
Myocardial Hypertrophy and Compensatory Increase in Systolic Function in a Mouse Model of Oxidative Stress


Abstract
1. Introduction
2. Results
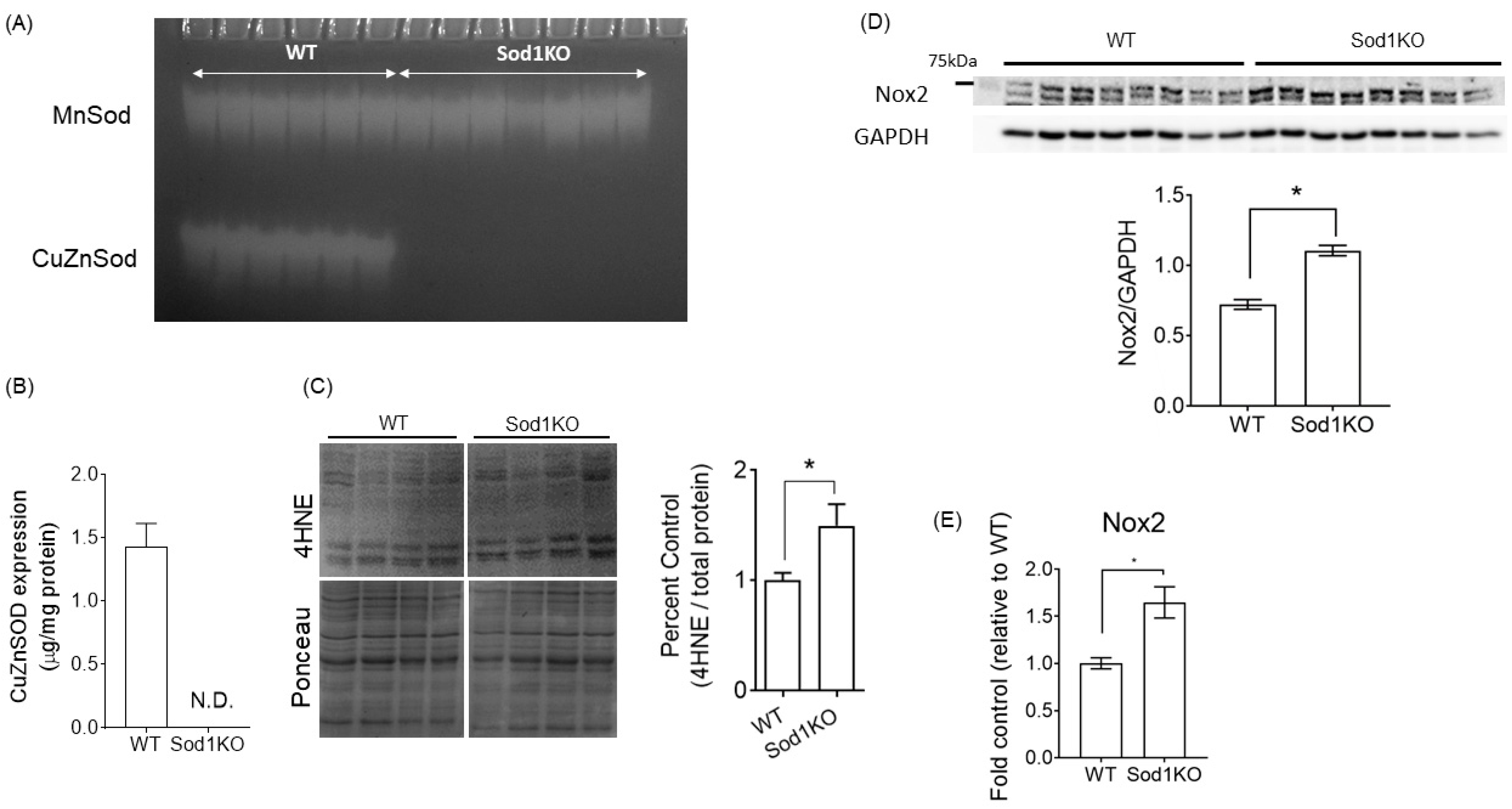
2.1. Sod1KO Mice Have Elevated Oxidative Modifications in the Heart
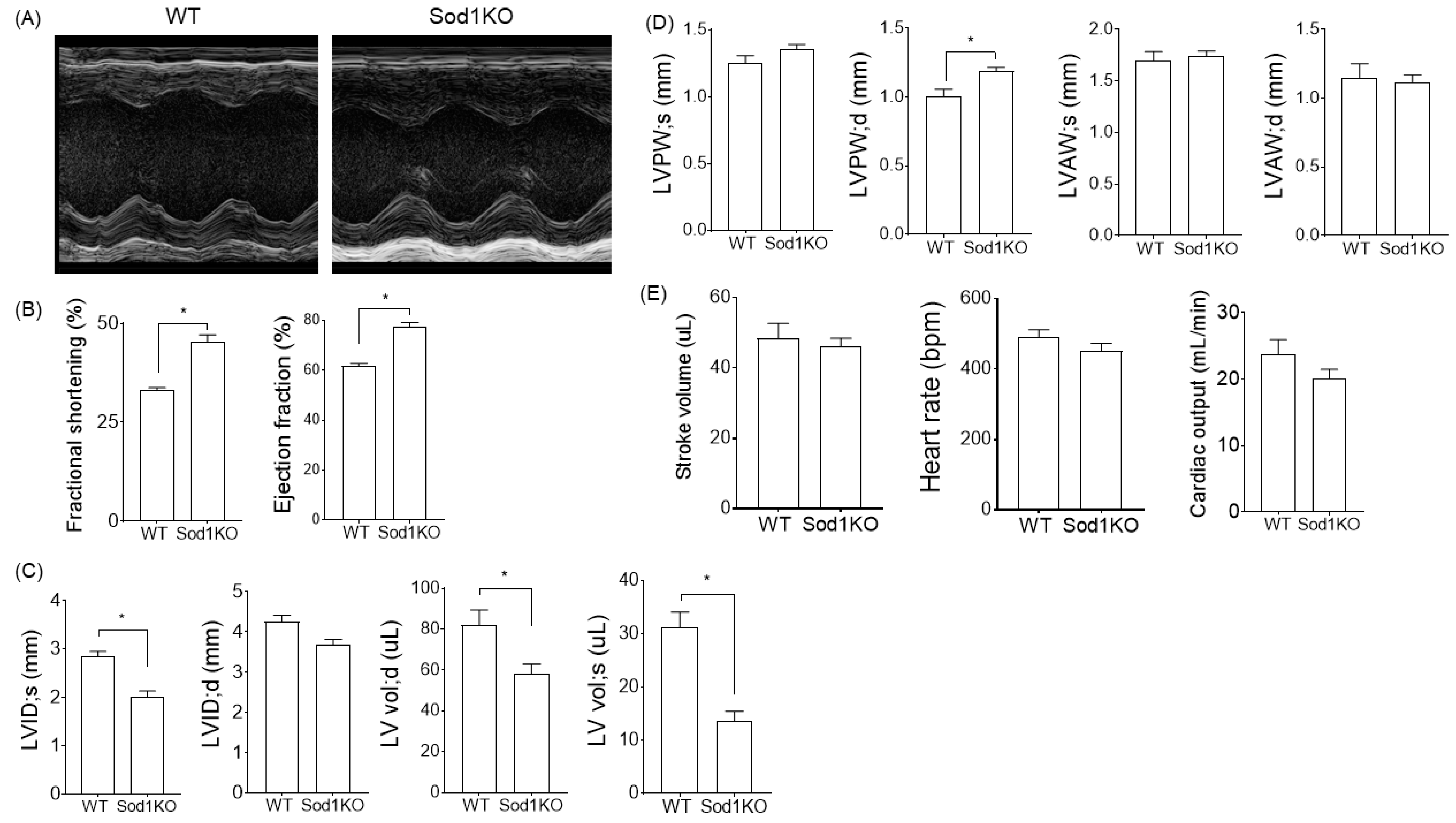
2.2. Sod1KO Mice Exhibit Increased Cardiac Systolic Function
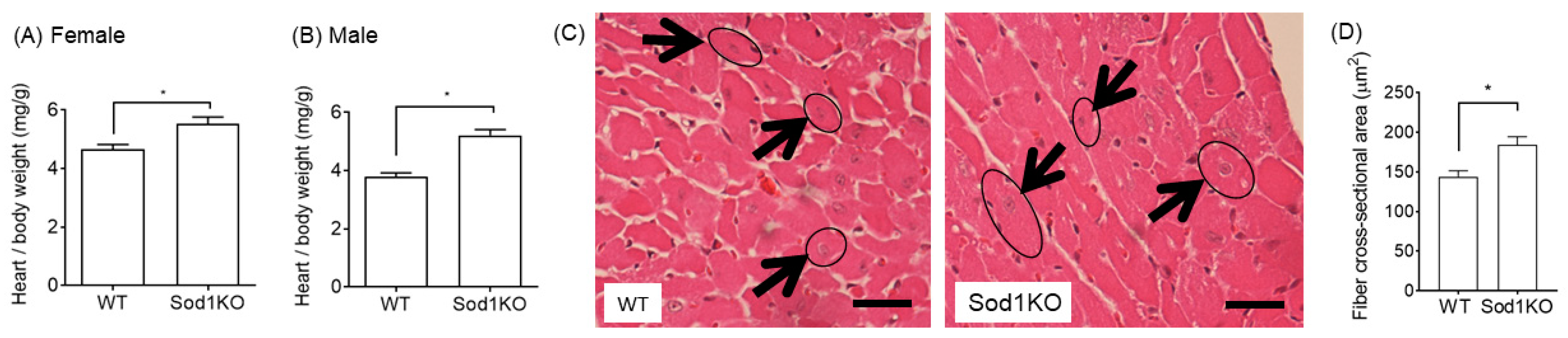
2.3. Increased Heart Weights Were Manifested in Male and Female Hearts
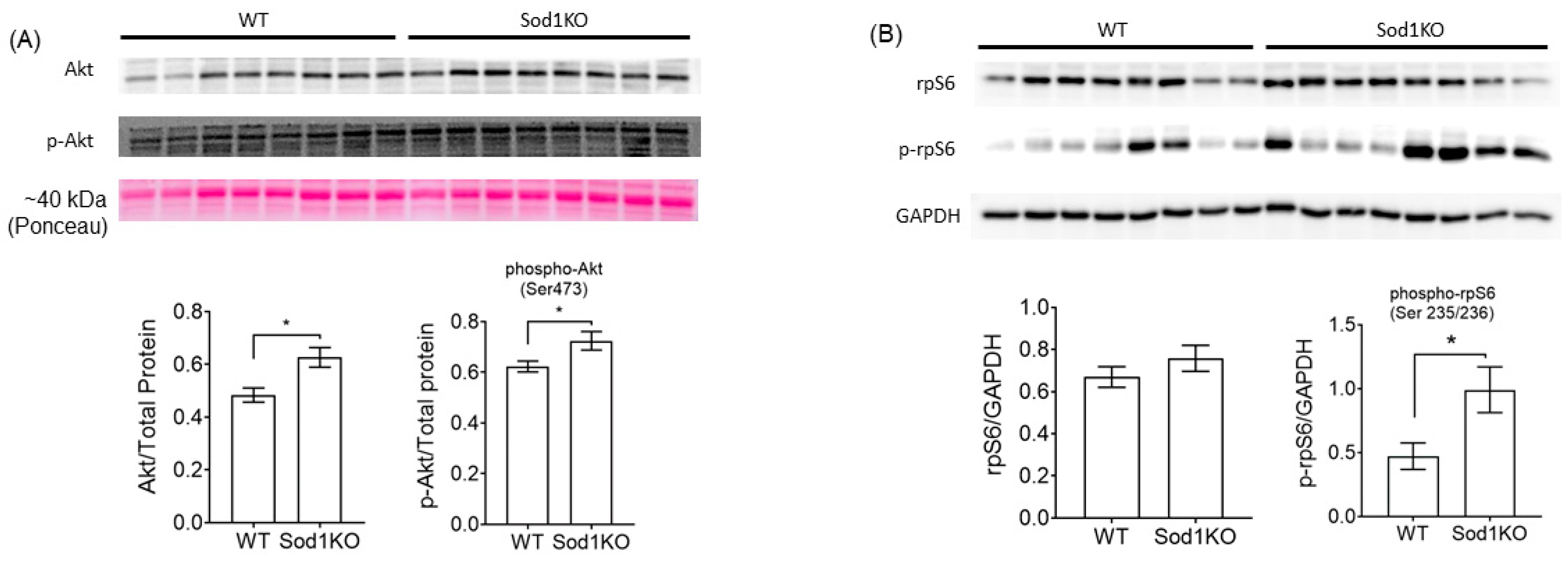
2.4. Activation of Akt Signaling and Increased Ribosomal Biogenesis
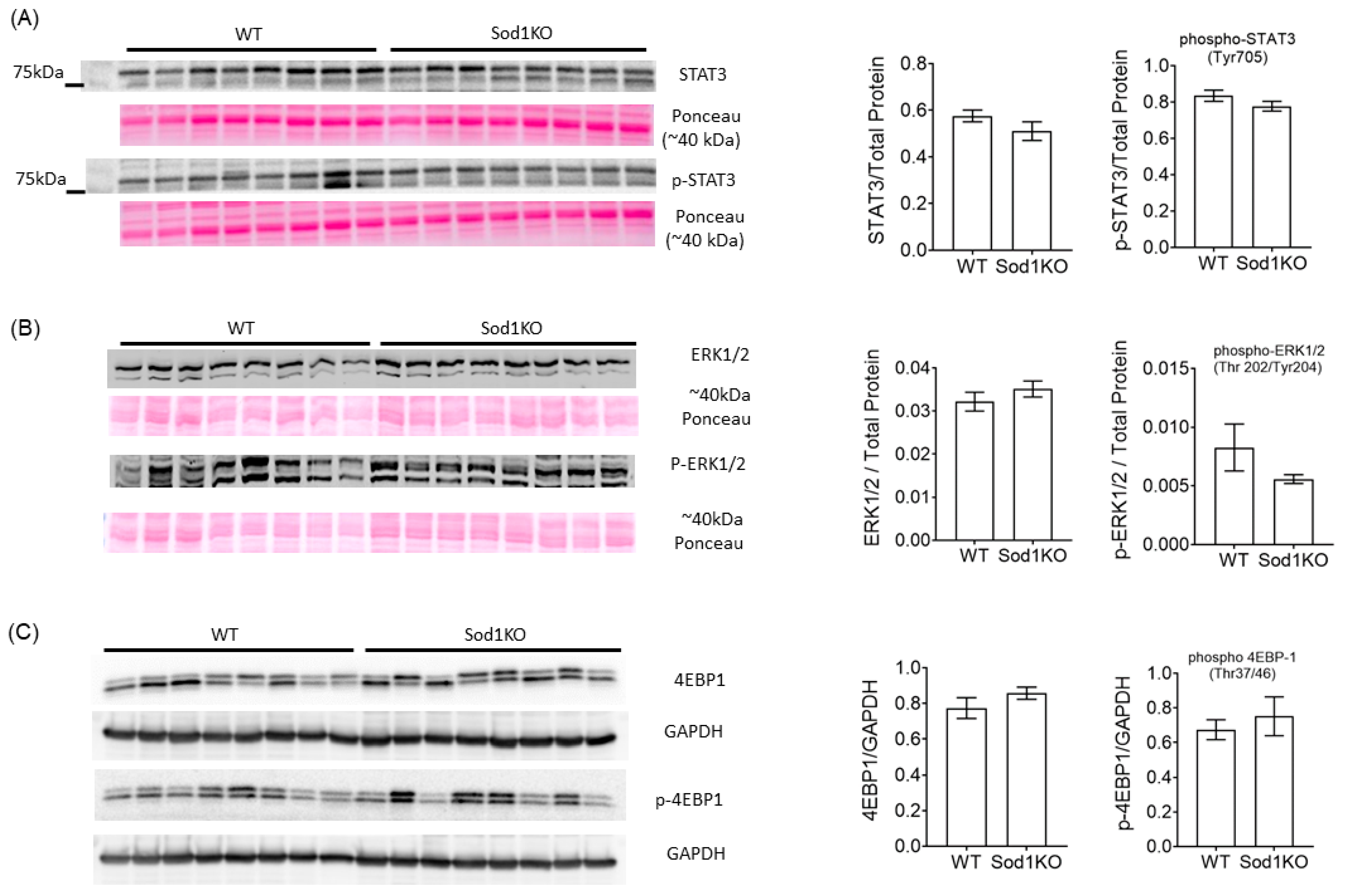
2.5. ERK and STAT3 Pathways Were Unchanged in Sod1KO Heart
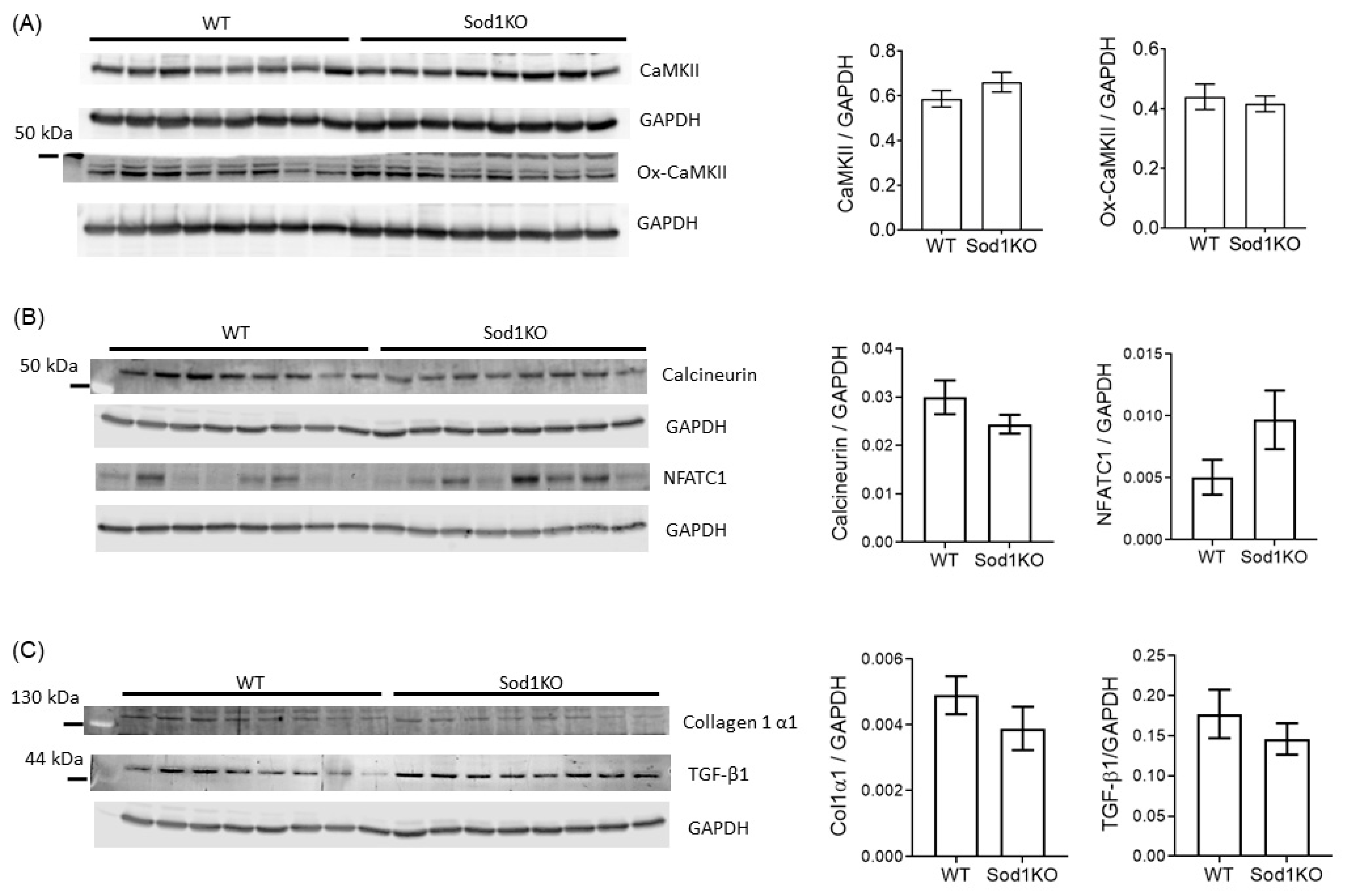
2.6. Lack of Pathological Hypertrophy and Fibrotic Response in the Sod1KO Hearts
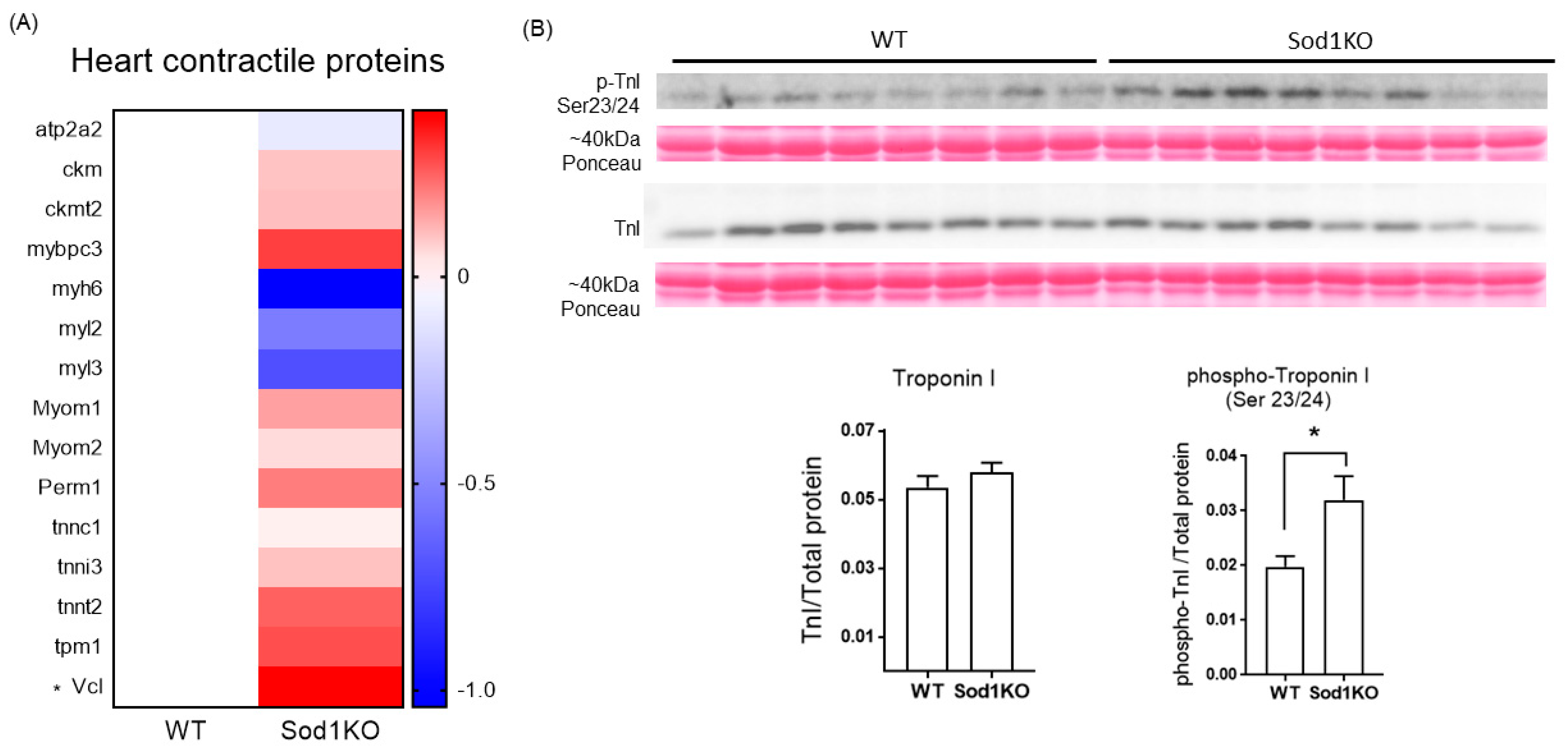
2.7. Expression and Post-Translational Modifications of the Heart Contractile Proteins Associated with Cardiac Systolic Function
3. Discussion
4. Materials and Methods
4.1. Animal Care and Sod1−/− Model
4.2. Activity Gel
4.3. Echocardiography
4.4. Immunohistochemistry
4.5. Immunoblotting
4.6. Real-Time PCR
4.7. Mass Spectrometry-Based Protein Analysis
4.8. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dyke, M.V. Heart Disease Death Rates among Blacks and Whites Aged ≥ 35 Years—United States, 1968–2015. MMWR Surveill. Summ. 2018, 67. [Google Scholar] [CrossRef]
- Martinelli, L.M.B.; Mizutani, B.M.; Mutti, A.; Dèlia, M.P.B.; Coltro, R.S.; Matsubara, B.B. Quality of Life and Its Association with Cardiovascular Risk Factors in a Community Health Care Program Population. Clinics 2008, 63, 783–788. [Google Scholar] [CrossRef]
- Hidalgo, C.; Saripalli, C.; Granzier, H.L. Effect of Exercise Training on Post-Translational and Post-Transcriptional Regulation of Titin Stiffness in Striated Muscle of Wild Type and IG KO Mice. Arch. Biochem. Biophys. 2014, 552–553, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Chiao, Y.A.; Rabinovitch, P.S. The Aging Heart. Cold Spring Harb. Perspect. Med. 2015, 5, a025148. [Google Scholar] [CrossRef]
- Bockus, L.B.; Humphries, K.M. CAMP-Dependent Protein Kinase (PKA) Signaling is Impaired in the Diabetic Heart. J. Biol. Chem. 2015, 290, 29250–29258. [Google Scholar] [CrossRef] [PubMed]
- Rain, S.; Handoko, M.L.; Trip, P.; Gan, C.T.-J.; Westerhof, N.; Stienen, G.J.; Paulus, W.J.; Ottenheijm, C.A.C.; Marcus, J.T.; Dorfmüller, P.; et al. Right Ventricular Diastolic Impairment in Patients with Pulmonary Arterial Hypertension. Circulation 2013, 128, 2016–2025. [Google Scholar] [CrossRef]
- Jones, D.P. Radical-Free Biology of Oxidative Stress. Am. J. Physiol. Cell Physiol. 2008, 295, C849–C868. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Chen, H.; Qin, H.; Sankarapandi, S.; Becher, M.W.; Wong, P.C.; Zweier, J.L. Overexpression of Human Copper, Zinc-Superoxide Dismutase (SOD1) Prevents Postischemic Injury. Proc. Natl. Acad. Sci. USA 1998, 95, 4556–4560. [Google Scholar] [CrossRef]
- Nojiri, H.; Shimizu, T.; Funakoshi, M.; Yamaguchi, O.; Zhou, H.; Kawakami, S.; Ohta, Y.; Sami, M.; Tachibana, T.; Ishikawa, H.; et al. Oxidative Stress Causes Heart Failure with Impaired Mitochondrial Respiration. J. Biol. Chem. 2006, 281, 33789–33801. [Google Scholar] [CrossRef]
- Chen, Z.; Siu, B.; Ho, Y.S.; Vincent, R.; Chua, C.C.; Hamdy, R.C.; Chua, B.H. Overexpression of MnSOD Protects against Myocardial Ischemia/Reperfusion Injury in Transgenic Mice. J. Mol. Cell. Cardiol. 1998, 30, 2281–2289. [Google Scholar] [CrossRef]
- Obal, D.; Dai, S.; Keith, R.; Dimova, N.; Kingery, J.; Zheng, Y.-T.; Zweier, J.; Velayutham, M.; Prabhu, S.D.; Li, Q.; et al. Cardiomyocyte-Restricted Overexpression of Extracellular Superoxide Dismutase Increases Nitric Oxide Bioavailability and Reduces Infarct Size after Ischemia/Reperfusion. Basic Res. Cardiol. 2012, 107, 305. [Google Scholar] [CrossRef]
- Yoshida, T.; Maulik, N.; Engelman, R.M.; Ho, Y.S.; Das, D.K. Targeted Disruption of the Mouse Sod I Gene Makes the Hearts Vulnerable to Ischemic Reperfusion Injury. Circ. Res. 2000, 86, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Otaki, Y.; Watanabe, T.; Nishiyama, S.; Takahashi, H.; Arimoto, T.; Shishido, T.; Miyamoto, T.; Konta, T.; Shibata, Y.; Sato, H.; et al. The Impact of Superoxide Dismutase-1 Genetic Variation on Cardiovascular and All-Cause Mortality in a Prospective Cohort Study: The Yamagata (Takahata) Study. PLoS ONE 2016, 11, e0164732. [Google Scholar] [CrossRef]
- Omar, B.A.; Gad, N.M.; Jordan, M.C.; Striplin, S.P.; Russell, W.J.; Downey, J.M.; McCord, J.M. Cardioprotection by Cu, Zn-Superoxide Dismutase is Lost at High Doses in the Reoxygenated Heart. Free Radic. Biol. Med. 1990, 9, 465–471. [Google Scholar] [CrossRef]
- Muller, F.L.; Song, W.; Liu, Y.; Chaudhuri, A.; Pieke-Dahl, S.; Strong, R.; Huang, T.-T.; Epstein, C.J.; Roberts, L.J.; Csete, M.; et al. Absence of CuZn Superoxide Dismutase Leads to Elevated Oxidative Stress and Acceleration of Age-Dependent Skeletal Muscle Atrophy. Free Radic. Biol. Med. 2006, 40, 1993–2004. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.C.; Lustgarten, M.S.; Liu, Y.; Muller, F.L.; Bhattacharya, A.; Liang, H.; Salmon, A.B.; Brooks, S.V.; Larkin, L.; Hayworth, C.R.; et al. Increased Superoxide in Vivo Accelerates Age-Associated Muscle Atrophy through Mitochondrial Dysfunction and Neuromuscular Junction Degeneration. FASEB J. 2010, 24, 1376–1390. [Google Scholar] [CrossRef]
- Larkin, L.M.; Davis, C.S.; Sims-Robinson, C.; Kostrominova, T.Y.; Van Remmen, H.; Richardson, A.; Feldman, E.L.; Brooks, S.V. Skeletal Muscle Weakness Due to Deficiency of CuZn-Superoxide Dismutase is Associated with Loss of Functional Innervation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301, R1400–R1407. [Google Scholar] [CrossRef] [PubMed]
- Persad, S.; Attwell, S.; Gray, V.; Mawji, N.; Deng, J.T.; Leung, D.; Yan, J.; Sanghera, J.; Walsh, M.P.; Dedhar, S. Regulation of Protein Kinase B/Akt-Serine 473 Phosphorylation by Integrin-Linked Kinase Critical Roles for Kinase Activity and Amino Acids Arginine 211 and Serine 343. J. Biol. Chem. 2001, 276, 27462–27469. [Google Scholar] [CrossRef]
- Pham, F.H.; Sugden, P.H.; Clerk, A. Regulation of Protein Kinase B and 4E-BP1 by Oxidative Stress in Cardiac Myocytes. Circ. Res. 2000, 86, 1252–1258. [Google Scholar] [CrossRef]
- Haghikia, A.; Stapel, B.; Hoch, M.; Hilfiker-Kleiner, D. STAT3 and Cardiac Remodeling. Heart Fail. Rev. 2011, 16, 35–47. [Google Scholar] [CrossRef]
- Pipicz, M.; Demján, V.; Sárközy, M.; Csont, T. Effects of Cardiovascular Risk Factors on Cardiac STAT3. Int. J. Mol. Sci. 2018, 19, 3572. [Google Scholar] [CrossRef]
- Tham, Y.K.; Bernardo, B.C.; Ooi, J.Y.Y.; Weeks, K.L.; McMullen, J.R. Pathophysiology of Cardiac Hypertrophy and Heart Failure: Signaling Pathways and Novel Therapeutic Targets. Arch. Toxicol. 2015, 89, 1401–1438. [Google Scholar] [CrossRef]
- Ahn, B.; Pharaoh, G.; Premkumar, P.; Huseman, K.; Ranjit, R.; Kinter, M.; Szweda, L.; Kiss, T.; Fulop, G.; Tarantini, S.; et al. Nrf2 Deficiency Exacerbates Age-Related Contractile Dysfunction and Loss of Skeletal Muscle Mass. Redox Biol. 2018, 17, 47–58. [Google Scholar] [CrossRef]
- Ahn, B.; Ranjit, R.; Premkumar, P.; Pharaoh, G.; Piekarz, K.M.; Matsuzaki, S.; Claflin, D.R.; Riddle, K.; Judge, J.; Bhaskaran, S.; et al. Mitochondrial Oxidative Stress Impairs Contractile Function but Paradoxically Increases Muscle Mass via Fibre Branching. J. Cachexia Sarcopenia Muscle 2019. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Chandrasekar, B.; Gu, Y.; Luo, J.; Hamid, T.; Hill, B.G.; Prabhu, S.D. Downregulation of CuZn-Superoxide Dismutase Contributes to Beta-Adrenergic Receptor-Mediated Oxidative Stress in the Heart. Cardiovasc. Res. 2007, 74, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Jones, D.P. Reactive Oxygen Species (ROS) as Pleiotropic Physiological Signalling Agents. Nat. Rev. Mol. Cell Biol. 2020, 1–21. [Google Scholar] [CrossRef]
- Bhaskaran, S.; Pollock, N.; Macpherson, P.C.; Ahn, B.; Piekarz, K.M.; Staunton, C.A.; Brown, J.L.; Qaisar, R.; Vasilaki, A.; Richardson, A.; et al. Neuron-Specific Deletion of CuZnSOD Leads to an Advanced Sarcopenic Phenotype in Older Mice. Aging Cell 2020, 19, e13225. [Google Scholar] [CrossRef]
- Li, Y.; Huang, T.T.; Carlson, E.J.; Melov, S.; Ursell, P.C.; Olson, J.L.; Noble, L.J.; Yoshimura, M.P.; Berger, C.; Chan, P.H.; et al. Dilated Cardiomyopathy and Neonatal Lethality in Mutant Mice Lacking Manganese Superoxide Dismutase. Nat. Genet. 1995, 11, 376–381. [Google Scholar] [CrossRef]
- Lebovitz, R.M.; Zhang, H.; Vogel, H.; Cartwright, J.; Dionne, L.; Lu, N.; Huang, S.; Matzuk, M.M. Neurodegeneration, Myocardial Injury, and Perinatal Death in Mitochondrial Superoxide Dismutase-Deficient Mice. Proc. Natl. Acad. Sci. USA 1996, 93, 9782–9787. [Google Scholar] [CrossRef] [PubMed]
- DeBosch, B.; Treskov, I.; Lupu, T.S.; Weinheimer, C.; Kovacs, A.; Courtois, M.; Muslin, A.J. Akt1 is Required for Physiological Cardiac Growth. Circulation 2006, 113, 2097–2104. [Google Scholar] [CrossRef]
- Weeks, K.L.; Bernardo, B.C.; Ooi, J.Y.Y.; Patterson, N.L.; McMullen, J.R. The IGF1-PI3K-Akt Signaling Pathway in Mediating Exercise-Induced Cardiac Hypertrophy and Protection. Adv. Exp. Med. Biol. 2017, 1000, 187–210. [Google Scholar] [CrossRef]
- Ikeda, M.; Ide, T.; Fujino, T.; Matsuo, Y.; Arai, S.; Saku, K.; Kakino, T.; Oga, Y.; Nishizaki, A.; Sunagawa, K. The Akt-MTOR Axis is a Pivotal Regulator of Eccentric Hypertrophy during Volume Overload. Sci. Rep. 2015, 5, 15881. [Google Scholar] [CrossRef]
- McMullen, J.R.; Sherwood, M.C.; Tarnavski, O.; Zhang, L.; Dorfman, A.L.; Shioi, T.; Izumo, S. Inhibition of MTOR Signaling with Rapamycin Regresses Established Cardiac Hypertrophy Induced by Pressure Overload. Circulation 2004, 109, 3050–3055. [Google Scholar] [CrossRef]
- Chen, L.; Zhao, L.; Samanta, A.; Mahmoudi, S.M.; Buehler, T.; Cantilena, A.; Vincent, R.J.; Girgis, M.; Breeden, J.; Asante, S.; et al. STAT3 Balances Myocyte Hypertrophy Vis-à-Vis Autophagy in Response to Angiotensin II by Modulating the AMPKα/MTOR Axis. PLoS ONE 2017, 12, e0179835. [Google Scholar] [CrossRef]
- Hingtgen, S.D.; Tian, X.; Yang, J.; Dunlay, S.M.; Peek, A.S.; Wu, Y.; Sharma, R.V.; Engelhardt, J.F.; Davisson, R.L. Nox2-Containing NADPH Oxidase and Akt Activation Play a Key Role in Angiotensin II-Induced Cardiomyocyte Hypertrophy. Physiol. Genom. 2006, 26, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Luczak, E.D.; Anderson, M.E. CaMKII Oxidative Activation and the Pathogenesis of Cardiac Disease. J. Mol. Cell Cardiol. 2014, 73, 112–116. [Google Scholar] [CrossRef]
- Richter, K.; Kietzmann, T. Reactive Oxygen Species and Fibrosis: Further Evidence of a Significant Liaison. Cell Tissue Res. 2016, 365, 591–605. [Google Scholar] [CrossRef]
- Zemljic-Harpf, A.; Manso, A.M.; Ross, R.S. Vinculin and Talin: Focus on the Myocardium. J. Investig. Med. 2009, 57, 849–855. [Google Scholar] [CrossRef] [PubMed]
- Zemljic-Harpf, A.E.; Ponrartana, S.; Avalos, R.T.; Jordan, M.C.; Roos, K.P.; Dalton, N.D.; Phan, V.Q.; Adamson, E.D.; Ross, R.S. Heterozygous Inactivation of the Vinculin Gene Predisposes to Stress-Induced Cardiomyopathy. Am. J. Pathol. 2004, 165, 1033–1044. [Google Scholar] [CrossRef]
- Kentish, J.C.; McCloskey, D.T.; Layland, J.; Palmer, S.; Leiden, J.M.; Martin, A.F.; Solaro, R.J. Phosphorylation of Troponin I by Protein Kinase a Accelerates Relaxation and Crossbridge Cycle Kinetics in Mouse Ventricular Muscle. Circ. Res. 2001, 88, 1059–1065. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.; Cheng, Y.; Lindert, S.; Wang, D.; Oxenford, L.; McCulloch, A.D.; McCammon, J.A.; Regnier, M. PKA Phosphorylation of Cardiac Troponin I Modulates Activation and Relaxation Kinetics of Ventricular Myofibrils. Biophys. J. 2014, 107, 1196–1204. [Google Scholar] [CrossRef]
- Vertechy, M.; Cooper, M.B.; Ghirardi, O.; Ramacci, M.T. Antioxidant Enzyme Activities in Heart and Skeletal Muscle of Rats of Different Ages. Exp. Gerontol. 1989, 24, 211–218. [Google Scholar] [CrossRef]
- Solaro, R.J.; Moir, A.J.; Perry, S.V. Phosphorylation of Troponin I and the Inotropic Effect of Adrenaline in the Perfused Rabbit Heart. Nature 1976, 262, 615–617. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Lopez, E.F.; Jin, Y.; Van Remmen, H.; Bauch, T.; Han, H.-C.; Lindsey, M.L. Age-Related Cardiac Muscle Sarcopenia: Combining Experimental and Mathematical Modeling to Identify Mechanisms. Exp. Gerontol. 2008, 43, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Kensler, R.W.; Craig, R.; Moss, R.L. Phosphorylation of Cardiac Myosin Binding Protein C Releases Myosin Heads from the Surface of Cardiac Thick Filaments. Proc. Natl. Acad. Sci. USA 2017, 114, E1355–E1364. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.T.; Yasunami, M.; Carlson, E.J.; Gillespie, A.M.; Reaume, A.G.; Hoffman, E.K.; Chan, P.H.; Scott, R.W.; Epstein, C.J. Superoxide-Mediated Cytotoxicity in Superoxide Dismutase-Deficient Fetal Fibroblasts. Arch. Biochem. Biophys. 1997, 344, 424–432. [Google Scholar] [CrossRef]
- Weydert, C.J.; Cullen, J.J. Measurement of Superoxide Dismutase, Catalase and Glutathione Peroxidase in Cultured Cells and Tissue. Nat. Protoc. 2010, 5, 51–66. [Google Scholar] [CrossRef] [PubMed]
- Newhardt, M.F.; Batushansky, A.; Matsuzaki, S.; Young, Z.T.; West, M.; Chin, N.C.; Szweda, L.I.; Kinter, M.; Humphries, K.M. Enhancing Cardiac Glycolysis Causes an Increase in PDK4 Content in Response to Short-Term High-Fat Diet. J. Biol. Chem. 2019, 294, 16831–16845. [Google Scholar] [CrossRef]
- MacLean, B.; Tomazela, D.M.; Shulman, N.; Chambers, M.; Finney, G.L.; Frewen, B.; Kern, R.; Tabb, D.L.; Liebler, D.C.; MacCoss, M.J. Skyline: An Open Source Document Editor for Creating and Analyzing Targeted Proteomics Experiments. Bioinformatics 2010, 26, 966–968. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (A) List of Primary Antibodies Used for Immunoblots | |||||
| Primary Antibody | Catalog Number | Source | Host | ||
| 4HNE | 393207 | EMD Millipore | Rabbit | ||
| Nox2 | Ab80508 | Abcam | Rabbit | ||
| Akt | 4691 | Cell Signaling Technologies | Rabbit | ||
| p-Akt | 4060 | Cell Signaling Technologies | Rabbit | ||
| ERK1/2 | 9102 | Cell Signaling Technologies | Rabbit | ||
| p-ERK1/2 | 4370 | Cell Signaling Technologies | Rabbit | ||
| STAT3 | 4904 | Cell Signaling Technologies | Rabbit | ||
| p-STAT3 | 9145 | Cell Signaling Technologies | Rabbit | ||
| rpS6 | 2217 | Cell Signaling Technologies | Rabbit | ||
| p-rpS6 | SC-293144 | Santa Cruz Biotechnology | Mouse | ||
| 4EBP1 | 9644 | Cell Signaling Technologies | Rabbit | ||
| p-4EBP1 | 2855 | Cell Signaling Technologies | Rabbit | ||
| GAPDH | G9545 | Sigma | Rabbit | ||
| CaMKII | Ab22609 | Abcam | Mouse | ||
| ox-CaMKII | 07-1387 | EMD Millipore | Rabbit | ||
| Calcineurin A | 2614 | Cell Signaling Technologies | Rabbit | ||
| NFATC1 | SC-7294 | Santa Cruz Biotechnology | Mouse | ||
| Collagen 1 α1 | Ab34710 | Abcam | Rabbit | ||
| TGF-β1 | AF-101-NA | R&D systems | Chicken | ||
| (B) List of Mouse Primers Used for qRT PCR | |||||
| Gene | Forward Primer | Reverse Primer | |||
| Tgf-b1 | CAA GGG CTA CCA TGC CAA CT | GTA CTG TGT GTC CAG GCT CCA A | |||
| Col1a1 | GAA GCA CGT CTG GTT TGG A | ACT CGA ACG GGA ATC CAT C | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varshney, R.; Ranjit, R.; Chiao, Y.A.; Kinter, M.; Ahn, B. Myocardial Hypertrophy and Compensatory Increase in Systolic Function in a Mouse Model of Oxidative Stress. Int. J. Mol. Sci. 2021, 22, 2039. https://doi.org/10.3390/ijms22042039
Varshney R, Ranjit R, Chiao YA, Kinter M, Ahn B. Myocardial Hypertrophy and Compensatory Increase in Systolic Function in a Mouse Model of Oxidative Stress. International Journal of Molecular Sciences. 2021; 22(4):2039. https://doi.org/10.3390/ijms22042039
Chicago/Turabian StyleVarshney, Rohan, Rojina Ranjit, Ying Ann Chiao, Michael Kinter, and Bumsoo Ahn. 2021. "Myocardial Hypertrophy and Compensatory Increase in Systolic Function in a Mouse Model of Oxidative Stress" International Journal of Molecular Sciences 22, no. 4: 2039. https://doi.org/10.3390/ijms22042039
APA StyleVarshney, R., Ranjit, R., Chiao, Y. A., Kinter, M., & Ahn, B. (2021). Myocardial Hypertrophy and Compensatory Increase in Systolic Function in a Mouse Model of Oxidative Stress. International Journal of Molecular Sciences, 22(4), 2039. https://doi.org/10.3390/ijms22042039