
One-Dimensional Organic–Inorganic Material (C6H9N2)2BiCl5: From Synthesis to Structural, Spectroscopic, and Electronic Characterizations

Abstract
1. Introduction
2. Experimental and Computational Methods
2.1. Synthesis of (C6H9N2)2BiCl5
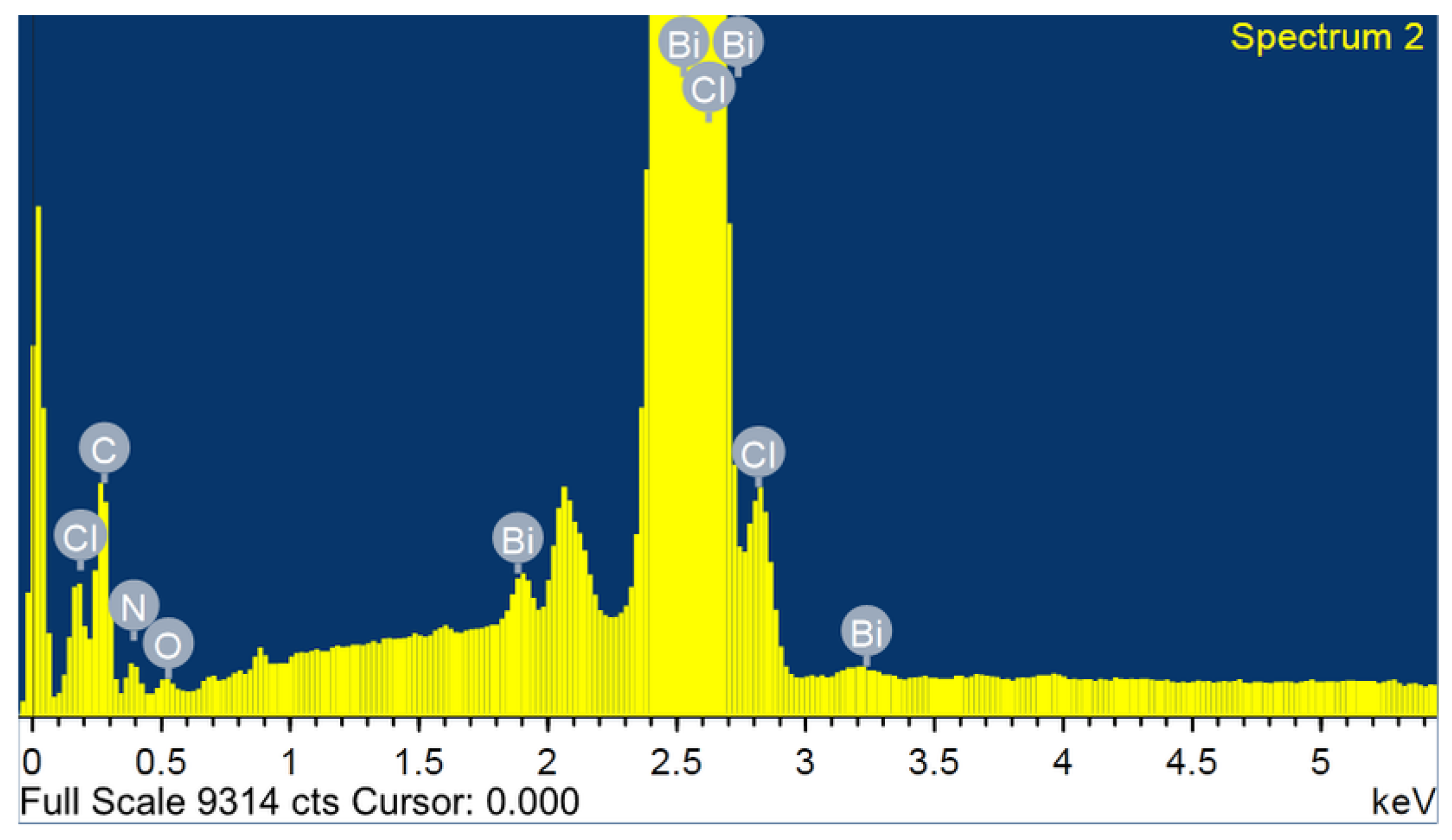
2.2. Surface Investigation by SEM/EDX
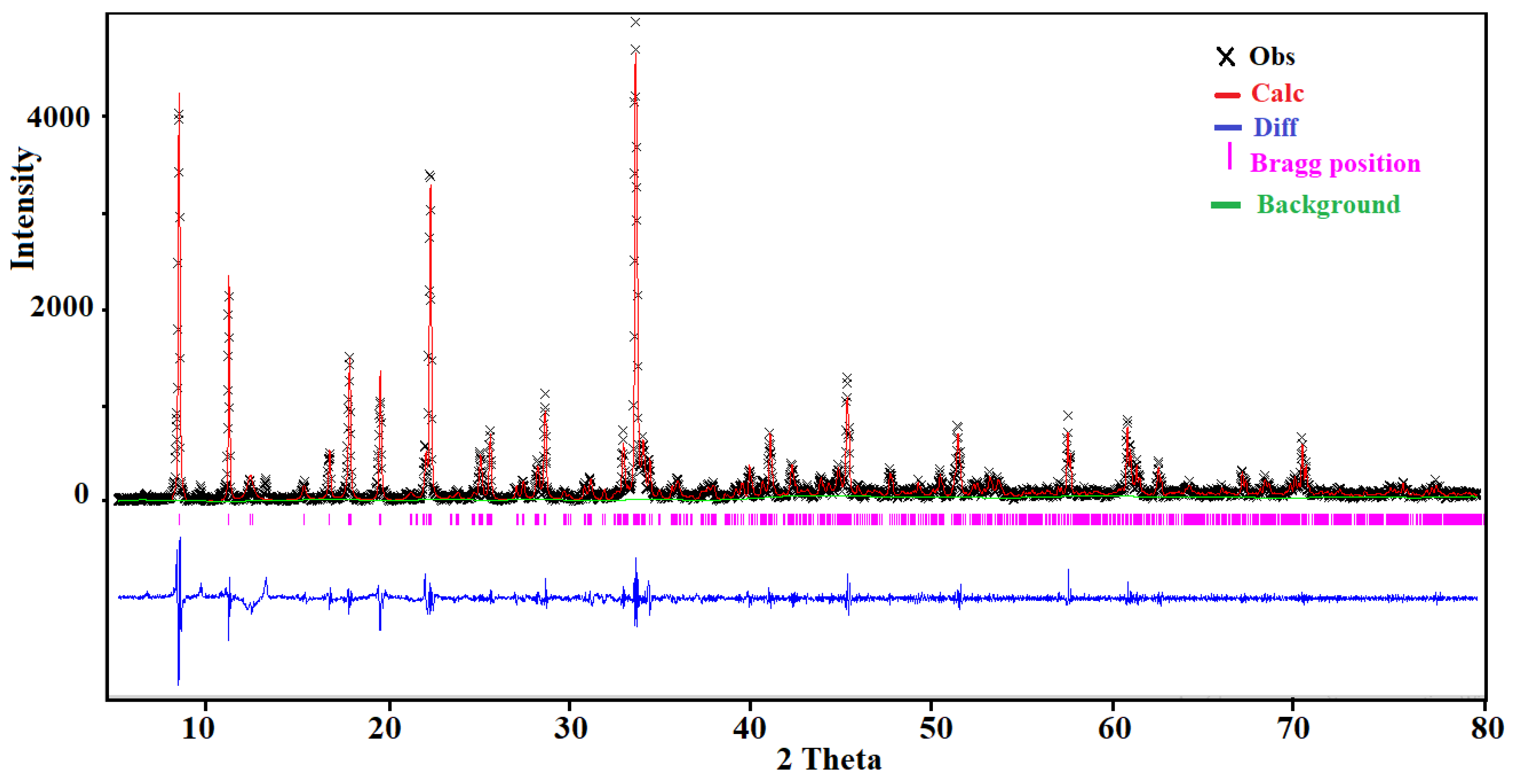
2.3. X-Ray Diffraction Analysis
2.4. Physical Measurements
2.5. Computational Methods
3. Discussion
3.1. Energy-Dispersive X-ray Analysis (EDX)
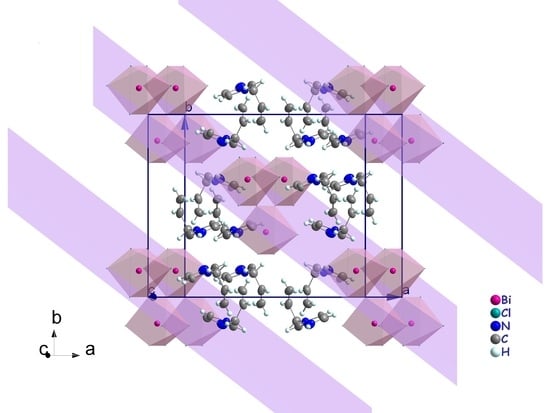
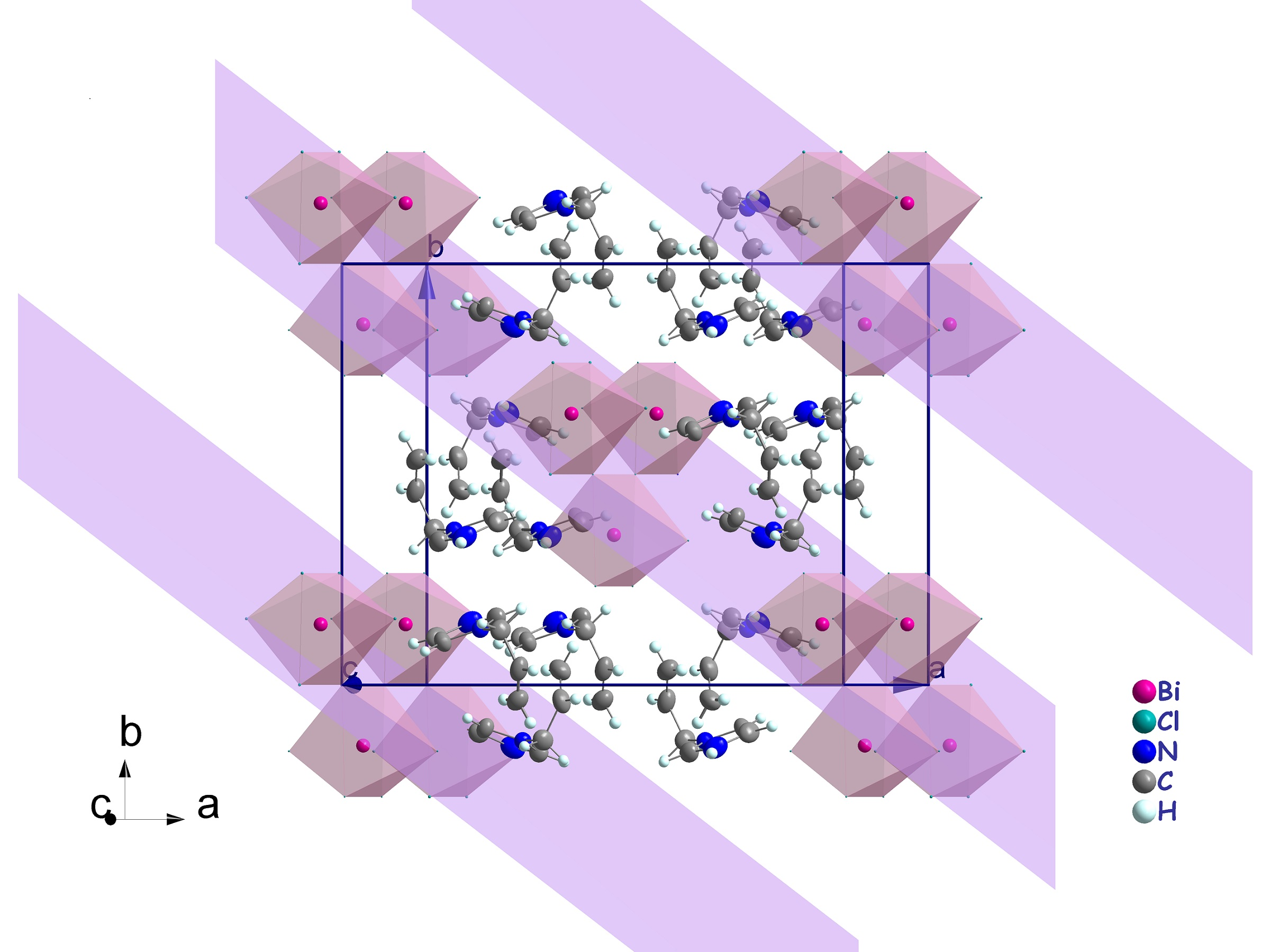
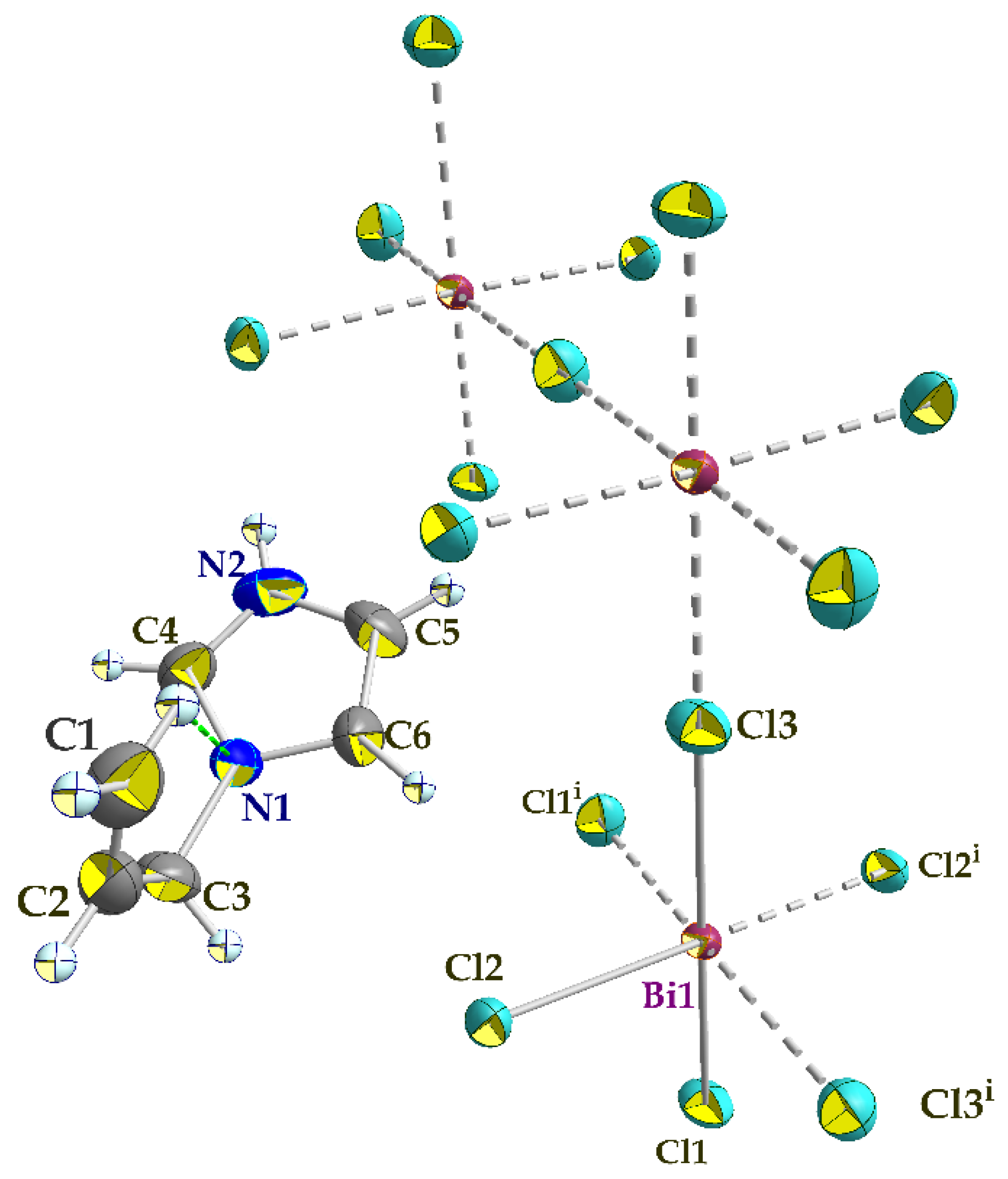
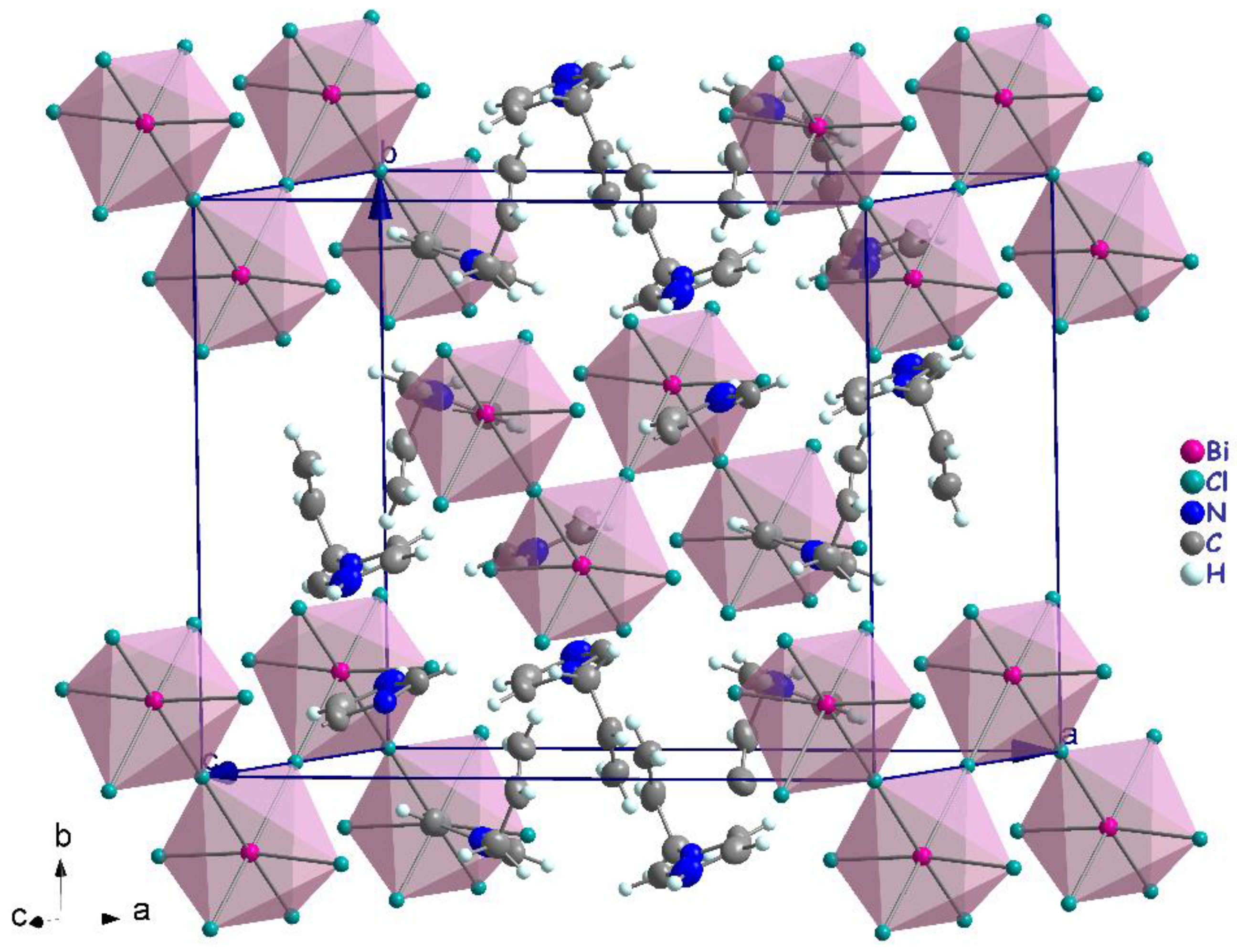
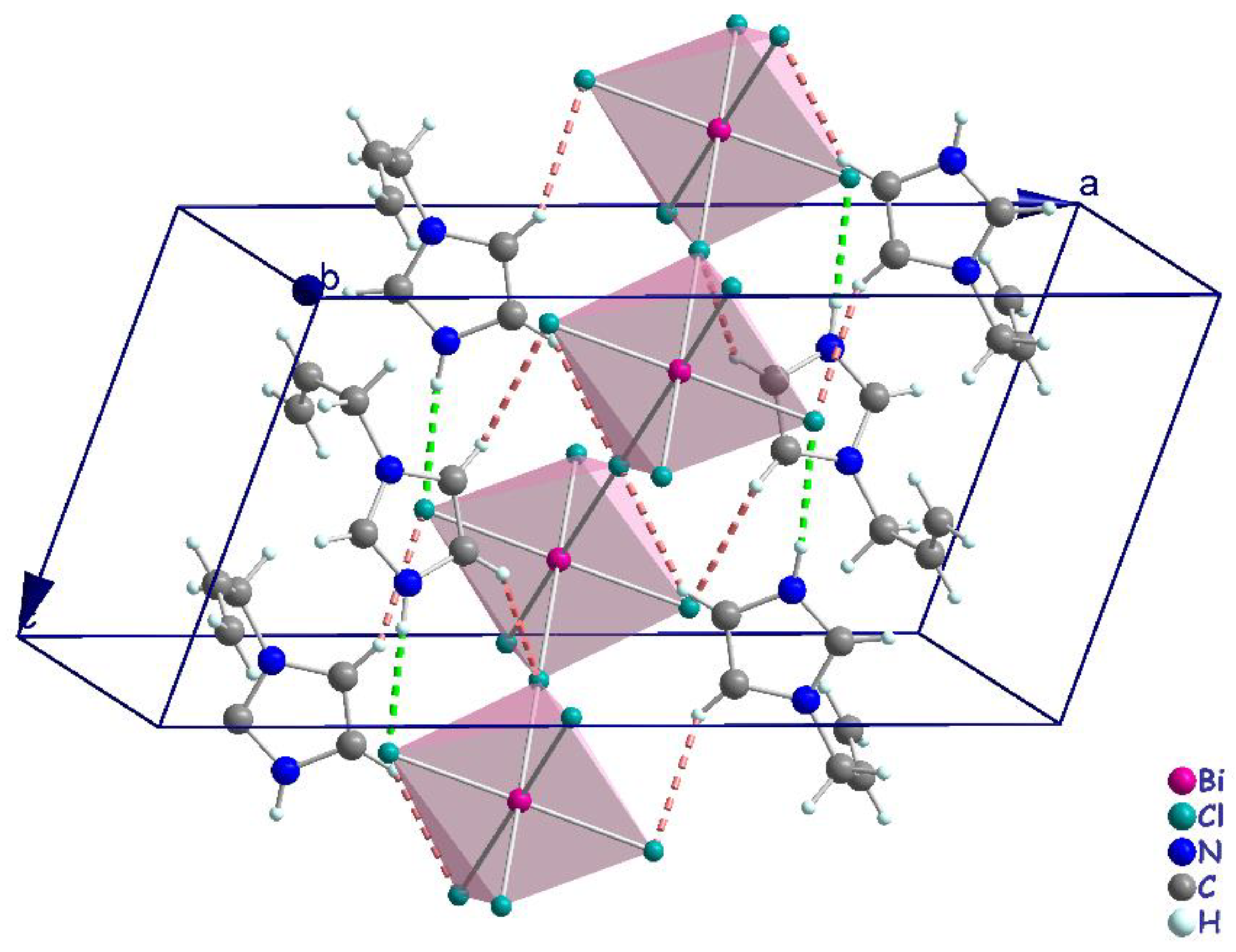
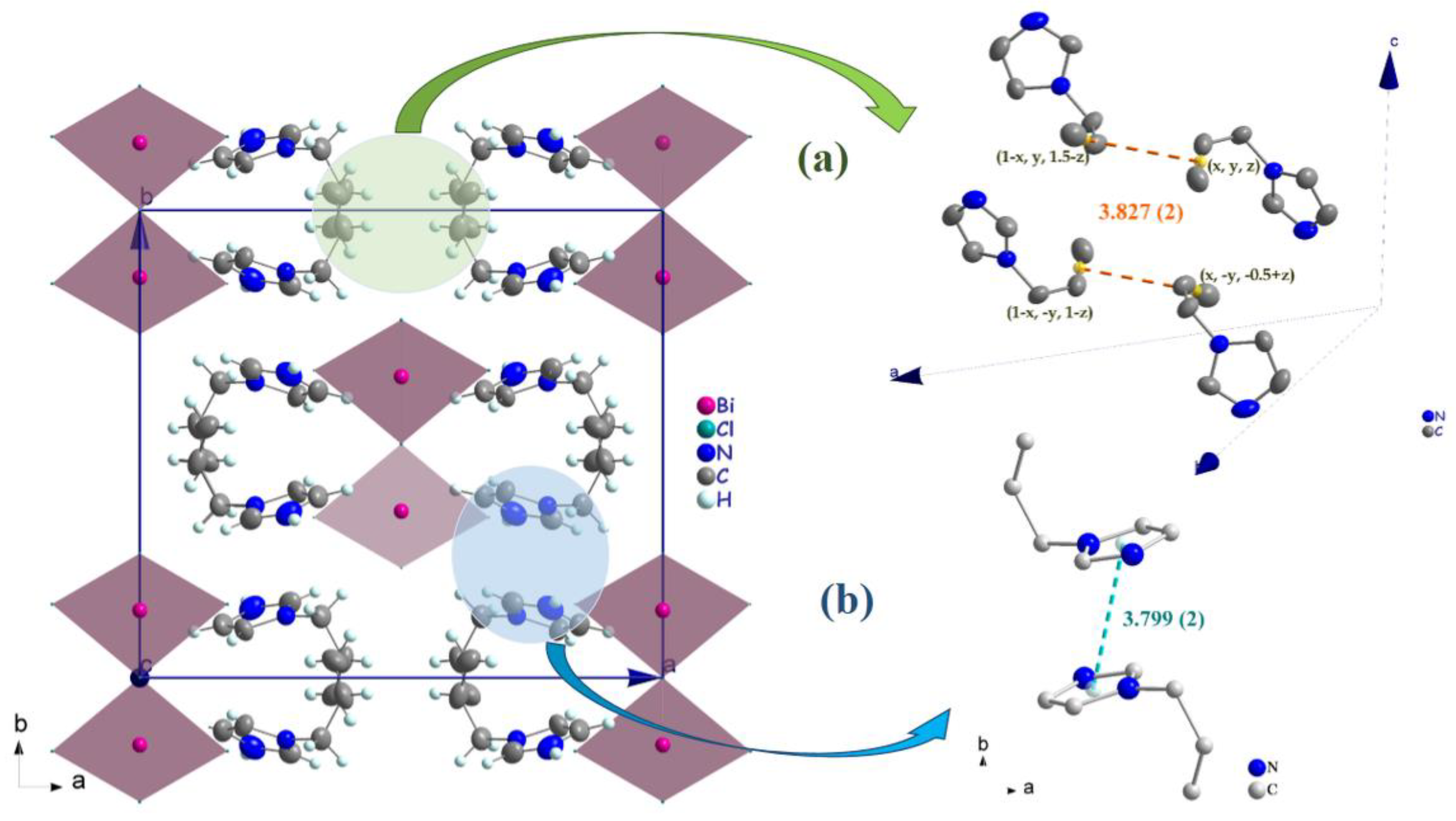
3.2. Crystal Structure Description
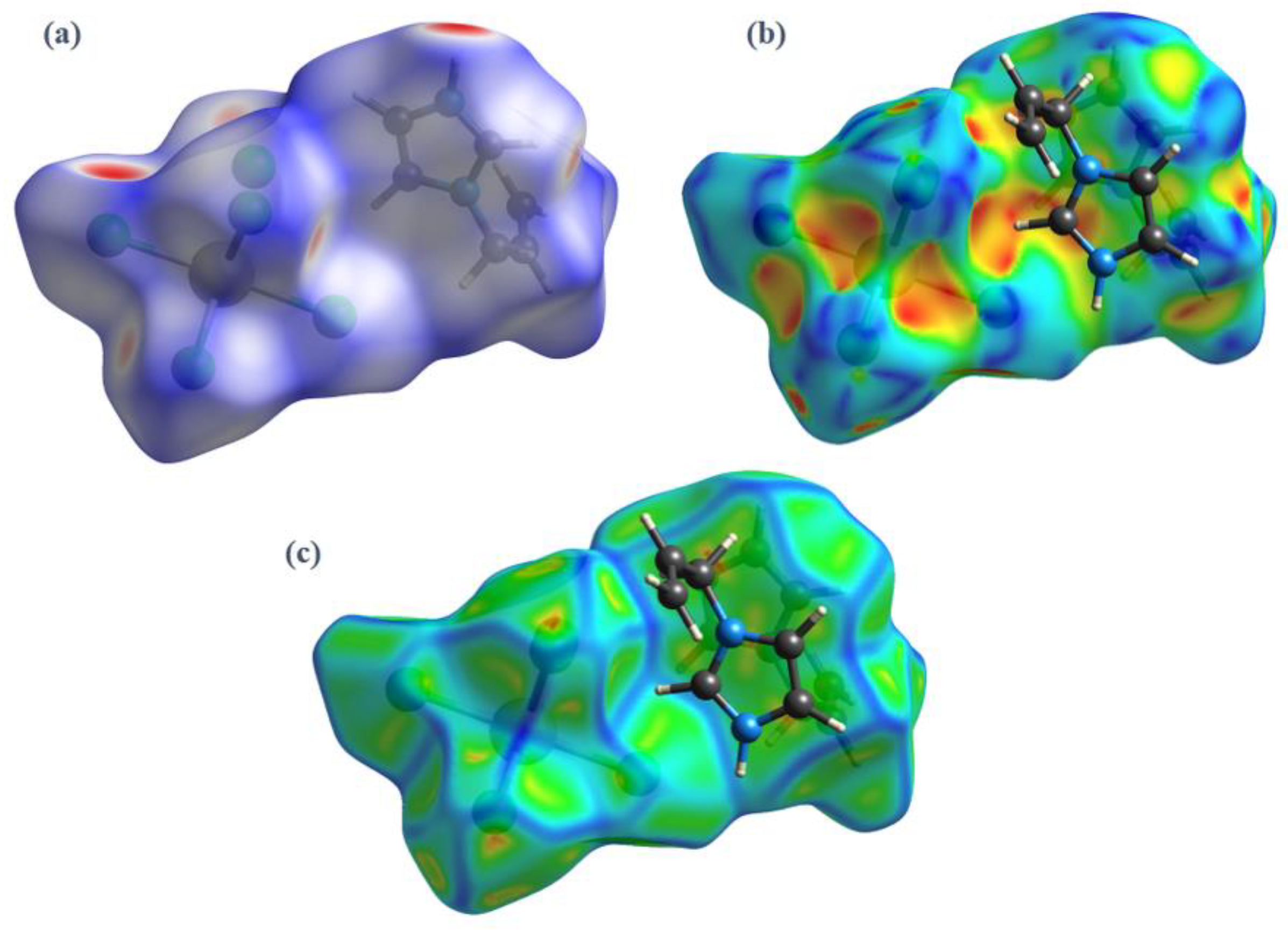
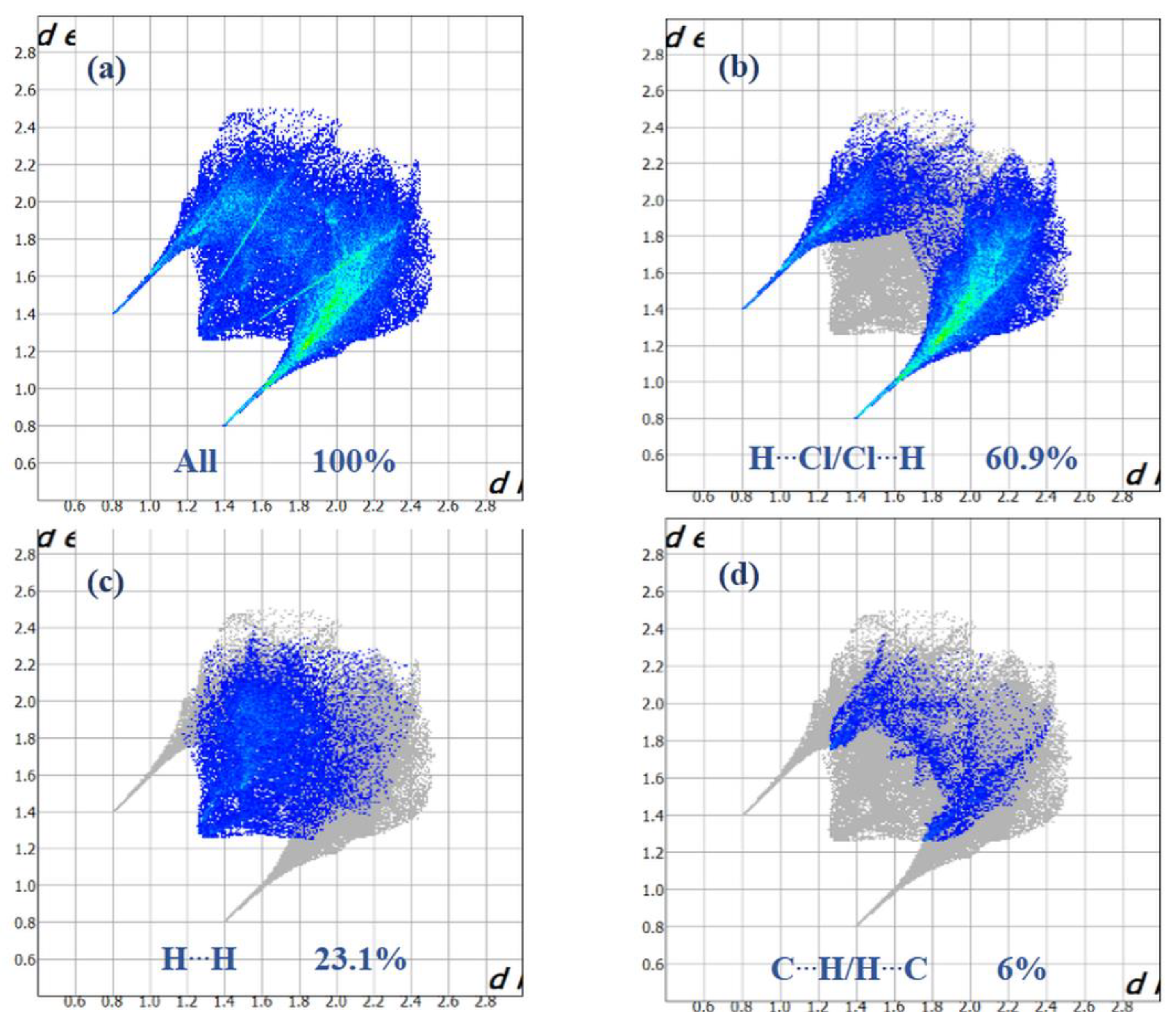
3.3. Hirshfeld Surface Analysis, Two-Dimensional Fingerprint Plots and Enrichment Ratios (EXY)
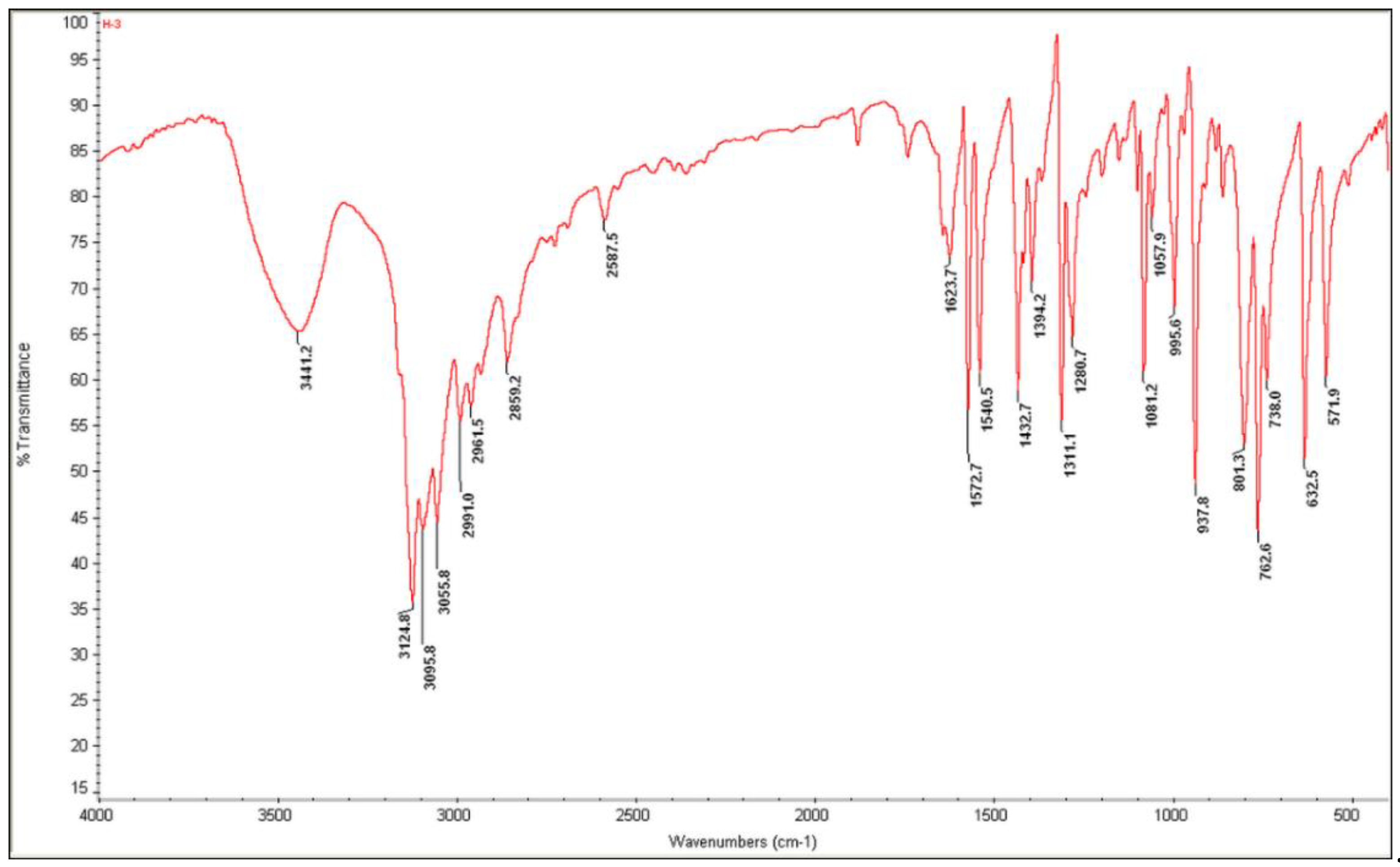
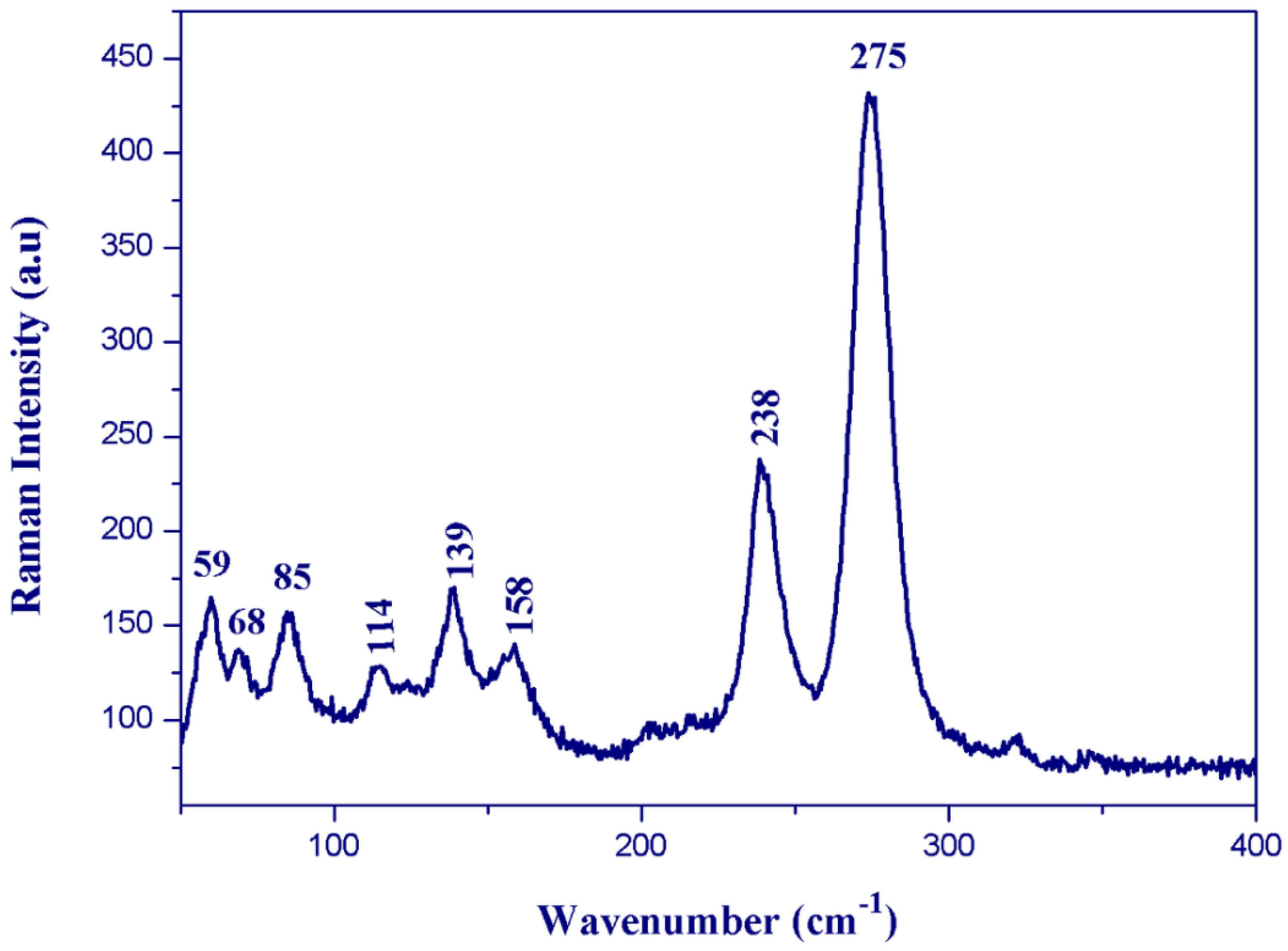
3.4. Vibrational Properties
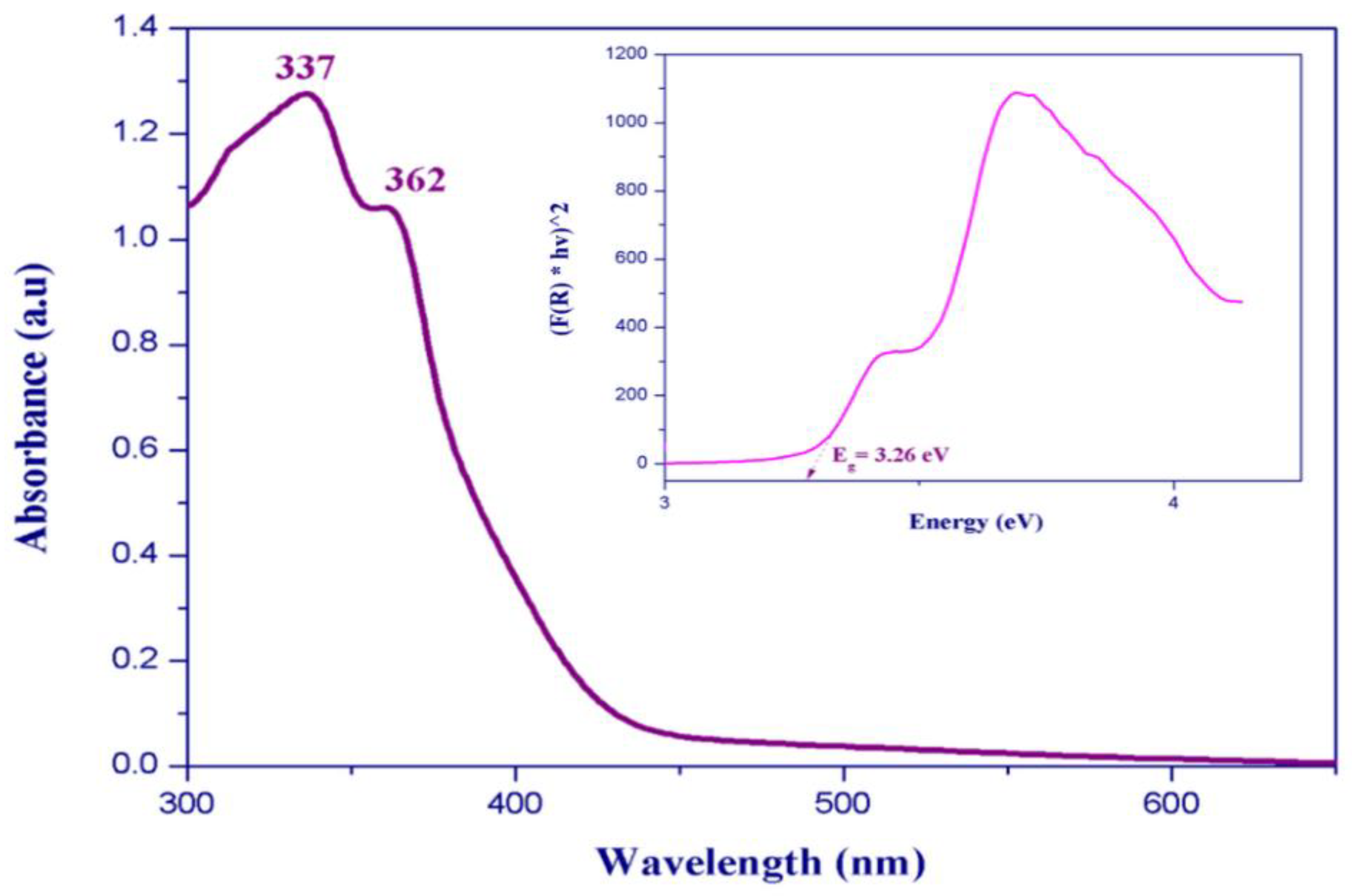
3.5. Optical Absorption
3.6. Density Functional Theory Calculations
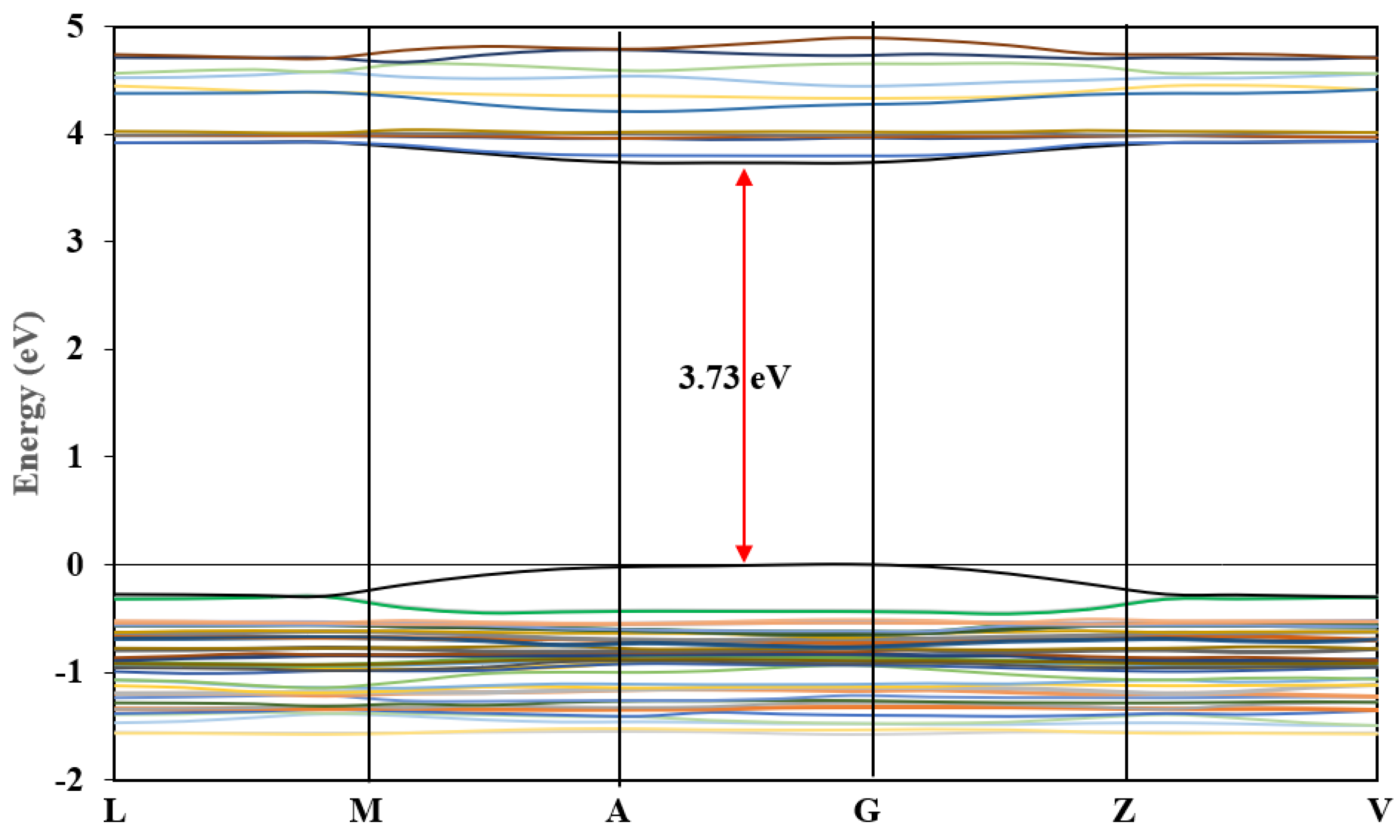
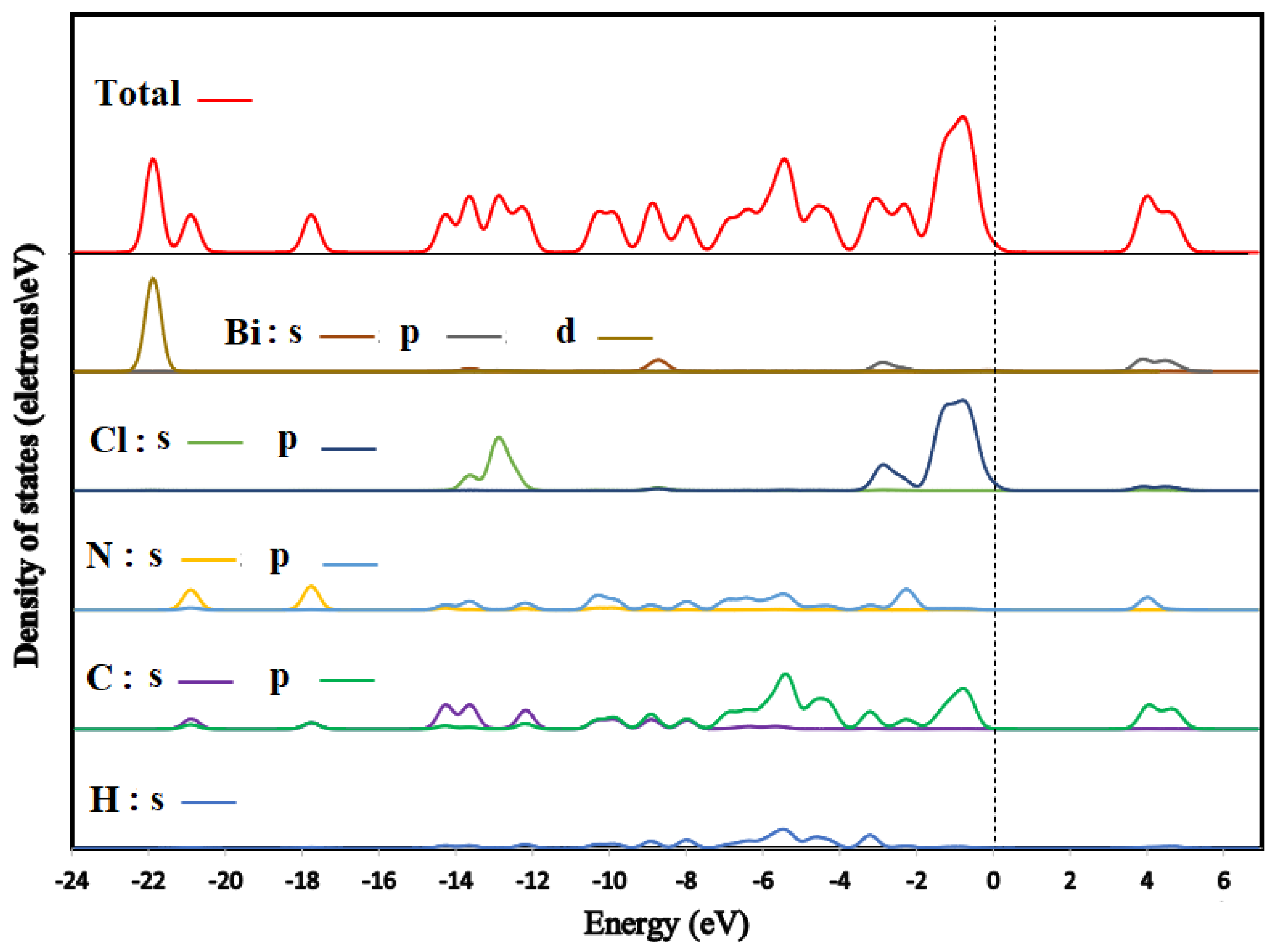
3.6.1. Band Structure and Density of States
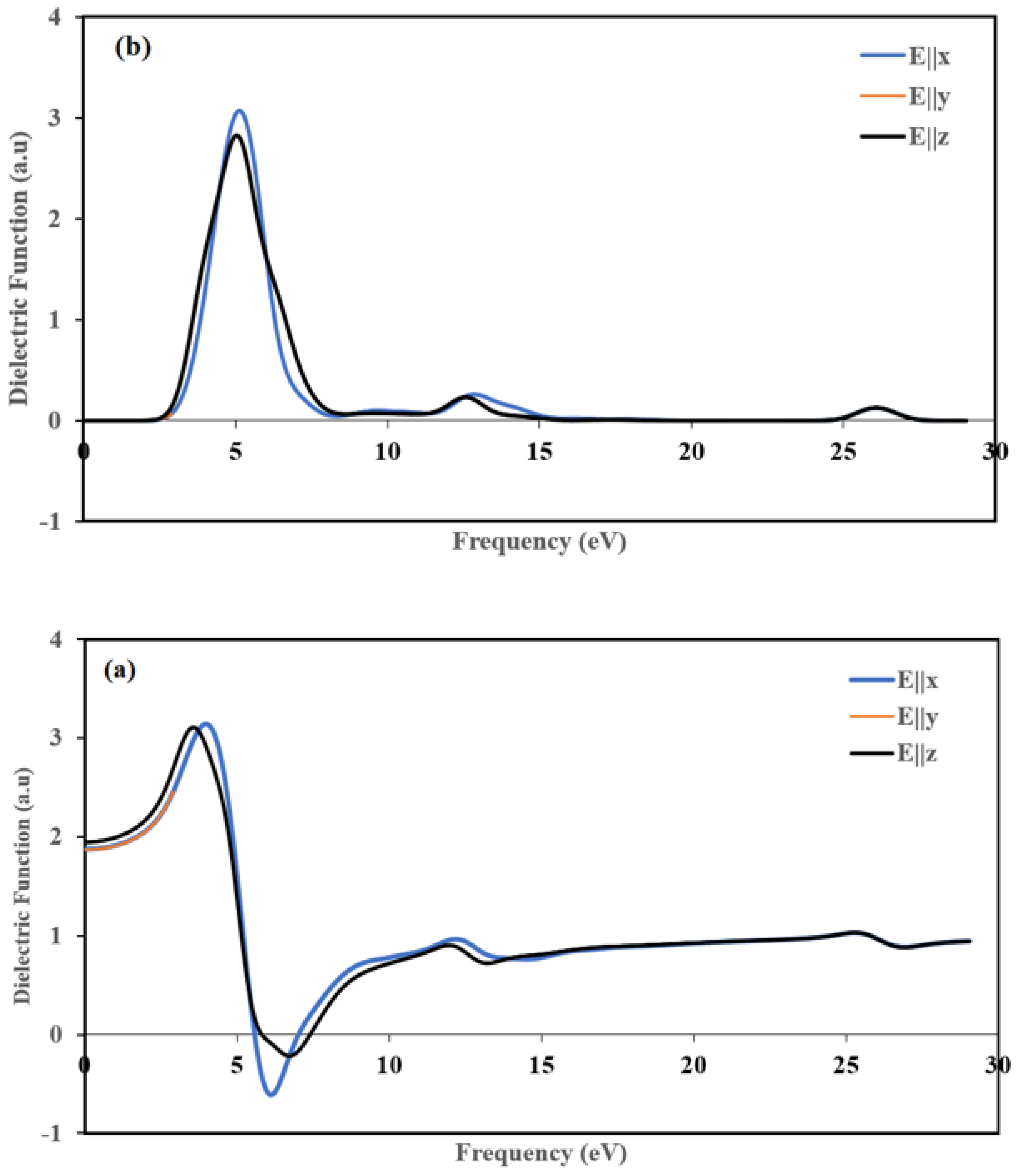
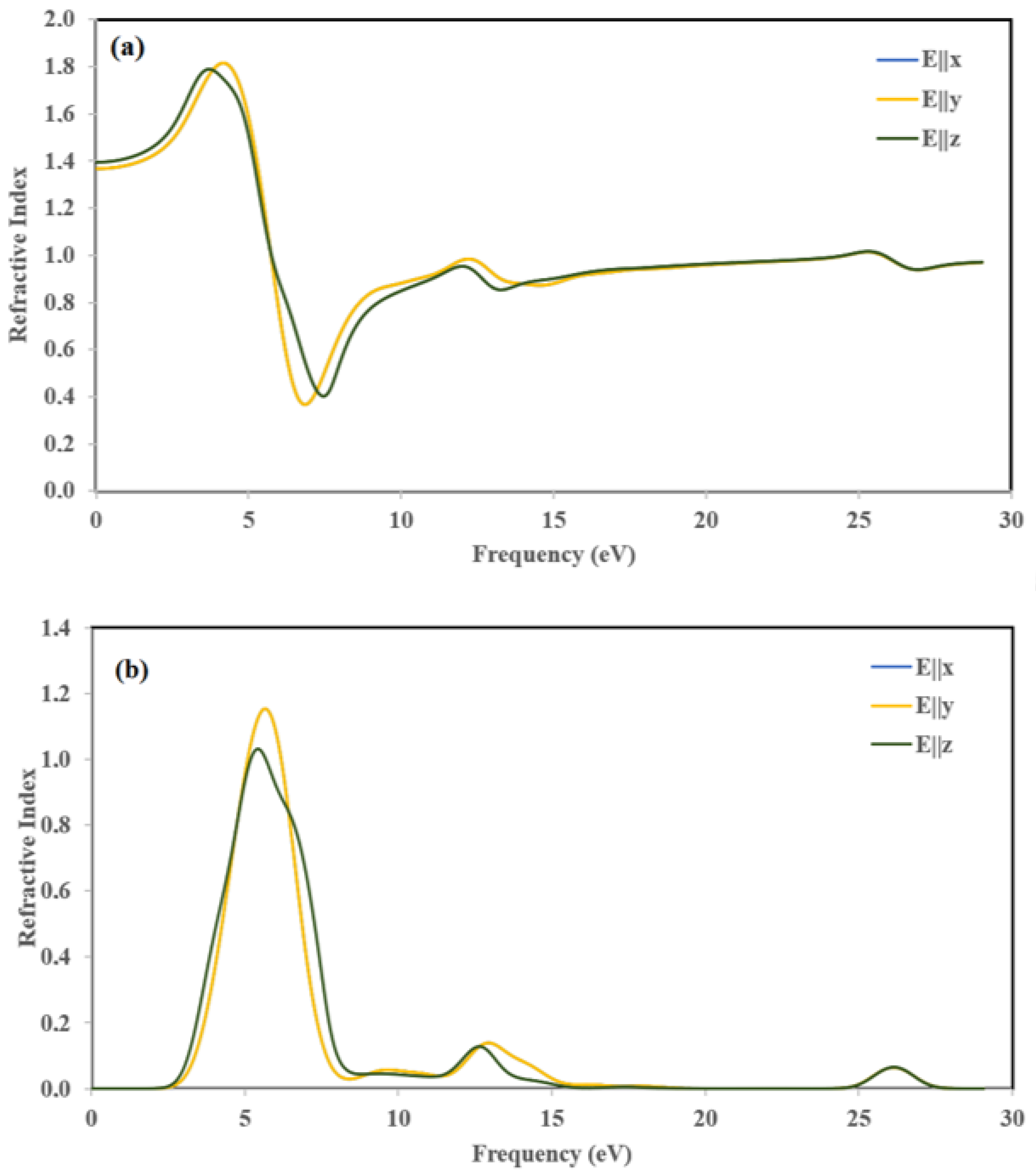
3.6.2. Optical Properties
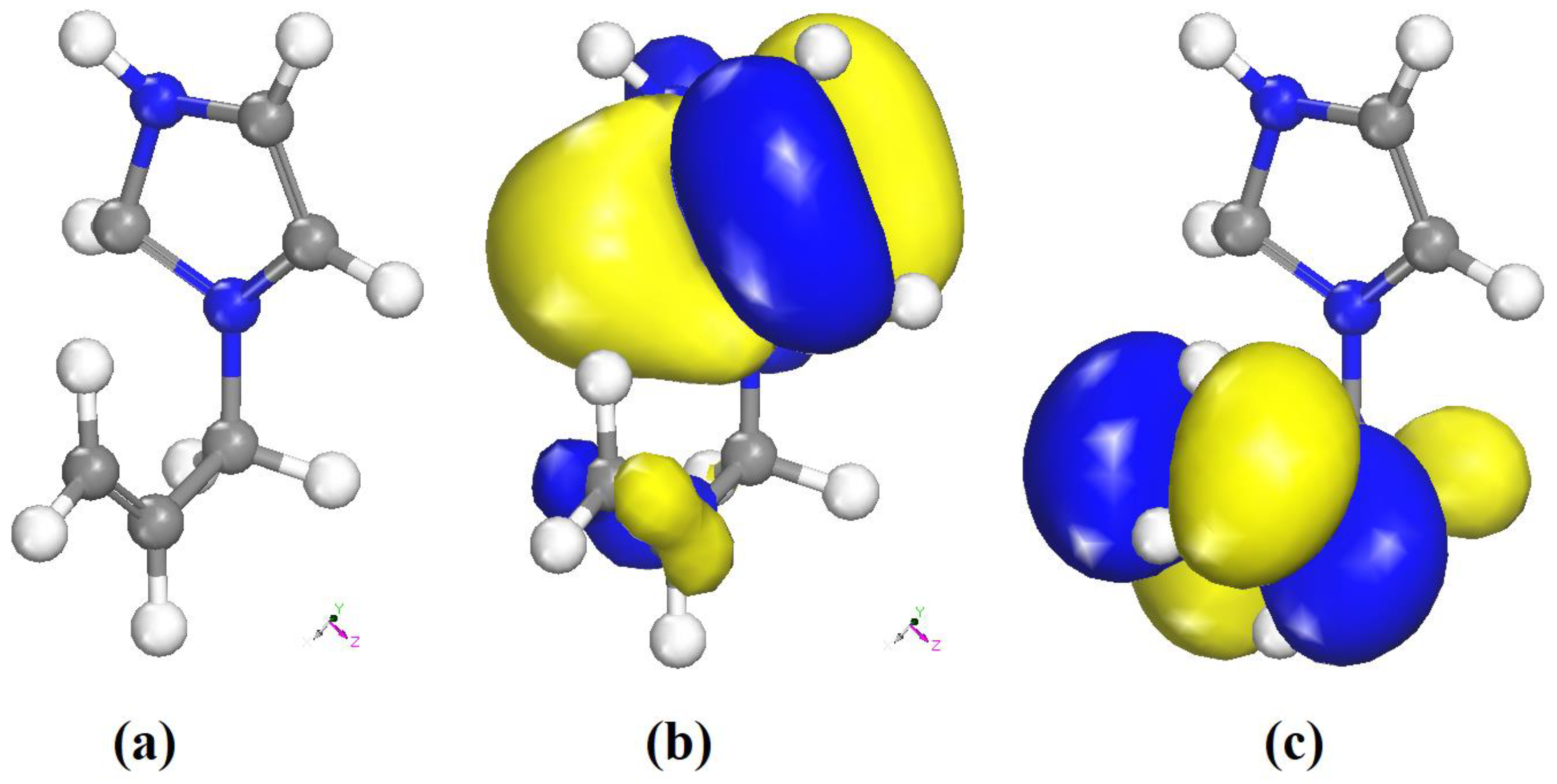
3.6.3. Frontier Molecular Orbital
3.6.4. Fukui Function
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References and Note
- Trabelsie, S.; Samet, A.; Dammak, H.; Michaud, F.; Santos, L.; Abid, Y.; Chaabouni, S. Optical properties of a new luminescent hybrid material [C6N2H5]3BiCl6 involving a resonance energy transfer (RET). Opt. Mater. 2019, 89, 355–360. [Google Scholar] [CrossRef]
- Barkaoui, H.; Abid, H.; Yangui, A.; Triki, S.; Boukheddaden, K.; Abid, Y. Yellowish White-Light Emission Involving Resonant Energy Transfer in a New One-Dimensional Hybrid Material: (C9H10N2)PbCl4. J. Phys. Chem. 2018, 122, 24253–24261. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, J.; Wu, J.; Mo, S.; Long, F.; Zou, Z.; Gao, Y. Crystal structure, optical behavior and electrical conduction of the new organic–inorganic compound CH3NH3CdI3. J. Mater. Sci. Mater. Electron. 2018, 29, 9821–9828. [Google Scholar] [CrossRef]
- Kotov, V.Y.; Ilyukhin, A.B.; Baranchikov, A.E.; Ishmetova, R.I.; Rusinov, G.L.; Kozyukhinv, S.A. Synthesis, crystal structure and optical properties of 1,1′-(1,n-alkanediyl)bis(3-methylimidazolium) halobismuthates. J. Mol. Struct. 2018, 1151, 186–190. [Google Scholar] [CrossRef]
- Essid, M.; Aloui, M.Z.; Ferretti, V.; Abid, S.; Lefebvre, F.; Rzaigui, M.; Ben Nasr, C. Crystal structure, Hirshfeld surface and spectroscopic studies of the noncentrosymmetric Bi(III) halide complex: [C8H12N]3BiCl6. Inorg. Chim. Acta 2017, 457, 122–129. [Google Scholar] [CrossRef]
- Hassen, S.; Chebbi, H.; Arfaoui, Y.; Robeyns, K.; Steenhaut, T.; Hermans, S.; Filinchuk, Y. Spectroscopic and structural studies, thermal characterization, optical proprieties and theoretical investigation of 2-aminobenzimidazolium tetrachlorocobaltate(II). Spectrochim. Acta A Mol. Biomol. Spectrosc. 2020, 240. [Google Scholar] [CrossRef]
- Adonin, S.A.; Rakhmanova, M.E.; Samsonenko, D.G.; Sokolov, M.N.; Fedin, V.P. Bi(III) halide complexes containing 4,4′-vinylenedipyridinium cation: Synthesis, structure and luminescence in solid state. Polyhedron 2015, 98, 1–4. [Google Scholar] [CrossRef]
- Lambarki, F.; Ouasri, A.; Zouihri, H.; Rhandour, A. Crystal structure, Hirshfeld and vibrational study at ambient temperature of propylammonium pentachlorobismuthate [n-C3H7NH3]2BiCl5. J. Mol. Struct. 2017, 1142, 275–284. [Google Scholar] [CrossRef]
- Chański, M.; Białońska, A.; Jakubas, R.; Piecha-Bisiorek, A. Structural characterization and properties of bis(1,4-H2-1,2,4-triazolium) pentachlorobismuthate (III) and cocrystal of ammonium chloride with tris(1,4-H2-1,2,4-triazolium) hexachlorobismuthate (III). Polyhedron 2014, 71, 69–74. [Google Scholar] [CrossRef]
- Ganguly, P. and Desiraju, G.R. Van der Waals and polar intermolecular contact distances: Quantifying supramolecular synthons. Chem. Asian J. 2012, 3, 868–880. [Google Scholar] [CrossRef]
- Ouerghi, Z.; Roisnel, T.; Fezai, R.; Kefi, R. Physico-chemical characterization, Hirshfeld surface analysis and opto-electric properties of a new hybrid material: Tris (2-amino-5-chloropyridinium) hexachlorobismuthate(III). J. Mol. Struct. 2018, 1173, 439–447. [Google Scholar] [CrossRef]
- Zhu, S.; Jiang, M.; Ye, J.; Xie, H.; Qiu, Y. Optical properties of photovoltaic materials: Organic-inorganic mixed halide perovskites CH3NH3Pb(I1-yXy)3 (X. = Cl, Br). Comput. Theor. Chem. 2018, 1144, 1–8. [Google Scholar] [CrossRef]
- Lyu, M.; Yun, J.-H.; Cai, M.; Jiao, Y.; Bernhardt, P.V.; Zhang, M.; Wang, Q.; Du, A.; Wang, H.; Liu, G.; et al. Organic–inorganic bismuth (III)-based material: A lead-free, air-stable and solution-processable light-absorber beyond organolead perovskites. Nano Res. 2016, 9, 692–702. [Google Scholar] [CrossRef]
- Adonin, S.A.; Sokolova, M.N.; Fedina, V.P. Polynuclear halide complexes of Bi(III): From structural diversity to the new properties. Coordin. Chem. Rev. 2016, 312, 1–21. [Google Scholar] [CrossRef]
- Qiu, Y.; Liu, W.; Chen, W.; Chen, W.; Zhou, G.; Hsu, P.-C.; Zhang, R.; Liang, Z.; Fan, S.; Zhang, Y.; et al. Efficient solar-driven water splitting by nanocone BiVO4-perovskite tandem cells. Sci. Adv. 2016, 2, e1501764. [Google Scholar] [CrossRef] [PubMed]
- Baklouti, Y.; Chaari, N.; Feki, H.; Chniba-Boudjada, N.; Zouari, F. Crystal structure, vibrational studies, optical properties and DFT calculations of 2-amino-5-diethyl-aminopentanium tetrachlorocadmate (II). Spectrochim. Acta A Mol. Biomol. Spectrosc. 2015, 136, 397–404. [Google Scholar] [CrossRef]
- Ben Rhaiem, T.; Elleuch, S.; Boughzala, H.; Abid, Y. A new luminescent organic-inorganic hybrid material based on cadmium iodide. Inorg. Chem. Commun. 2019, 109, 107572. [Google Scholar] [CrossRef]
- Ferjani, H.; Bechaieb, R.; El-Fattah, W.A.; Fettouhi, M. Broad-band luminescence involving fluconazole antifungal drug in a lead-free bismuth iodide perovskite: Combined experimental and computational insights. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2020, 237, 118354. [Google Scholar] [CrossRef]
- APEX3, Bruker; Bruker AXS Inc.: Madison, WI, USA, 2017.
- S.A.I.N.T. Bruker; Bruker AXS Inc.: Madison, WI, USA, 2017.
- Sheldrick, G.M. SADABS; Bruker AXS Inc.: Madison, WI, USA, 2017. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, 71, 3–8. [Google Scholar]
- Brandenburg, K. DIAMOND; Crystal Impact GbR: Bonn, Germany, 2006. [Google Scholar]
- Larson, A.C.; Von Dreele, R.B. General Structure Analysis System (GSAS); Report LAUR 86-748; Los Alamos National Laboratory: Los Alamos, NM, USA, 2000. [Google Scholar]
- Brian, H.T.J. EXPGUI, a graphical user interface for GSAS. Appl. Crystallogr. 2001, 34, 210–213. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865e3868. [Google Scholar] [CrossRef]
- Materials Studio CASTEP Manual, Accelrys, 2010, 261–262.
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Delley, B. From molecules to solids with the DMol3 approach. J. Chem. Phys. 2000, 113, 7756. [Google Scholar] [CrossRef]
- Mulliken, R.S. Electronic Population Analysis on LCAO–MO Molecular Wave Functions. J. Chem. Phys. 1955, 23, 1833. [Google Scholar] [CrossRef]
- Delley, B. Ground-State Enthalpies: Evaluation of Electronic Structure Approaches with Emphasis on the Density Functional Method. J. Phys. Chem. A 2006, 110, 13632–13639. [Google Scholar] [CrossRef]
- Dewar, M.J.S.; Thiel, W. Ground states of molecules. 38. The MNDO method. Approximations and parameters. J. Am. Chem. Soc. 1977, 99, 4899–4907. [Google Scholar] [CrossRef]
- Singh, R.N.; Kumar, A.; Tiwari, R.K.; Rawat, P. A combined experimental and theoretical (DFT and AIM) studies on synthesis, molecular structure, spectroscopic properties and multiple interactions analysis in a novel Ethyl-4-[2-(thiocarbamoyl)hydrazinylidene]-3,5-dimethyl-1H-pyrrole-2-carboxylate and its dimer. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2013, 112, 182–190. [Google Scholar]
- Hirshfeld, H.L. Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. Cryst. Eng. Comm. 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. Crystal Explorer17; The University of Western Australia: Crawley, Australia, 2017. [Google Scholar]
- Jelsch, C.; Ejsmont, K.; Huder, L. The enrichment ratio of atomic contacts in crystals, an indicator derived from the Hirshfeld surface analysis. IUCr J. 2014, 1, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Jelsch, C.; Soudani, S.; Ben Nasr, C. Likelihood of atom–atom contacts in crystal structures of halogenated organic compounds. IUCr J. 2015, 2, 327–340. [Google Scholar] [CrossRef]
- Ferjani, H.; Boughzala, H.; Driss, A. Poly[bis(1-carbamoylguanidinium) [tri-lchlorido-dichloridobismuthate(III)]]. Acta Cryst. 2012, E68, m615. [Google Scholar]
- Ouasri, A.; Jeghnou, H.; Rhandour, A.; Roussel, P. Structures and phases transition in hexylenediammonium pentachlorobismuthate (III) [NH3(CH2)6NH3]BiCl5 crystal. J. Solid State Chem. 2013, 200, 22–29. [Google Scholar] [CrossRef]
- Ferjani, H. Crystal structure, optical property and Hirshfeld surface analysis of bis [1-(prop-2-en-1-yl)-1Himidazol-3-ium] hexachloridostannate(IV). Acta Cryst. 2020, E76, 1624–1628. [Google Scholar]
- Ferjani, H. Structural, Hirshfeld Surface Analysis, Morphological Approach, and Spectroscopic Study of New Hybrid Iodobismuthate Containing Tetranuclear 0D Cluster Bi4I16·4(C6H9N2)2(H2O). Crystals 2020, 10, 397. [Google Scholar] [CrossRef]
- Bofill, L.; Prohens, R.; Barbas, R.; Frontera, A. DFT Analysis of Uncommon π···H-Bond Array Interaction in a New Pterostilbene/Theophylline Cocrystal. Cryst. Growth Des. 2020, 20, 6691–6698. [Google Scholar] [CrossRef]
- Renjith, R.; Mary, Y.S.; Panicker, C.Y.; Varghese, H.T.; Pakosinska-Parys, M.; Alsenoy, C.V.; Manojkumar, T.K. Spectroscopic (FT-IR, FT-Raman), First Order Hyperpolarizability, NBO Analysis, HOMO and LUMO Analysis of 1,7,8,9-Tetrachloro-10,10-Dimethoxy-4-[3-(4-Phenylpiperazin-1-Yl)Propyl]-4-Azatricyclo[5.2.1.02,6]Dec-8-Ene-3,5-Dione by Density Functional Methods. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2014, 124, 500–513. [Google Scholar] [CrossRef] [PubMed]
- Yıldırıma, İ.; Fırıncıb, R.; Günayb, M.E.; Özdemirc, N. Synthesis, Spectroscopy, X-ray Crystallography, and DFT Studies of Dichlorobis[1-(allyl)-1H-imidazole-κN3]copper(II). Russ. J. Phys. Chem. A 2019, 93, 2758–2764. [Google Scholar]
- Anbarasan, P.M.; Meenakshi, G.; Jeyapriya, K.; Subramarnan, M.K.; Subramani, K. Growth, structural and optical studies on organic single crystal imidazole. Indian J. Phys. 2008, 82, 1473–1483. [Google Scholar]
- Bukvetskii, B.V.; Sedakova, T.V.; Mirochnik, A.G. Crystal structure and luminescence of antimony(III) bromide with aniline. J. Struct. Chem. 2009, 50, 322–327. [Google Scholar] [CrossRef]
- Rao, A.S.; Baruah, U.; Das, S.K. Stabilization of [BiCl6]3− and [Bi2Cl10]4− with various organic precursors as cations leading to inorganic-organic supramolecular adducts: Syntheses, crystal structures and properties of [C5H7N2]3[BiCl6], [C5H7N2][C5H8N2][BiCl6] and [C10H10N2]2[Bi2Cl10]. J. Inorg. Chim. Acta 2011, 371, 206–212. [Google Scholar] [CrossRef]
- Oldenburg, K.; Vogler, A.; Miko, I.; Horvath, O. Photoredox decomposition of tin(II), lead(II), antimony(III) and bismuth(III) iodide complexes in solution. J. Inorg. Chem. Acta 1996, 248, 107–110. [Google Scholar] [CrossRef]
- Vogler, A.; Paukner, A.; Kunkely, H. Photochemistry of coordination compounds of the main group metals. J. Coord. Chem. Rev. 1990, 97, 285–297. [Google Scholar] [CrossRef]
- Vogler, A.; Nikol, H. Photochemistry and photophysics of coordination compounds of the main group metals. J. Pure. Appl. Chem. 1992, 1311, 64–69. [Google Scholar] [CrossRef]
- Ruiz-Fuertes, J.; Lopez-Moreno, S.; Lopez-Solano, J.L.; Errandonea, D.; Segura, A.; Perales, R.L.; Munoz, A.; Radescu, S.; Hernandez, P.R.; Gospodinov, M.; et al. Pressure effects on the electronic and optical properties of AWO4 wolframites (A=Cd, Mg, Mn, and Zn): The distinctive behavior of multiferroic MnWO4. Phys. Rev. B 2012, 86, 125202–125211. [Google Scholar] [CrossRef]
- Panchal, V.; Errandonea, D.; Segura, A.; Rodrıguez-Hernandez, P.; Munoz, A.; Lopez-Moreno, S.; Bettinelli, M. The electronic structure of zircon-type orthovanadates: Effects of high-pressure and cation substitution. J. Appl. Phys. 2011, 110, 043723–043733. [Google Scholar] [CrossRef]
- Haj Lakhdar, M.; Ben Smida, Y.; Amlouk, M. Synthesis, optical characterization and DFT calculations of electronic structure of Sb2O3 films obtained by thermal oxidation of Sb2S3. J. Alloys Comp. 2016, 681, 197–204. [Google Scholar] [CrossRef]
- Yaseen, M.; Murtaza, G.; Khalil, R.M.A. Ab-initio study of Li based chalcopyrite compounds LiGaX2 (X=S, Se, Te) in tetragonal symmetry: A class of future materials for optoelectronic applications. Curr. Appl. Phys. 2018, 18, 1113–1121. [Google Scholar] [CrossRef]
- Osuwa, J.; Oriaku, C.; Atuloma, C. Study of Physical Properties of Ternary Cu11Cd40S49 Thin Film Glasses. Chalcogenide Lett. 2010, 7, 383–388. [Google Scholar]
- Osuwa, J.; Oriaku, C.; Uko, O. Compositional and Optical Band Gap of Ternary Cd0.47Al0.05S0.48 Glassy Thin Film. Chalcogenide Lett. 2010, 7, 449–453. [Google Scholar]
- Iglesias, J.; Pachali, K.; Steinfink, H. Structural Chemistry of Ba2CdS3, Ba2CdSe3, Bacds2, BaCu2S2 and BaCu2Se2. J. Solid State Chem. 1974, 9, 6–14. [Google Scholar] [CrossRef]
- Thahirunnisa, S.; Banu, I.S. Optical properties of novel ASiP2 (A=Ca,Sr) chalcopyrites: First-principle study. Appl. Phys. A 2018, 124, 801. [Google Scholar] [CrossRef]
- Delin, A.; Eriksson, A.O.; Ahuja, R.; Johansson, B.; Brooks, M.S.; Gasche, T. Optical properties of the group-IVB refractory metal compounds. Phys. Rev. B 1996, 54, 1673. [Google Scholar] [CrossRef]
- Lewis, F.V.; Ioannides, C.; Parke, D.V. Interaction of a series of nitriles with the alcohol-inducible isoform of P450: Computer analysis of structure-activity relationships. Xenobiotica 1994, 24, 401–408. [Google Scholar] [CrossRef]
- James, C.; Amal Raj, A.; Reghunathan, R.V.; Jayakumar, S.; Habent Joe, I. Structural conformation and vibrational spectroscopic studies of 2,6-bis(p-N,N-dimethyl benzylidene)cyclohexanone using density functional theory. J. Raman Spectrosc. 2006, 37, 1381–1392. [Google Scholar] [CrossRef]
- Gholami, M.; Danaee, I.; Maddahy, M.H.; RashvandAvei, M. Correlated ab Initio and Electroanalytical Study on Inhibition Behavior of 2-Mercaptobenzothiazole and Its Thiole–Thione Tautomerism Effect for the Corrosion of Steel (API5LX52) in Sulphuric Acid Solution. Ind. Eng. Chem. Res. 2013, 52, 14875. [Google Scholar] [CrossRef]
- Pearson, R.G. Absolute electronegativity and hardness: Application to inorganic chemistry. Inorg. Chem. 1988, 27, 734–740. [Google Scholar] [CrossRef]
- Sastri, V. Perumareddi, Molecular Orbital Theoretical Studies of Some Organic Corrosion Inhibitors. J. Corros. 1997, 53, 617. [Google Scholar] [CrossRef]
- Lgaz, H.; Salghi, R.; Ali, I.H. Corrosion Inhibition Behavior of 9-Hydroxyrisperidone as a Green Corrosion Inhibitor for Mild Steel in Hydrochloric Acid: Electrochemical, DFT and MD Simulations Studies. Int. J. Electrochem. Sci. 2018, 13, 250. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Empirical Formula | BiCl5·2(C6H9N2) |
|---|---|
| Formula weight (g/mol) | 604.53 |
| Crystal system, space group | Monoclinic, C2/c |
| a (Å) | 17.0571 (10) |
| b (Å) | 14.3209 (9) |
| c (Å) | 8.5420 (6) |
| β (°) | 109.760 (2) |
| V (Å3) | 1963.7 (2) |
| Z | 4 |
| µ (mm−1) | 9.66 |
| Dx (Mg∙m−3) | 2.045 |
| F(000) | 1144 |
| Crystal size (mm) | 0.42 × 0.11 × 0.08 |
| Crystal habit | column, colorless |
| θmin/θmax (°) | 2.538/28.344 |
| Measured reflections | 44657 |
| Independent reflections | 2454 |
| Observed reflections with I > 2σ(I) | 2362 |
| Rint | 0.053 |
| Data/restraints/parameters | 2454/0/103 |
| R[F2 > 2σ(F2)] | 0.015 |
| wR(F2) | 0.037 |
| GooF = S | 1.15 |
| Δρmax/Δρmin (e Å−3) | 1.60/−0.68 |
| Bond Distances (Å) | |
| Bi1–Cl1 | 2.559 (6) |
| Bi1–Cl2 | 2.697 (7) |
| Bi1–Cl3 | 2.968 (1) |
| Bond Angles (°) | |
| Cl1i–Bi1–Cl1 | 94.27 (4) |
| Cl1–Bi1–Cl2i | 87.61 (2) |
| Cl1–Bi1–Cl2 | 86.85 (2) |
| Cl2i–Bi1–Cl2 | 171.86 (3) |
| Cl1i–Bi1–Cl3 | 86.85 (2) |
| Cl2–Bi1–Cl3 | 93.13 (2) |
| Cl1i–Bi1–Cl3i | 178.88 (2) |
| Cl1–Bi1–Cl3i | 93.13 (2) |
| Cl2–Bi1–Cl3i | 86.85 (2) |
| Cl3–Bi1–Cl3i | 92.025(6) |
| Bi1ii–Cl3–Bi1 | 180 |
| D–H···A | D–H | H···A | D···A | D–H···A |
|---|---|---|---|---|
| N2–H7···Cl2 iii | 0.86 | 2.35 | 3.190 (3) | 165 |
| C4–H4···Cl1 iv | 0.93 | 2.75 | 3.611 (4) | 155 |
| C5–H5···Cl3 v | 0.93 | 2.77 | 3.557 (3) | 143 |
| C1–H1A···N1 | 0.93 | 2.53 | 2.853(5) | 101 |
| C6–H6···Cl2 | 0.93 | 2.84 | 3.528 (3) | 132 |
| Atoms | H | C | N | Cl | Bi |
|---|---|---|---|---|---|
| Surface (%) | 57.9 | 4.6 | 1.7 | 33.45 | 2.15 |
| H | 33.52 | - | - | % contacts | - |
| C | 5.33 | 0.21 | - | - | - |
| N | 1.97 | 0.16 | 0.03 | - | - |
| Cl | 38.85 | 3.09 | 1.14 | 11.19 | - |
| Bi | 2.49 | 0.20 | 0.07 | 1.44 | 0.05 |
| H | 0.69 | - | - | Enrichment | - |
| C | 1.13 | 2.85 | - | - | - |
| N | 0.96 | 6.87 | 6.66 | 0.0 | - |
| Cl | 1.57 | 0.29 | 0.0 | 0.07 | - |
| Bi | 0.32 | 0.0 | 0.0 | 2.43 | 0.0 |
| N1 | 0.045 | 0.044 | 0.045 |
| C6 | 0.071 | 0.093 | 0.082 |
| H6 | 0.064 | 0.061 | 0.063 |
| C1 | −0.011 | 0.017 | 0.003 |
| H1A | 0.022 | 0.024 | 0.023 |
| H1B | 0.025 | 0.018 | 0.021 |
| C2 | 0.011 | −0.003 | 0.004 |
| H2 | 0.036 | 0.031 | 0.033 |
| C3 | −0.011 | −0.012 | −0.011 |
| H3A | 0.039 | 0.040 | 0.040 |
| H3B | 0.049 | 0.038 | 0.043 |
| C5 | 0.247 | 0.237 | 0.242 |
| H5 | 0.147 | 0.134 | 0.141 |
| N2 | 0.069 | 0.104 | 0.087 |
| H7 | 0.074 | 0.063 | 0.068 |
| C4 | 0.057 | 0.053 | 0.055 |
| H4 | 0.065 | 0.057 | 0.061 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferjani, H.; Chebbi, H.; Fettouhi, M. One-Dimensional Organic–Inorganic Material (C6H9N2)2BiCl5: From Synthesis to Structural, Spectroscopic, and Electronic Characterizations. Int. J. Mol. Sci. 2021, 22, 2030. https://doi.org/10.3390/ijms22042030
Ferjani H, Chebbi H, Fettouhi M. One-Dimensional Organic–Inorganic Material (C6H9N2)2BiCl5: From Synthesis to Structural, Spectroscopic, and Electronic Characterizations. International Journal of Molecular Sciences. 2021; 22(4):2030. https://doi.org/10.3390/ijms22042030
Chicago/Turabian StyleFerjani, Hela, Hammouda Chebbi, and Mohammed Fettouhi. 2021. "One-Dimensional Organic–Inorganic Material (C6H9N2)2BiCl5: From Synthesis to Structural, Spectroscopic, and Electronic Characterizations" International Journal of Molecular Sciences 22, no. 4: 2030. https://doi.org/10.3390/ijms22042030
APA StyleFerjani, H., Chebbi, H., & Fettouhi, M. (2021). One-Dimensional Organic–Inorganic Material (C6H9N2)2BiCl5: From Synthesis to Structural, Spectroscopic, and Electronic Characterizations. International Journal of Molecular Sciences, 22(4), 2030. https://doi.org/10.3390/ijms22042030