
Enhanced Production of the Mical Redox Domain for Enzymology and F-actin Disassembly Assays


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Cold Adapted Chaperonins Alone Do Not Work Well for Expressing High-Levels of MicalRedox Protein
2.2. The Use and Removal of Solubility Tags Destabilizes MicalRedox Protein
2.3. Enhanced Expression and Solubility of MicalRedox Protein Using Low-Temperature Expression, Chaperonins and No Solubility Tag
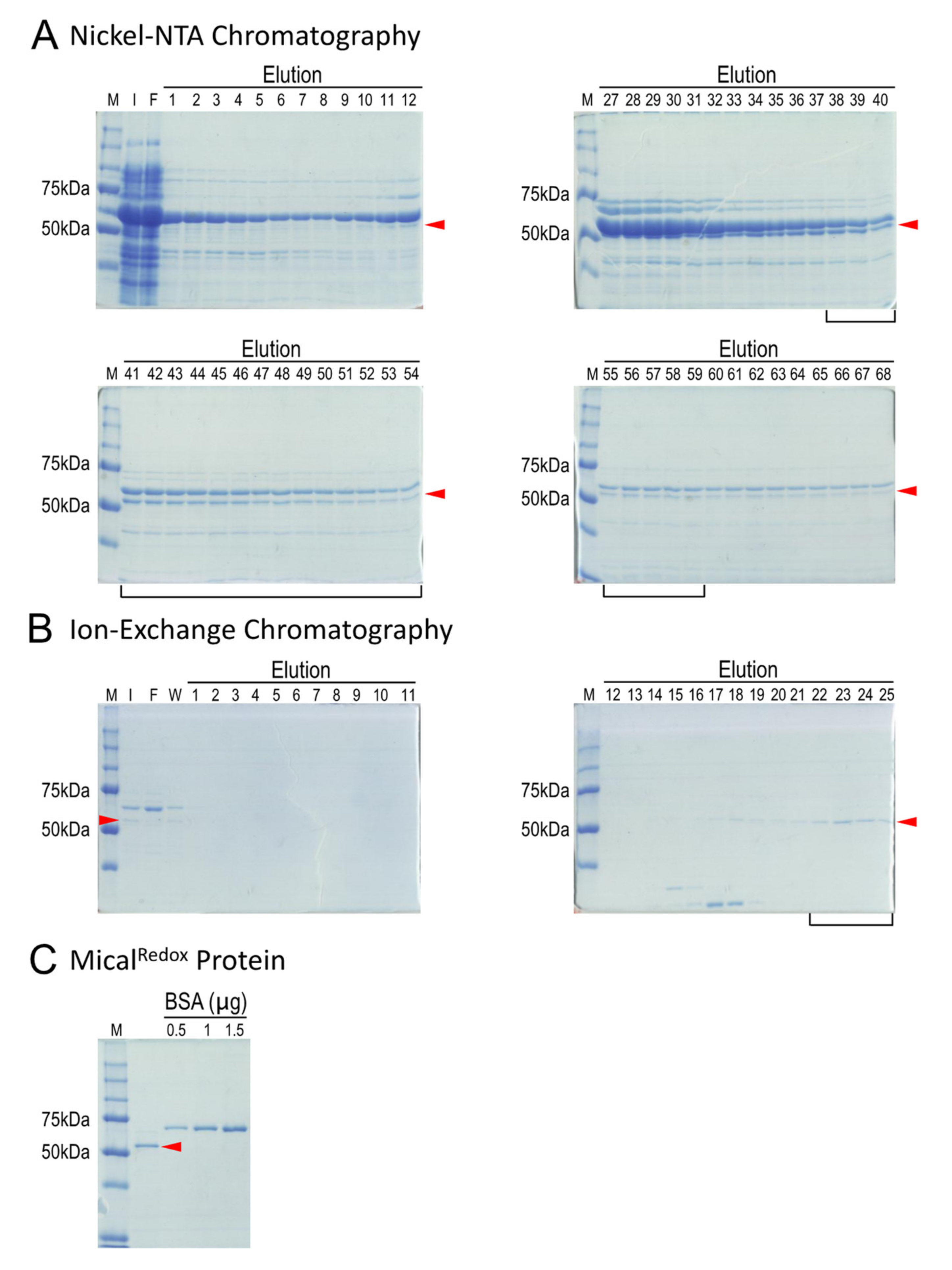
2.4. Simple Two-Step Purification Strategy for Obtaining MicalRedox Protein
2.5. Biochemical Analysis of Purified MicalRedox Protein: F-actin Triggered Catalytic Activity of MicalRedox
2.6. Biochemical Analysis of Purified MicalRedox Protein: F-actin Disassembly Activity of MicalRedox
3. Discussion
4. Materials and Methods
4.1. Chemicals and Chromatography Reagents
4.2. Molecular Biology and Protein Expression
4.3. Homogenization and Clarification of Cell Lysates
4.4. Ni2+-NTA Chromatography and Examination of Fractions
4.5. Ion Exchange Chromatography and Examination of Fractions
4.6. Buffer Exchange, Sample Concentration, Protein Quantification and Western Blotting
4.7. Analysis of MicalRedox Protein: Catalytic (NADPH Consumption) Assays
4.8. Analysis of MicalRedox Protein: F-actin Disassembly Assays
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pollard, T.D. Actin and Actin-Binding Proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, a018226. [Google Scholar] [CrossRef]
- Goodson, H.V.; Jonasson, E.M. Microtubules and Microtubule-Associated Proteins. Cold Spring Harb. Perspect. Biol. 2018, 10, a022608. [Google Scholar] [CrossRef]
- Terman, J.R.; Kashina, A. Post-translational modification and regulation of actin. Curr. Opin. Cell Biol. 2013, 25, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Varland, S.; Vandekerckhove, J.; Drazic, A. Actin Post-translational Modifications: The Cinderella of Cytoskeletal Control. Trends Biochem. Sci. 2019, 44, 502–516. [Google Scholar] [CrossRef] [PubMed]
- Hung, R.-J.; Terman, J.R. Extracellular inhibitors, repellents and Semaphorin/Plexin/MICAL-mediated actin filament disassembly. Cytoskeleton 2011, 68, 415–433. [Google Scholar] [CrossRef]
- Rich, S.K.; Terman, J.R. Axon formation, extension and navigation: Only a neuroscience phenomenon? Curr. Opin. Neurobiol. 2018, 53, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Alto, L.T.; Terman, J.R. Semaphorins and their Signaling Mechanisms. Methods Mol. Biol. 2017, 1493, 1–25. [Google Scholar]
- Terman, J.R.; Mao, T.; Pasterkamp, R.J.; Yu, H.H.; Kolodkin, A.L. MICALs, a family of conserved flavoprotein oxidoreductases, function in plexin-mediated axonal repulsion. Cell 2002, 109, 887–900. [Google Scholar] [CrossRef]
- Alto, L.T.; Terman, J.R. MICALs. Curr. Biol. 2018, 28, R538–R541. [Google Scholar] [CrossRef] [PubMed]
- Vanoni, M.A. Structure-function studies of MICAL, the unusual multidomain flavoenzyme involved in actin cytoskeleton dynamics. Arch. Biochem. Biophys. 2017, 632, 118–141. [Google Scholar] [CrossRef] [PubMed]
- Fremont, S.; Romet-Lemonne, G.; Houdusse, A.; Echard, A. Emerging roles of MICAL family proteins—from actin oxidation to membrane trafficking during cytokinesis. J. Cell Sci. 2017, 130, 1509–1517. [Google Scholar] [CrossRef]
- Manta, B.; Gladyshev, V.N. Regulated methionine oxidation by monooxygenases. Free Radic. Biol. Med. 2017, 109, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Hung, R.J.; Yazdani, U.; Yoon, J.; Wu, H.; Yang, T.; Gupta, N.; Huang, Z.; van Berkel, W.J.; Terman, J.R. Mical links semaphorins to F-actin disassembly. Nature 2010, 463, 823–827. [Google Scholar] [CrossRef] [PubMed]
- Hung, R.J.; Pak, C.W.; Terman, J.R. Direct redox regulation of F-actin assembly and disassembly by Mical. Science 2011, 334, 1710–1713. [Google Scholar] [CrossRef]
- Hung, R.J.; Spaeth, C.S.; Yesilyurt, H.G.; Terman, J.R. SelR reverses Mical-mediated oxidation of actin to regulate F-actin dynamics. Nat. Cell Biol. 2013, 15, 1445–1454. [Google Scholar] [CrossRef]
- Grintsevich, E.E.; Ge, P.; Sawaya, M.R.; Yesilyurt, H.G.; Terman, J.R.; Zhou, Z.H.; Reisler, E. Catastrophic disassembly of actin filaments via Mical-mediated oxidation. Nat. Commun. 2017, 8, 2183. [Google Scholar] [CrossRef]
- Lee, B.C.; Peterfi, Z.; Hoffmann, F.W.; Moore, R.E.; Kaya, A.; Avanesov, A.; Tarrago, L.; Zhou, Y.; Weerapana, E.; Fomenko, D.E.; et al. MsrB1 and MICALs Regulate Actin Assembly and Macrophage Function via Reversible Stereoselective Methionine Oxidation. Mol. Cell 2013, 51, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Grigoriev, I.; Yu, K.L.; Martinez-Sanchez, E.; Serra-Marques, A.; Smal, I.; Meijering, E.; Demmers, J.; Peranen, J.; Pasterkamp, R.J.; van der Sluijs, P.; et al. Rab6, Rab8 and MICAL3 Cooperate in Controlling Docking and Fusion of Exocytotic Carriers. Curr. Biol. 2011, 21, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Grintsevich, E.E.; Yesilyurt, H.G.; Rich, S.K.; Hung, R.J.; Terman, J.R.; Reisler, E. F-actin dismantling through a redox-driven synergy between Mical and cofilin. Nat. Cell Biol. 2016, 18, 876–885. [Google Scholar] [CrossRef]
- Yoon, J.; Kim, S.B.; Ahmed, G.; Shay, J.W.; Terman, J.R. Amplification of F-Actin Disassembly and Cellular Repulsion by Growth Factor Signaling. Dev. Cell 2017, 42, 117–129. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fremont, S.; Hammich, H.; Bai, J.; Wioland, H.; Klinkert, K.; Rocancourt, M.; Kikuti, C.; Stroebel, D.; Romet-Lemonne, G.; Pylypenko, O.; et al. Oxidation of F-actin controls the terminal steps of cytokinesis. Nat. Commun. 2017, 8, 14528. [Google Scholar] [CrossRef] [PubMed]
- Orr, B.O.; Fetter, R.D.; Davis, G.W. Retrograde semaphorin-plexin signalling drives homeostatic synaptic plasticity. Nature 2017, 550, 109–113. [Google Scholar] [CrossRef]
- Tominaga, K.; Minato, H.; Murayama, T.; Sasahara, A.; Nishimura, T.; Kiyokawa, E.; Kanauchi, H.; Shimizu, S.; Sato, A.; Nishioka, K.; et al. Semaphorin signaling via MICAL3 induces symmetric cell division to expand breast cancer stem-like cells. Proc. Natl. Acad. Sci. USA 2019, 116, 625–630. [Google Scholar] [CrossRef]
- Hamdan, H.; Lim, B.C.; Torii, T.; Joshi, A.; Konning, M.; Smith, C.; Palmer, D.J.; Ng, P.; Leterrier, C.; Oses-Prieto, J.A.; et al. Mapping axon initial segment structure and function by multiplexed proximity biotinylation. Nat. Commun. 2020, 11, 100. [Google Scholar] [CrossRef]
- Wu, H.; Yesilyurt, H.G.; Yoon, J.; Terman, J.R. The MICALs are a Family of F-actin Dismantling Oxidoreductases Conserved from Drosophila to Humans. Sci. Rep. 2018, 8, 937. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Terman, J.R. Characterization of MICAL flavoprotein oxidoreductases: Expression and solubility of different truncated forms of MICAL. In Flavins and Flavoproteins 2008 (Proceedings of the 16th International Symposium on Flavins and Flavoproteins); Frago, S., Gomez-Moreno, C., Medina, M., Eds.; Prensas Universitarias De Zaragoza: Zaragoza, Spain, 2008; pp. 345–350. [Google Scholar]
- Wu, H.; Hung, R.J.; Terman, J.R. A simple and efficient method for generating high-quality recombinant Mical enzyme for in vitro assays. Protein Expr. Purif. 2016, 127, 116–124. [Google Scholar] [CrossRef]
- Ferrer, M.; Chernikova, T.N.; Yakimov, M.M.; Golyshin, P.N.; Timmis, K.N. Chaperonins govern growth of Escherichia coli at low temperatures. Nat. Biotechnol. 2003, 21, 1266–1267. [Google Scholar] [CrossRef]
- Costa, S.; Almeida, A.; Castro, A.; Domingues, L. Fusion tags for protein solubility, purification and immunogenicity in Escherichia coli: The novel Fh8 system. Front. Microbiol. 2014, 5, 63. [Google Scholar] [CrossRef] [PubMed]
- Terpe, K. Overview of tag protein fusions: From molecular and biochemical fundamentals to commercial systems. Appl. Microbiol. Biotechnol. 2003, 60, 523–533. [Google Scholar] [CrossRef]
- Davis, G.D.; Elisee, C.; Newham, D.M.; Harrison, R.G. New fusion protein systems designed to give soluble expression in Escherichia coli. Biotechnol. Bioeng. 1999, 65, 382–388. [Google Scholar] [CrossRef]
- Harrison, R.G. Expression of soluble heterologous proteins via fusion with NusA protein. InNovation 2000, 11, 4–7. [Google Scholar]
- Kohl, T.; Schmidt, C.; Wiemann, S.; Poustka, A.; Korf, U. Automated production of recombinant human proteins as resource for proteome research. Proteome Sci. 2008, 6, 4. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Wu, H.; Terman, J.R. Data presenting a modified bacterial expression vector for expressing and purifying Nus solubility-tagged proteins. Data Brief 2016, 8, 1227–1231. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Harlow, E.; Lane, D. Bradford assay. CSH Protoc. 2006, 2006. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.A. Actin filament assembly and organization in vitro. In The Cytoskeleton: A Practical Approach; Carraway, K.L., Carraway, C.A.C., Eds.; Oxford University Press: New York, NY, USA, 1992; pp. 47–71. [Google Scholar]
- Demain, A.L.; Vaishnav, P. Production of recombinant proteins by microbes and higher organisms. Biotechnol. Adv. 2009, 27, 297–306. [Google Scholar] [CrossRef]
- Kamionka, M. Engineering of therapeutic proteins production in Escherichia coli. Curr. Pharm. Biotechnol. 2011, 12, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Bussow, K.; Scheich, C.; Sievert, V.; Harttig, U.; Schultz, J.; Simon, B.; Bork, P.; Lehrach, H.; Heinemann, U. Structural genomics of human proteins--target selection and generation of a public catalogue of expression clones. Microb. Cell Factories 2005, 4, 21. [Google Scholar] [CrossRef]
- Pacheco, B.; Crombet, L.; Loppnau, P.; Cossar, D. A screening strategy for heterologous protein expression in Escherichia coli with the highest return of investment. Protein Expr. Purif. 2012, 81, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Terpe, K. Overview of bacterial expression systems for heterologous protein production: From molecular and biochemical fundamentals to commercial systems. Appl. Microbiol. Biotechnol. 2006, 72, 211–222. [Google Scholar] [CrossRef]
- Stoscheck, C.M. Quantitation of Protein. Method Enzymol. 1990, 182, 50–68. [Google Scholar]
- Mahmood, T.; Yang, P.C. Western blot: Technique, theory and trouble shooting. N. Am. J. Med. Sci. 2012, 4, 429–434. [Google Scholar] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoon, J.; Wu, H.; Hung, R.-J.; Terman, J.R. Enhanced Production of the Mical Redox Domain for Enzymology and F-actin Disassembly Assays. Int. J. Mol. Sci. 2021, 22, 1991. https://doi.org/10.3390/ijms22041991
Yoon J, Wu H, Hung R-J, Terman JR. Enhanced Production of the Mical Redox Domain for Enzymology and F-actin Disassembly Assays. International Journal of Molecular Sciences. 2021; 22(4):1991. https://doi.org/10.3390/ijms22041991
Chicago/Turabian StyleYoon, Jimok, Heng Wu, Ruei-Jiun Hung, and Jonathan R. Terman. 2021. "Enhanced Production of the Mical Redox Domain for Enzymology and F-actin Disassembly Assays" International Journal of Molecular Sciences 22, no. 4: 1991. https://doi.org/10.3390/ijms22041991
APA StyleYoon, J., Wu, H., Hung, R.-J., & Terman, J. R. (2021). Enhanced Production of the Mical Redox Domain for Enzymology and F-actin Disassembly Assays. International Journal of Molecular Sciences, 22(4), 1991. https://doi.org/10.3390/ijms22041991