, C.; Usein, C.; Gassama-Sow, A.; Wane, A.A.; Gouali, M.; Damian, M.; Aïdara-Kane, A.; Germani, Y.; et al. Role of deoxyribose catabolism in colonization of the murine intestine by pathogenic Escherichia coli strains. Infect. Immun. 2009, 77, 1442–1450. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Wannemuehler, Y.; Dell’anna, G.; Nicholson, B.; Barbieri, N.L.; Kariyawasam, S.; Feng, Y.; Logue, C.M.; Nolan, L.K.; Li, G. A novel two-component signaling system facilitates uropathogenic Escherichia coli’s ability to exploit abundant host metabolites. PLoS Pathog. 2013, 9, e1003428. [Google Scholar] [CrossRef]
- Martin, M.; Ferrier, B.; Baverel, G. Transport and utilization of a-ketoglutarate by the rat kidney in vivo. Pflugers Arch. 1989, 413, 217–224. [Google Scholar] [CrossRef]
- Iuchi, S.; Lin, E.C. arcA (dye), a global regulatory gene in Escherichia coli mediating repression of enzymes in aerobic pathways. Proc. Natl. Acad. Sci. USA 1988, 85, 1888–1892. [Google Scholar] [CrossRef]
- Neu, J.; Shenoy, V.; Chakrabarti, R. Glutamine nutrition and metabolism: Where do we go from here? FASEB J. 1996, 10, 829–837. [Google Scholar] [CrossRef]
- Filip, R.; Pierzynowski, S. The role of glutamine and α-ketoglutarate in gut metabolism and the potential application in medicine and nutrition. J. Pre-Clin. Clin. Res. 2007, 1, 9–15. [Google Scholar]
- Billig, S.; Schneefeld, M.; Huber, C.; Grassl, G.A.; Eisenreich, W.; Bange, F.C. Lactate oxidation facilitates growth of Mycobacterium tuberculosis in human macrophages. Sci. Rep. 2017, 7, 6484. [Google Scholar] [CrossRef] [PubMed]
- Iuchi, S.; Aristarkhov, A.; Dong, J.M.; Taylor, J.S.; Lin, E.C. Effects of nitrate respiration on expression of the Arc-controlled operons encoding succinate dehydrogenase and flavin-linked L-lactate dehydrogenase. J. Bacteriol. 1994, 176, 1695–1701. [Google Scholar] [CrossRef]
- Peekhaus, N.; Conway, T. What’s for dinner?: Entner-Doudoroff metabolism in Escherichia coli. J. Bacteriol. 1998, 180, 3495–3502. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Gunzer, F.; Westendorf, A.M.; Buer, J.; Scharfe, M.; Jarek, M.; Gössling, F.; Blöcker, H.; Zeng, A.P. Genomic peculiarity of coding sequences and metabolic potential of probiotic Escherichia coli strain Nissle 1917 inferred from raw genome data. J. Biotechnol. 2005, 117, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Best, A.; Abu Kwaik, Y. Nutrition and bipartite metabolism of intracellular pathogens. Trends Microbiol. 2019, 27, 550–561. [Google Scholar] [CrossRef] [PubMed]
- Revelles, O.; Millard, P.; Nougayrède, J.P.; Dobrindt, U.; Oswald, E.; Létisse, F.; Portais, J.C. The carbon storage regulator (Csr) system exerts a nutrient-specific control over central metabolism in Escherichia coli strain Nissle 1917. PLoS ONE 2013, 8, e66386. [Google Scholar] [CrossRef] [PubMed]
- Behnsen, J.; Deriu, E.; Sassone-Corsi, M.; Raffatellu, M. Probiotics: Properties, examples, and specific applications. Cold Spring Harb. Perspect. Med. 2013, 3, a010074. [Google Scholar] [CrossRef] [PubMed]
- Sonnenborn, U.; Schulze, J. The non-pathogenic Escherichia coli strain Nissle 1917—Features of a versatile probiotic. Microb. Ecol. Health Dis. 2009, 21, 122–158. [Google Scholar]
- Blom, J.; Kreis, J.; Spänig, S.; Juhre, T.; Bertelli, C.; Ernst, C.; Goesmann, A. EDGAR 2.0: An enhanced software platform for comparative gene content analyses. Nucleic Acids Res. 2016, 44, W22–W28. [Google Scholar] [CrossRef] [PubMed]
- King, Z.A.; Lu, J.; Drager, A.; Miller, P.; Federowicz, S.; Lerman, J.A.; Ebrahim, A.; Palsson, B.O.; Lewis, N.E. BiGG models: A platform for integrating, standardizing and sharing genome-scale models. Nucleic Acids Res. 2016, 44, D515–D522. [Google Scholar] [CrossRef]
- Wang, H.; Marcišauskas, S.; Sánchez, B.J.; Domenzain, I.; Hermansson, D.; Agren, R.; Nielsen, J.; Kerkhoven, E.J. RAVEN 2.0: A versatile toolbox for metabolic network reconstruction and a case study on Streptomyces coelicolor. PLoS Comput. Biol. 2018, 14, e1006541. [Google Scholar] [CrossRef]
- Vaas, L.A.; Sikorski, J.; Hofner, B.; Fiebig, A.; Buddruhs, N.; Klenk, H.P.; Goker, M. opm: An R package for analysing OmniLog® phenotype microarray data. Bioinformatics 2013, 29, 1823–1824. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrates | PM a | Prediction b (Draft/iDK1463) | Associated Reactions c | Modification |
|---|---|---|---|---|
| Carbon Sources | ||||
| acetoacetic acid | - | +/− | ACACtex | Deletion |
| γ-amino-N-butyric acid | - | +/− | ABUTtex | Deletion |
| butyric acid | - | +/− | BUTt2rpp | Deletion |
| D-cellobiose | - | +/− | CELLBpts | Deactivated |
| 1,2-propanediol | - | +/− | LCARS | Deactivated |
| L-ornithine | - | +/− | PTRCORNt7pp | Deactivated |
| L-sorbose | + | −/+ | EX_srb__L_e, SRBtex, SRBptspp, SRB1PR | Addition |
| PTS sugars d | + | −/+ | ACGAptspp, ACMANAptspp, FRUptspp, SBTptspp, MNLptspp, MANptspp, GAMptspp | Addition |
| D-tartaric acid | - | +/− | DTARTD, SUCTARTtpp | Deactivated |
| L-tartaric acid | - | +/− | TARTD, TARTRt7pp | Deactivated |
| Nitrogen Sources | ||||
| ethanolamine | - | +/− | ETHAtex | Deletion |
| inosine | - | +/− | URIC | Deletion |
| uric acid | - | +/− | URIC | Deletion |
| xanthine | - | +/− | XANtex | Deletion |
| Metabolic Model | iDK1463 |
|---|---|
| Genes | 1463 |
| Metabolic reactions | 2984 |
| Enzymatic reactions | 1633 |
| Transport reactions | 903 |
| Exchange reactions | 441 |
| Demand reactions | 7 |
| Gene-reaction association | 2986 |
| Gene-associated reactions | 2368 |
| Not gene-associated reactions | 578 |
| Spontaneous reactions | 40 |
| Metabolites | 2112 |
| Cytoplasmic | 1160 |
| Periplasmic | 509 |
| Extracellular | 443 |
| Unique metabolites | 1313 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, D.; Kim, Y.; Yoon, S.H. Development of a Genome-Scale Metabolic Model and Phenome Analysis of the Probiotic Escherichia coli Strain Nissle 1917. Int. J. Mol. Sci. 2021, 22, 2122. https://doi.org/10.3390/ijms22042122
Kim D, Kim Y, Yoon SH. Development of a Genome-Scale Metabolic Model and Phenome Analysis of the Probiotic Escherichia coli Strain Nissle 1917. International Journal of Molecular Sciences. 2021; 22(4):2122. https://doi.org/10.3390/ijms22042122
Chicago/Turabian StyleKim, Dohyeon, Youngshin Kim, and Sung Ho Yoon. 2021. "Development of a Genome-Scale Metabolic Model and Phenome Analysis of the Probiotic Escherichia coli Strain Nissle 1917" International Journal of Molecular Sciences 22, no. 4: 2122. https://doi.org/10.3390/ijms22042122
APA StyleKim, D., Kim, Y., & Yoon, S. H. (2021). Development of a Genome-Scale Metabolic Model and Phenome Analysis of the Probiotic Escherichia coli Strain Nissle 1917. International Journal of Molecular Sciences, 22(4), 2122. https://doi.org/10.3390/ijms22042122