
A New Intronic Variant in ECEL1 in Two Patients with Distal Arthrogryposis Type 5D

 , and
, and
Abstract
1. Introduction
2. Results
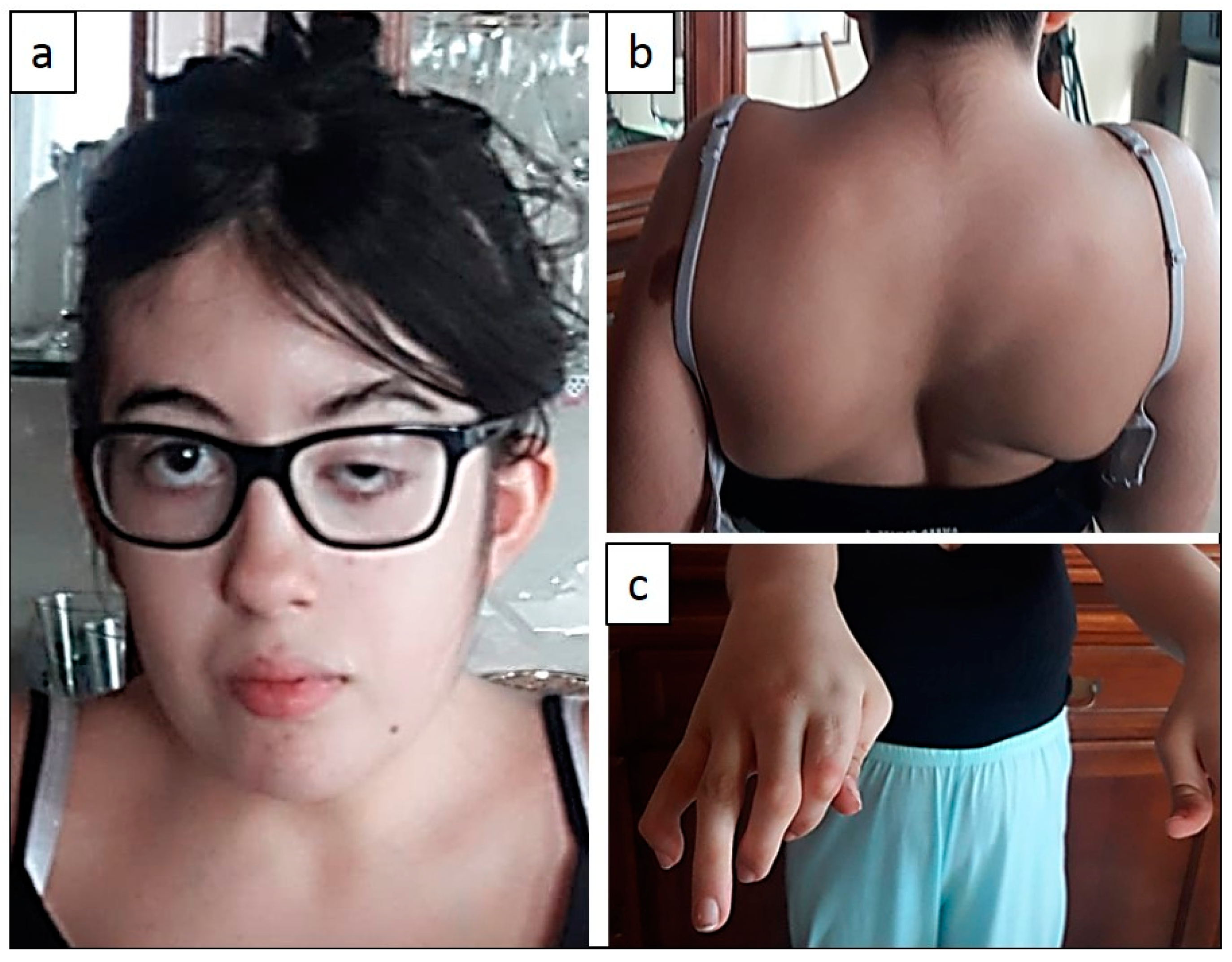
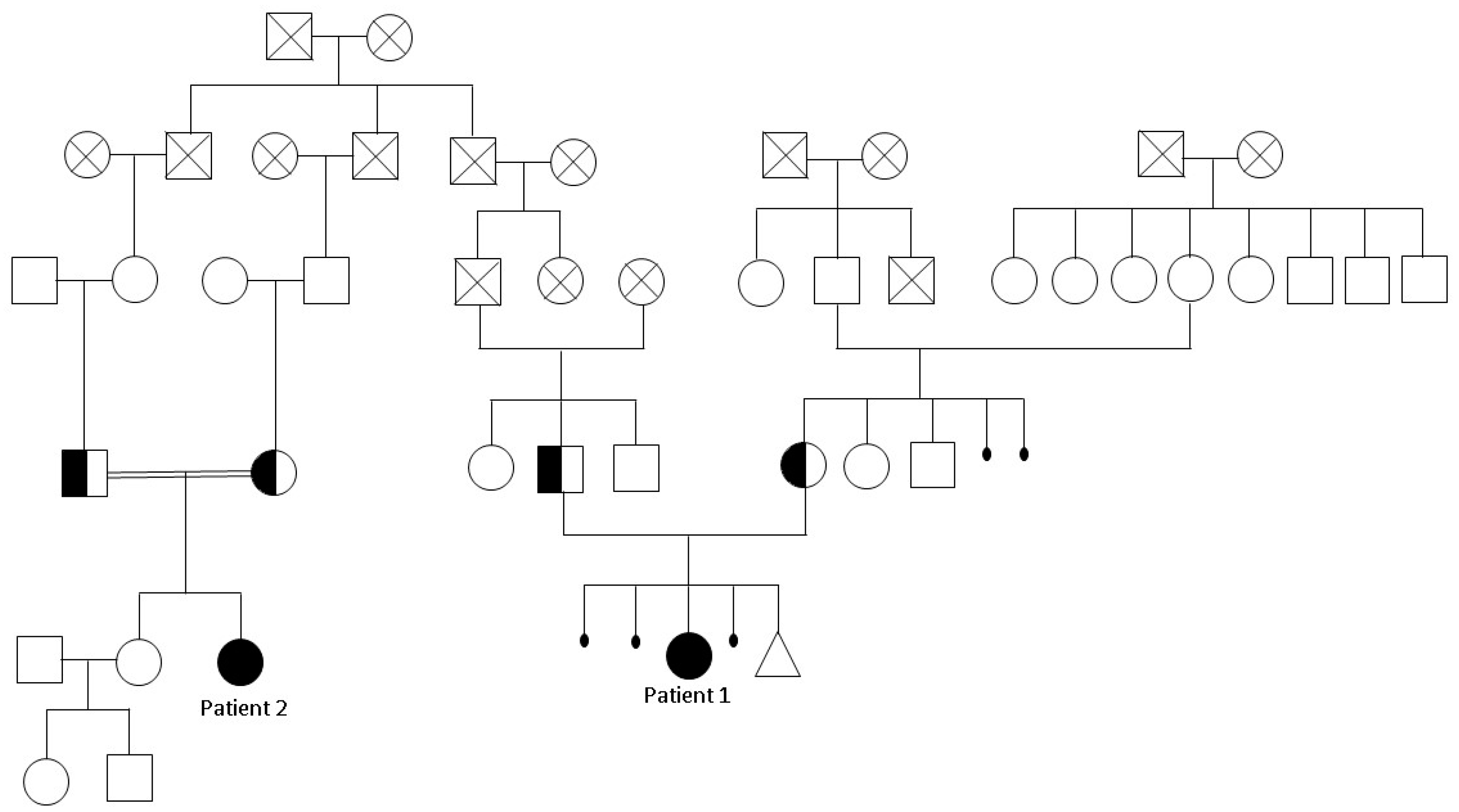
2.1. Clinical Features
2.2. Chromosomal Microarray Analysis and Exome Sequencing
2.3. ECEL1 Variants
2.4. In Silico Analysis
3. Discussion
4. Materials and Methods
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lowry, R.B.; Sibbald, B.; Bedard, T.; Hall, J.G. Prevalence of multiple congenital contractures including arthrogryposis multiplex congenita in Alberta, Canada, and a strategy for classification and coding. Birth Defects Res. A Clin. Mol. Teratol. 2010, 88, 1057–1061. [Google Scholar] [CrossRef] [PubMed]
- Bayram, Y.; Karaca, E.; Coban Akdemir, Z.; Yilmaz, E.O.; Tayfun, G.A.; Aydin, H.; Torun, D.; Bozdogan, S.T.; Gezdirici, A.; Isikay, S.; et al. Molecular etiology of arthrogryposis in multiple families of mostly Turkish origin. J. Clin. Investig. 2016, 126, 762–778. [Google Scholar] [CrossRef] [PubMed]
- Ullmann, U.; D’Argenzio, L.; Mathur, S.; Whyte, T.; Quinlivan, R.; Longman, C.; Farrugia, M.E.; Manzur, A.; Willis, T.; Jungbluth, H.; et al. ECEL1 gene related contractural syndrome: Long-term follow-up and update on clinical and pathological aspects. Neuromuscul. Disord. 2018, 28, 741–749. [Google Scholar] [CrossRef] [PubMed]
- Benoit, A.; Vargas, M.A.; DesGroseillers, L.; Boileau, G. Endothelin-converting enzyme-like 1 (ECEL1) is present both in the plasma membrane and in the endoplasmic reticulum. Biochem. J. 2004, 380, 881–888. [Google Scholar] [CrossRef] [PubMed]
- Nagata, K.; Kiryu-Seo, S.; Maeda, M.; Yoshida, K.; Morita, T.; Kiyama, H. Damage-induced neuronal endopeptidase is critical for presynaptic for- mation of neuromuscular junctions. J. Neurosci. 2010, 30, 6954–6962. [Google Scholar] [CrossRef] [PubMed]
- Valdenaire, O.; Richards, J.G.; Faull, R.L.; Schweizer, A. XCE, a new mem- ber of the endothelin-converting enzyme and neutral endopeptidase family, is preferentially expressed in the CNS. Brain Res. Mol. Brain Res. 1999, 64, 211–221. [Google Scholar] [CrossRef]
- Yauy, K.; Baux, D.; Pegeot, H.; Van Goethem, C.; Mathieu, C.; Guignard, T.; Juntas Morales, R.; Lacourt, D.; Krahn, M.; Lehtokari, V.L.; et al. MoBiDiC Prioritization Algorithm, a Free, Accessible, and Efficient Pipeline for Single-Nucleotide Variant Annotation and Prioritization for Next-Generation Sequencing Routine Molecular Diagnosis. J. Mol. Diagn. 2018, 20, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Jian, X.; Boerwinkle, E.; Liu, X. In silico prediction of splice-altering single nucleotide variants in the human genome. Nucleic Acids Res. 2014, 42, 13534–13544. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Barnett, C.P.; Todd, E.J.; Ong, R.; Davis, M.R.; Atkinson, V.; Allcock, R.; Laing, N.; Ravenscroft, G. Distal arthrogryposis type 5D with novel clinical features and compound heterozygous mutations in ECEL1. Am. J. Med. Genet. A 2014, 164A, 1846–1849. [Google Scholar] [CrossRef] [PubMed]
- Dieterich, K.; Quijano-Roy, S.; Monnier, N.; Zhou, J.; Fauré, J.; Smirnow, D.A.; Carlier, R.; Laroche, C.; Marcorelles, P.; Mercier, S.; et al. The neuronal endopeptidase ECEL1 is associated with a distinct form of recessive distal arthrogryposis. Hum. Mol. Genet 2013, 22, 1483–1492. [Google Scholar] [CrossRef] [PubMed]
- Dohrn, N.; Le, V.Q.; Petersen, A.; Skovbo, P.; Pedersen, I.S.; Ernst, A.; Krarup, H.; Petersen, M.B. ECEL1 mutation causes fetal arthrogryposis multiplex congenita. Am. J. Med. Genet. A 2015, 167, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Hamzeh, A.R.; Nair, P.; Mohamed, M.; Saif, F.; Tawfiq, N.; Khalifa, M.; Al-Ali, M.T.; Bastaki, F. A Novel Variant in the Endothelin-Converting Enzyme-Like 1 (ECEL1) Gene in an Emirati Child. Med. Princ. Pract. 2017, 26, 195–198. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.Y.; Liu, D.Y.; Jiao, Z.J.; Dong, Y.; Li, J.; Xiang, R. The Novel Compound Heterozygous Mutations of ECEL1 Identified in a Family with Distal Arthrogryposis Type 5D. Biomed. Res. Int. 2020, 2020, 2149342. [Google Scholar] [CrossRef] [PubMed]
- McMillin, M.J.; Below, J.E.; Shively, K.M.; Beck, A.E.; Gildersleeve, H.I.; Pinner, J.; Gogola, G.R.; Hecht, J.T.; Grange, D.K.; Harris, D.J.; et al. Mutations in ECEL1 cause distal arthrogryposis type 5D. Am. J. Hum. Genet. 2013, 92, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.J.; Rai, G.K.; Bhat, V.; Ramesh, V.A.; Nagarajaram, H.A.; Matalia, J.; Phadke, S.R. Distal arthrogryposis type 5D with a novel ECEL1 gene mutation. Am. J. Med. Genet. A 2014, 164A, 2857–2862. [Google Scholar] [CrossRef] [PubMed]
- Shaaban, S.; Duzcan, F.; Yildirim, C.; Chan, W.M.; Andrews, C.; Akarsu, N.A.; Engle, E.C. Expanding the phenotypic spectrum of ECEL1-related congenital contracture syndromes. Clin. Genet. 2014, 85, 562–567. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, R.; Al-Owain, M.; Khan, A.O.; Zaki, M.S.; Hossni, H.A.; Al-Tassan, R.; Eyaid, W.; Alkuraya, F.S. Identification of three novel ECEL1 mutations in three families with distal arthrogryposis type 5D. Clin. Genet. 2014, 85, 568–572. [Google Scholar] [CrossRef] [PubMed]
- Stattin, E.L.; Johansson, J.; Gudmundsson, S.; Ameur, A.; Lundberg, S.; Bondeson, M.L.; Wilbe, M. A novel ECEL1 mutation expands the phenotype of distal arthrogryposis multiplex congenita type 5D to include pretibial vertical skin creases. J. Med. Genet A 2018, 176, 1405–1410. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Clinical Features Reported for DA5D | Patient 1 | Patient 2 | |
|---|---|---|---|
| PRENATAL | Diminished fetal movements | yes | yes |
| Intrauterine growth restriction | no | no | |
| Diminished facial expression | no | yes | |
| Micrognathia | yes | yes | |
| Mouth held open | yes | yes | |
| Ptosis, unilateral or more severe on one side | yes | no | |
| Strabismus | yes | no | |
| ophthalmoplegia | no | yes | |
| HEAD AND NECK | Refractive errors | yes | yes |
| Central tongue atrophy | no | yes | |
| Furrowed tongue | no | yes | |
| Speech difficulties | no | yes | |
| Nasal voice | yes | yes | |
| Cleft palate | no | no | |
| Short neck | yes | yes | |
| Neck contractures | yes | yes | |
| Scoliosis | yes | yes | |
| Hyperlordosis | yes | yes | |
| Dislocated hips | no | yes | |
| Contractures of shoulders | yes | yes | |
| Contractures of elbows | no | yes | |
| SKELETAL AND MUSCLE | Contractures of wrists | yes | yes |
| Extension contractures of knees | yes | yes | |
| Severe camptodactyly | yes | yes | |
| Adducted thumbs | yes | yes | |
| Adducted wrists | no | yes | |
| Clubfoot | yes | yes | |
| Calcaneovalgus deformity | yes | no | |
| Decreased muscle mass | no | yes | |
| Pterygia of neck | yes | yes | |
| SKIN | Pterygia of axillae | yes | yes |
| Pterygia of elbows | no | yes | |
| Pterygia of groin | no | yes | |
| OTHER | Restrictive lung disease | no | yes |
| Paroxysmal tachycardia | no | yes |
| Reference | Variation (NM_004826.4) | Protein Consequence | Status | Type | |
|---|---|---|---|---|---|
| 1 | Present study | c.[1507-9G>A] | - | hom | activation of an intronic cryptic acceptor site. potential alteration of splicing |
| 2 | Jin et al., (2020) | c.[69C>A];c.[1810G>A] | p.(Cys23Ter);p.(Gly604Arg) | comp het | nonsense + missense |
| 3 | Umair et al., (2019) | c.[158C>A] | p.(Pro53Leu) | hom | missense |
| 4 | Ullmann et al., (2018) | c.[2005_2006delAC] | p.(Thr669fs) | hom | frameshift |
| 5 | Ullmann et al., (2018) | c.[1470G>A] | p.(Trp490Ter) | hom | nonsense |
| 6 | Stattin et al., (2018) | c.[1163T>C] | p.(Leu388Pro) | hom | missense |
| 7 | Ullmann et al., (2018) | c.[589G>A] | p.(Gly197Ser) | hom | missense |
| 8 | Hamzeh et al., (2017), McMillin et al., (2013) | c.[1184G>A] | p.(Arg395Gln) | hom | missense |
| 9 | Bayram et al., (2016) | c.[1147C>T] | p.(Gln383Ter) | NA | nonsense |
| 10 | Patil et al., (2014), Dohrn et al., (2015), Bayram et al., (2016) | c.[2023G>A] | p.(Ala675Thr) | hom | missense |
| 11 | Shaaban et al., (2014) | c.1819G>A | p.(Ser607Gly) | hom | missense |
| 12 | Barnett et al., (2014) | c.[1797-1G];[1531G>A] | p.(Gly511Ser) | comp het | splice site + missense |
| 13 | Shaheen et al., (2014) | c.1221_1223dup | - | hom | in frame duplication |
| 14 | Shaheen et al., (2014) | c.[1210C>T] | p.(Arg404Cys) | hom | missense |
| 15 | Shaheen et al., (2014) | c.[1057dupC] | - | hom | frameshift |
| 16 | Dieterich et al., (2013) | c.[2278T>C] | p.(Cys760Arg) | hom | missense |
| 17 | Dieterich et al., (2013) | c.[1685+1G>T] | p.(Lys552AlafsX33) | hom | splice donor (introducing a premature termination) |
| 18 | Dieterich et al., (2013) | c.[1649C>G] | p.(Ser550Ter) | hom | nonsense |
| 19 | Dieterich et al., (2013) | c.[1470G>A];[997C>T] | p.(Trp490Ter);(Arg333Ter) | comp het | nonsense + nonsense |
| 20 | McMillin et al., (2013) | c.[1252C>T];[590G>A] | p.(Arg418Cys);(Gly197Asp) | comp het | missense + missense |
| 21 | McMillin et al., (2013) | c.[1252C>A];[1184+3A>T] | p.(Arg418Ser) | comp het | missense + splice site |
| 22 | Dieterich et al., (2013) | c.[966+1G>A] | p.(Asp559AlafsX33) | hom | splice donor (introducing a premature termination) |
| 23 | Dieterich et al., (2013) | c.[874delG] | p.[Val292CysfsTer51] | hom | frameshift (premature truncation) |
| 24 | McMillin et al., (2013) | c.[869A>G];[797_801delinsGCT] | p.(Try290Cys);(p.Asp266Glyfs15) | comp het | missense + frameshift |
| 25 | McMillin et al., (2013) | c.[716dupA] | p.(Tyr239Ter) | hom | frameshift (premature truncation) |
| 26 | McMillin et al., (2013) | c.[716dupA];[344_355del] | p.(Tyr239Ter);(Asn115_Ala118del) | comp het | frameshift + in frame deletion |
| 27 | reported as path in ClinVar | c.[509del] | p.(Gly170fs) | NA | frameshift |
| 28 | reported as path in ClinVar | c.[110_155del] | p.(Phe37fs) | NA | frameshift |
| 29 | reported as path in ClinVar | c.[2151+2T>A] | - | NA | splice donor |
| 30 | reported as path in ClinVar | c.[1581+1G>A] | - | NA | splice donor |
| 31 | reported as path in ClinVar | c.[1506+1G>A] | - | NA | splice donor |
| 32 | reported as path in ClinVar | c.[505_529del] | p.(Gly169fs) | NA | frameshift |
| 33 | reported as path in ClinVar | c.[278del] | p.(Gly93fs) | NA | frameshift |
| 34 | reported as path in ClinVar | c.[4G>T] | p.(Glu2Ter) | NA | nonsense |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alesi, V.; Sessini, F.; Genovese, S.; Calvieri, G.; Sallicandro, E.; Ciocca, L.; Mingoia, M.; Novelli, A.; Moi, P. A New Intronic Variant in ECEL1 in Two Patients with Distal Arthrogryposis Type 5D. Int. J. Mol. Sci. 2021, 22, 2106. https://doi.org/10.3390/ijms22042106
Alesi V, Sessini F, Genovese S, Calvieri G, Sallicandro E, Ciocca L, Mingoia M, Novelli A, Moi P. A New Intronic Variant in ECEL1 in Two Patients with Distal Arthrogryposis Type 5D. International Journal of Molecular Sciences. 2021; 22(4):2106. https://doi.org/10.3390/ijms22042106
Chicago/Turabian StyleAlesi, Viola, Francesca Sessini, Silvia Genovese, Giusy Calvieri, Ester Sallicandro, Laura Ciocca, Maura Mingoia, Antonio Novelli, and Paolo Moi. 2021. "A New Intronic Variant in ECEL1 in Two Patients with Distal Arthrogryposis Type 5D" International Journal of Molecular Sciences 22, no. 4: 2106. https://doi.org/10.3390/ijms22042106
APA StyleAlesi, V., Sessini, F., Genovese, S., Calvieri, G., Sallicandro, E., Ciocca, L., Mingoia, M., Novelli, A., & Moi, P. (2021). A New Intronic Variant in ECEL1 in Two Patients with Distal Arthrogryposis Type 5D. International Journal of Molecular Sciences, 22(4), 2106. https://doi.org/10.3390/ijms22042106