
Cracking the Breast Cancer Glyco-Code through Glycan-Lectin Interactions: Targeting Immunosuppressive Macrophages





{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
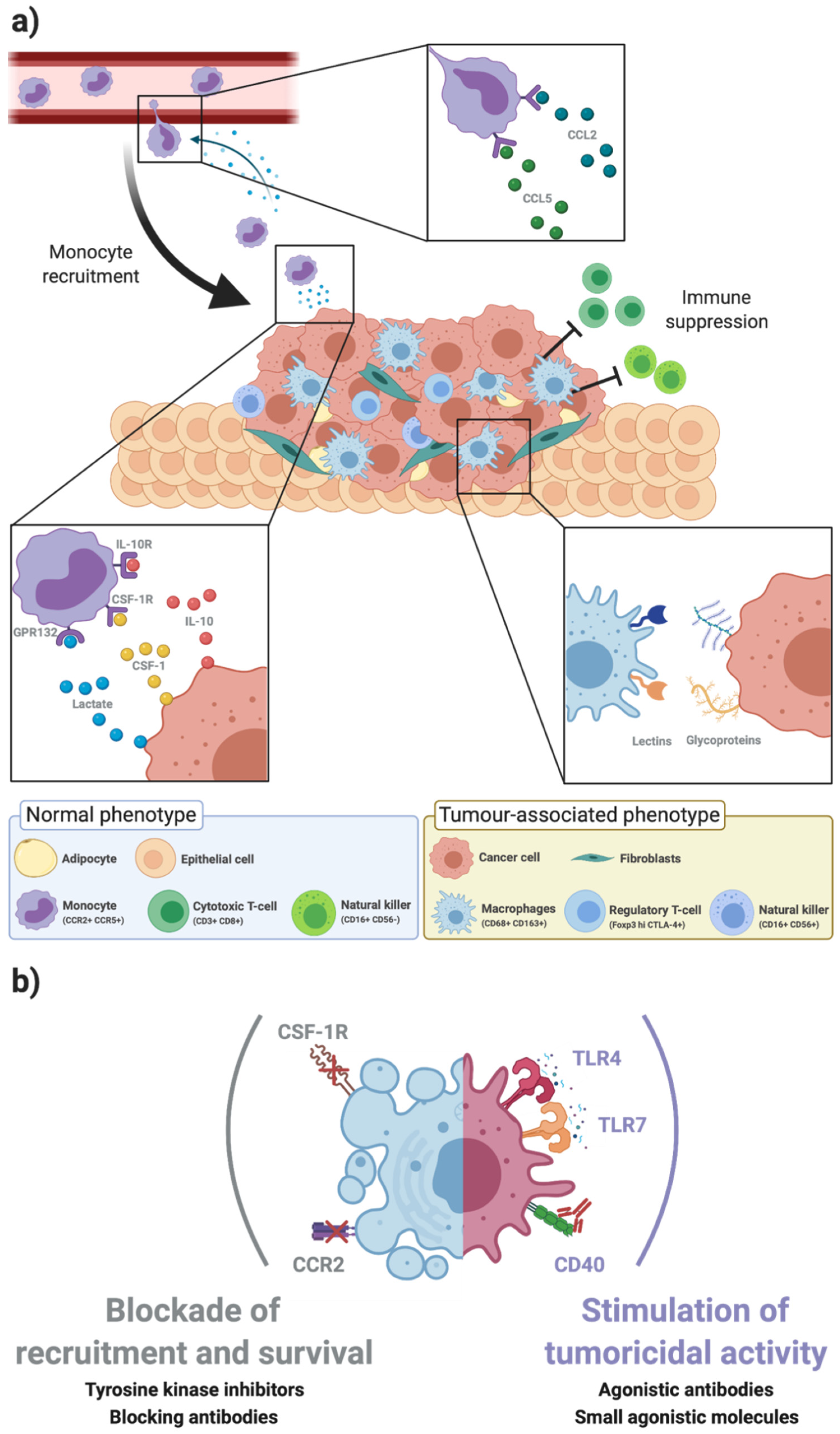
2. Tumour-Associated Macrophages within the Breast Cancer Microenvironment
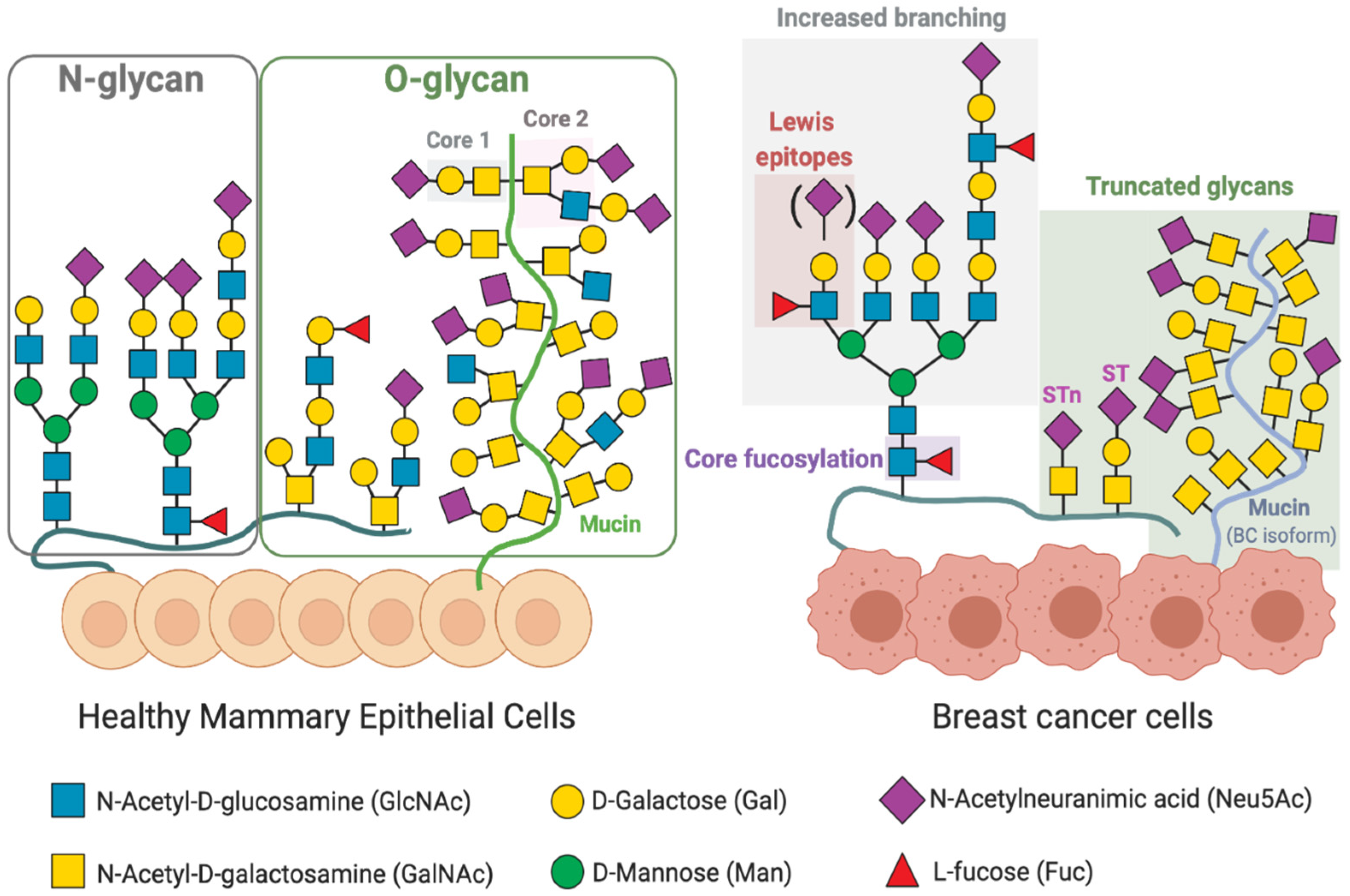
3. Altered Glycosylation Patterns in Breast Cancer
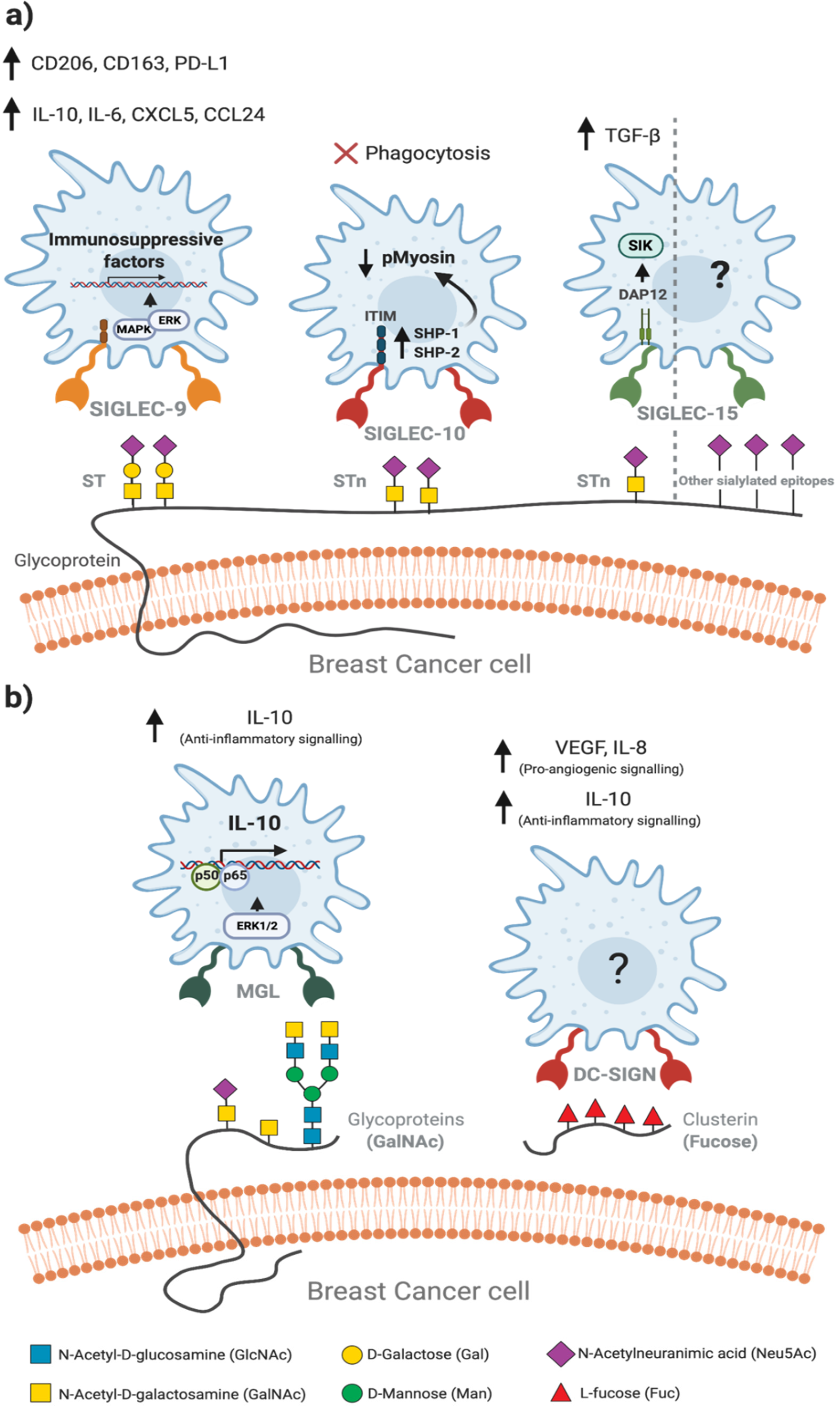
4. Tumour-Associated Macrophages Recognise Altered Glycosylation Patterns on Breast Cancer Cells
5. Glycan-Lectin Interactions: Novel Targets for Tumour-Associated Macrophage-Based Immunotherapy in Breast Cancer?
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cancer Today. Available online: https://gco.iarc.fr/today/online-analysis-pie?v=2020&mode=cancer&mode_population=continents&population=900&populations=900&key=total&sex=2&cancer=39&type=0&statistic=5&prevalence=0&population_group=0&ages_group%5B%5D=0&ages_group%5B%5D=17&nb_items=7&group_cancer=1&include_nmsc=1&include_nmsc_other=1&half_pie=0&donut=0 (accessed on 28 December 2020).
- Pusztai, L.; Karn, T.; Safonov, A.; Abu-Khalaf, M.M.; Bianchini, G. New Strategies in Breast Cancer: Immunotherapy. Clin. Cancer Res. 2016, 22, 2105–2110. [Google Scholar] [CrossRef]
- Gonzalez-Angulo, A.M.; Morales-Vasquez, F.; Hortobagyi, G.N. Overview of Resistance to Systemic Therapy in Patients with Breast Cancer. In Breast Cancer Chemosensitivity; Yu, D., Hung, M.-C., Eds.; Springer: New York, NY, USA, 2007; pp. 1–22. [Google Scholar]
- Quail, D.F.; Joyce, J.A. Microenvironmental Regulation of Tumor Progression and Metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Chrétien, S.; Zerdes, I.; Bergh, J.; Matikas, A.; Foukakis, T. Beyond PD-1/PD-L1 Inhibition: What the Future Holds for Breast Cancer Immunotherapy. Cancers 2019, 11, 628. [Google Scholar] [CrossRef]
- Pinho, S.S.; Reis, C.A. Glycosylation in Cancer: Mechanisms and Clinical Implications. Nat. Rev. Cancer 2015, 15, 540–555. [Google Scholar] [CrossRef] [PubMed]
- Burchell, J.M.; Beatson, R.; Graham, R.; Taylor-Papadimitriou, J.; Tajadura-Ortega, V. O-Linked Mucin-Type Glycosylation in Breast Cancer. Biochem. Soc. Trans. 2018, 46, 779–788. [Google Scholar] [CrossRef] [PubMed]
- Cornelissen, L.; Van Vliet, S. A Bitter Sweet Symphony: Immune Responses to Altered O-Glycan Epitopes in Cancer. Biomolecules 2016, 6, 26. [Google Scholar] [CrossRef]
- RodrÍguez, E.; Schetters, S.T.T.; van Kooyk, Y. The Tumour Glyco-Code as a Novel Immune Checkpoint for Immunotherapy. Nat. Rev. Immunol. 2018, 18, 204–211. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, Y. Tumor-Associated Macrophages: From Basic Research to Clinical Application. J. Hematol. Oncol. 2017, 10, 58. [Google Scholar] [CrossRef] [PubMed]
- Nagae, M.; Yamaguchi, Y. Sugar Recognition and Protein–Protein Interaction of Mammalian Lectins Conferring Diverse Functions. Curr. Opin. Struct. Biol. 2015, 34, 108–115. [Google Scholar] [CrossRef]
- Brown, G.D.; Crocker, P.R. Lectin Receptors Expressed on Myeloid Cells. Microbiol. Spectr. 2016, 4, 5. [Google Scholar] [CrossRef]
- Drickamer, K.; Taylor, M.E. Recent Insights into Structures and Functions of C-Type Lectins in the Immune System. Curr. Opin. Struct. Biol. 2015, 34, 26–34. [Google Scholar] [CrossRef]
- Eiro, N.; Gonzalez, L.O.; Fraile, M.; Cid, S.; Schneider, J.; Vizoso, F.J. Breast Cancer Tumor Stroma: Cellular Components, Phenotypic Heterogeneity, Intercellular Communication, Prognostic Implications and Therapeutic Opportunities. Cancers 2019, 11, 664. [Google Scholar] [CrossRef] [PubMed]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the Tumor Immune Microenvironment (TIME) for Effective Therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Goff, S.L.; Danforth, D.N. The Role of Immune Cells in Breast Tissue and Immunotherapy for the Treatment of Breast Cancer. Clin. Breast Cancer 2020, S1526820920301555. [Google Scholar] [CrossRef]
- Wagner, J.; Rapsomaniki, M.A.; Chevrier, S.; Anzeneder, T.; Langwieder, C.; Dykgers, A.; Rees, M.; Ramaswamy, A.; Muenst, S.; Soysal, S.D.; et al. A Single-Cell Atlas of the Tumor and Immune Ecosystem of Human Breast Cancer. Cell 2019, 177, 1330–1345.e18. [Google Scholar] [CrossRef]
- Savas, P.; Salgado, R.; Denkert, C.; Sotiriou, C.; Darcy, P.K.; Smyth, M.J.; Loi, S. Clinical Relevance of Host Immunity in Breast Cancer: From TILs to the Clinic. Nat. Rev. Clin. Oncol. 2016, 13, 228–241. [Google Scholar] [CrossRef] [PubMed]
- Hammerl, D.; Smid, M.; Timmermans, A.M.; Sleijfer, S.; Martens, J.W.M.; Debets, R. Breast Cancer Genomics and Immuno-Oncological Markers to Guide Immune Therapies. Semin. Cancer Biol. 2018, 52, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Jézéquel, P.; Loussouarn, D.; Guérin-Charbonnel, C.; Campion, L.; Vanier, A.; Gouraud, W.; Lasla, H.; Guette, C.; Valo, I.; Verrièle, V.; et al. Gene-Expression Molecular Subtyping of Triple-Negative Breast Cancer Tumours: Importance of Immune Response. Breast Cancer Res. 2015, 17, 43. [Google Scholar] [CrossRef] [PubMed]
- Allavena, P.; Sica, A.; Solinas, G.; Porta, C.; Mantovani, A. The Inflammatory Micro-Environment in Tumor Progression: The Role of Tumor-Associated Macrophages. Crit. Rev. Oncol. Hematol. 2008, 66, 1–9. [Google Scholar] [CrossRef]
- Bin Fang, W.; Yao, M.; Brummer, G.; Acevedo, D.; Alhakamy, N.; Berkland, C.; Cheng, N. Targeted Gene Silencing of CCL2 Inhibits Triple Negative Breast Cancer Progression by Blocking Cancer Stem Cell Renewal and M2 Macrophage Recruitment. Oncotarget 2016, 7. [Google Scholar] [CrossRef]
- Bronte, V.; Murray, P.J. Understanding Local Macrophage Phenotypes In Disease: Modulating Macrophage Function to Treat Cancer. Nat. Med. 2015, 21, 117–119. [Google Scholar] [CrossRef] [PubMed]
- Gaynor, N.; Crown, J.; Collins, D.M. Immune Checkpoint Inhibitors: Key Trials and an Emerging Role in Breast Cancer. Semin. Cancer Biol. 2020. [Google Scholar] [CrossRef]
- Kwa, M.J.; Adams, S. Checkpoint Inhibitors in Triple-Negative Breast Cancer (TNBC): Where to Go from Here: Checkpoint Inhibitors in TNBC. Cancer 2018, 124, 2086–2103. [Google Scholar] [CrossRef]
- Masoud, V.; Pagès, G. Targeted Therapies in Breast Cancer: New Challenges to Fight against Resistance. World J. Clin. Oncol. 2017, 8, 120. [Google Scholar] [CrossRef]
- Hickey, R.M.; Kulik, L.M.; Nimeiri, H.; Kalyan, A.; Kircher, S.; Desai, K.; Riaz, A.; Lewandowski, R.J.; Salem, R. Immuno-Oncology and Its Opportunities for Interventional Radiologists: Immune Checkpoint Inhibition and Potential Synergies with Interventional Oncology Procedures. J. Vasc. Interv. Radiol. 2017, 28, 1487–1494. [Google Scholar] [CrossRef]
- Planes-Laine, G.; Rochigneux, P.; Bertucci, F.; Chrétien, A.S.; Viens, P.; Sabatier, R.; Gonçalves, A. PD-1/PD-L1 Targeting in Breast Cancer: The First Clinical Evidences Are Emerging. A Literature Review. Cancers 2019, 11, 1033. [Google Scholar] [CrossRef]
- Schmid, P.; Cortes, J.; Pusztai, L.; McArthur, H.; Kümmel, S.; Bergh, J.; Denkert, C.; Park, Y.H.; Hui, R.; Harbeck, N.; et al. Pembrolizumab for Early Triple-Negative Breast Cancer. N. Engl. J. Med. 2020, 382, 810–821. [Google Scholar] [CrossRef] [PubMed]
- De Melo Gagliato, D.; Buzaid, A.C.; Perez-Garcia, J.; Cortes, J. Immunotherapy in Breast Cancer: Current Practice and Clinical Challenges. BioDrugs 2020, 34, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Pathria, P.; Louis, T.L.; Varner, J.A. Targeting Tumor-Associated Macrophages in Cancer. Trends Immunol. 2019, 40, 310–327. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.-Q.; Waaijer, S.J.H.; Zwager, M.C.; de Vries, E.G.E.; van der Vegt, B.; Schröder, C.P. Tumor-Associated Macrophages in Breast Cancer: Innocent Bystander or Important Player? Cancer Treat. Rev. 2018, 70, 178–189. [Google Scholar] [CrossRef]
- Melgarejo, E.; Medina, M.Á.; Sánchez-Jiménez, F.; Urdiales, J.L. Monocyte Chemoattractant Protein-1: A Key Mediator in Inflammatory Processes. Int. J. Biochem. Cell Biol. 2009, 41, 998–1001. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, A.; Brouillard, A.; Kumar, S.; Nandi, D.; Kulkarni, A. Dual Inhibition of CSF1R and MAPK Pathways Using Supramolecular Nanoparticles Enhances Macrophage Immunotherapy. Biomaterials 2020, 227, 119559. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Mantovani, A. Macrophage Plasticity and Polarization: In Vivo Veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Biswas, S.K.; Mantovani, A. Macrophage Plasticity and Interaction with Lymphocyte Subsets: Cancer as a Paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Cazet, A.; Julien, S.; Bobowski, M.; Burchell, J.; Delannoy, P. Tumour-Associated Carbohydrate Antigens in Breast Cancer. Breast Cancer Res. 2010, 12, 204. [Google Scholar] [CrossRef]
- Hakomori, S.-I.; Kanagi, R. Glycosphingolipids as Tumor-Associated and Differentiation Markers. J. Natl. Cancer Inst. 1983, 71, 231–251. [Google Scholar] [PubMed]
- Scott, D.A.; Drake, R.R. Glycosylation and Its Implications in Breast Cancer. Expert Rev. Proteom. 2019, 16, 665–680. [Google Scholar] [CrossRef]
- Peixoto, A.; Relvas-Santos, M.; Azevedo, R.; Santos, L.L.; Ferreira, J.A. Protein Glycosylation and Tumor Microenvironment Alterations Driving Cancer Hallmarks. Front. Oncol. 2019, 9, 380. [Google Scholar] [CrossRef]
- Varki, A.; Cummings, R.D.; Aebi, M.; Packer, N.H.; Seeberger, P.H.; Esko, J.D.; Stanley, P.; Hart, G.; Darvill, A.; Kinoshita, T.; et al. Symbol Nomenclature for Graphical Representations of Glycans. Glycobiology 2015, 25, 1323–1324. [Google Scholar] [CrossRef]
- Burchell, J.M.; Mungul, A.; Taylor-Papadimitriou, J. O-Linked Glycosylation in the Mammary Gland: Changes That Occur During Malignancy. J. Mammary Gland Biol. Neoplasia 2001, 6, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Brockhausen, I. Mucin-type O-glycans in Human Colon and Breast Cancer: Glycodynamics and Functions. EMBO Rep. 2006, 7, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Hanish, F.-G.; Uhlenbruck, G.; Peter-Katalinic, J.; Egge, H.; Dabrowski, J.; Dabrowski, U. Structures of Neutral O-Linked Polylactosaminoglycans on Human Skim Milk Mucins: Another Type of Linear Extended Poly-N-Acetyllactosamine Backbone with Galp(1-4)GlcNAcp(1-6) Repeating Units. J. Biol. Chem. 1989, 2, 872–883. [Google Scholar] [CrossRef]
- Croce, M.V.; Isla-Larrain, M.T.; Demichelis, S.O.; Segal-Eiras, A.; Gori, J.R.; Price, M.R. Tissue and Serum MUC1 Mucin Detection in Breast Cancer Patients. Breast Cancer Res. Treat. 2003, 81, 195–207. [Google Scholar] [CrossRef]
- Tajadura-Ortega, V.; Gambardella, G.; Skinner, A.; Halim, A.; Van Coillie, J.; Schjoldager, K.T.-B.G.; Beatson, R.; Graham, R.; Achkova, D.; Taylor-Papadimitriou, J.; et al. O-Linked Mucin-Type Glycosylation Regulates the Transcriptional Programme Downstream of EGFR. Glycobiology 2020, cwaa075. [Google Scholar] [CrossRef]
- Beatson, R.; Tajadura-Ortega, V.; Achkova, D.; Picco, G.; Tsourouktsoglou, T.-D.; Klausing, S.; Hillier, M.; Maher, J.; Noll, T.; Crocker, P.R.; et al. The Mucin MUC1 Modulates the Tumor Immunological Microenvironment through Engagement of the Lectin Siglec-9. Nat. Immunol. 2016, 17, 1273–1281. [Google Scholar] [CrossRef]
- Bennett, E.P.; Mandel, U.; Clausen, H.; Gerken, T.A.; Fritz, T.A.; Tabak, L.A. Control of Mucin-Type O-Glycosylation: A Classification of the Polypeptide GalNAc-Transferase Gene Family. Glycobiology 2012, 22, 736–756. [Google Scholar] [CrossRef] [PubMed]
- Freire, T.; Berois, N.; Sóñora, C.; Varangot, M.; Barrios, E.; Osinaga, E. UDP-N-Acetyl-D-Galactosamine:PolypeptideN-Acetylgalactosaminyltransferase 6 (PpGalNAc-T6) MRNA as a Potential New Marker for Detection of Bone Marrow-Disseminated Breast Cancer Cells. Int. J. Cancer 2006, 119, 1383–1388. [Google Scholar] [CrossRef]
- Wu, C.; Guo, X.; Wang, W.; Wang, Y.; Shan, Y.; Zhang, B.; Song, W.; Ma, S.; Ge, J.; Deng, H.; et al. N-Acetylgalactosaminyltransferase-14 as a Potential Biomarker for Breast Cancer by Immunohistochemistry. BMC Cancer 2010, 10, 123. [Google Scholar] [CrossRef]
- Park, J.-H.; Nishidate, T.; Kijima, K.; Ohashi, T.; Takegawa, K.; Fujikane, T.; Hirata, K.; Nakamura, Y.; Katagiri, T. Critical Roles of Mucin 1 Glycosylation by Transactivated Polypeptide N-Acetylgalactosaminyltransferase 6 in Mammary Carcinogenesis. Cancer Res. 2010, 70, 2759–2769. [Google Scholar] [CrossRef] [PubMed]
- Hakomori, S. Glycosylation Defining Cancer Malignancy: New Wine in an Old Bottle. Proc. Natl. Acad. Sci. USA 2002, 99, 10231–10233. [Google Scholar] [CrossRef]
- Dennis, J.W.; Laferte, S.; Waghorne, C.; Breitman, M.L.; Kerbel, R.S. Β1-6 Branching of Asn-Linked Oligosaccharides Is Directly Associated with Metastasis. Science 1987, 236, 4. [Google Scholar] [CrossRef]
- Seberger, P.J.; Chaney, W.G. Control of Metastasis by Asn-Linked, Β1–6 Branched Oligosaccharides in Mouse Mammary Cancer Cells. Glycobiology 1999, 9, 235–241. [Google Scholar] [CrossRef][Green Version]
- Scott, D.A.; Casadonte, R.; Cardinali, B.; Spruill, L.; Mehta, A.S.; Carli, F.; Simone, N.; Kriegsmann, M.; Del Mastro, L.; Kriegsmann, J.; et al. Increases in Tumor N-Glycan Polylactosamines Associated with Advanced HER2-Positive and Triple-Negative Breast Cancer Tissues. PROTEOM. Clin. Appl. 2019, 13, 1800014. [Google Scholar] [CrossRef]
- De Leoz, M.L.A.; Young, L.J.T.; An, H.J.; Kronewitter, S.R.; Kim, J.; Miyamoto, S.; Borowsky, A.D.; Chew, H.K.; Lebrilla, C.B. High-Mannose Glycans Are Elevated during Breast Cancer Progression. Mol. Cell. Proteom. 2010, 10, M110.002717. [Google Scholar] [CrossRef]
- Hasegawa, Y.; Takahashi, M.; Ariki, S.; Asakawa, D.; Tajiri, M.; Wada, Y.; Yamaguchi, Y.; Nishitani, C.; Takamiya, R.; Saito, A.; et al. Surfactant Protein D Suppresses Lung Cancer Progression by Downregulation of Epidermal Growth Factor Signaling. Oncogene 2015, 34, 838–845. [Google Scholar] [CrossRef]
- Johns, T.G.; Mellman, I.; Cartwright, G.A.; Ritter, G.; Old, L.J.; Burgess, A.W.; Scott, A.M. The Antitumor Monoclonal Antibody 806 Recognizes a High-mannose Form of the EGF Receptor That Reaches the Cell Surface When Cells Over-express the Receptor. FASEB J. 2005, 19, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Lomax-Browne, H.J.; Robertson, C.; Antonopoulos, A.; Leathem, A.J.C.; Haslam, S.M.; Dell, A.; Dwek, M.V. Serum IgA1 Shows Increased Levels of α 2,6-Linked Sialic Acid in Breast Cancer. Interface Focus 2019, 9, 20180079. [Google Scholar] [CrossRef]
- Scott, D.A.; Norris-Caneda, K.; Spruill, L.; Bruner, E.; Kono, Y.; Angel, P.M.; Mehta, A.S.; Drake, R.R. Specific N-Linked Glycosylation Patterns in Areas of Necrosis in Tumor Tissues. Int. J. Mass Spectrom. 2019, 437, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Scott, E.; Elliott, D.J.; Munkley, J. Tumour Associated Glycans: A Route to Boost Immunotherapy? Clin. Chim. Acta 2020, 502, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Listinsky, J.J.; Siegal, G.P.; Listinsky, C.M. The Emerging Importance of α-L-Fucose in Human Breast Cancer: A Review. Am. J. Transl. Res. 2011, 3, 292. [Google Scholar]
- Liu, H.; Ma, L.; Lin, J.; Cao, B.; Qu, D.; Luo, C.; Huang, W.; Han, L.; Xu, H.; Wu, Z.; et al. Advances in Molecular Mechanisms of Drugs Affecting Abnormal Glycosylation and Metastasis of Breast Cancer. Pharmacol. Res. 2020, 155, 104738. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-C.; Yen, H.-Y.; Chen, C.-Y.; Chen, C.-H.; Cheng, P.-F.; Juan, Y.-H.; Chen, C.-H.; Khoo, K.-H.; Yu, C.-J.; Yang, P.-C.; et al. Sialylation and Fucosylation of Epidermal Growth Factor Receptor Suppress Its Dimerization and Activation in Lung Cancer Cells. Proc. Natl. Acad. Sci. USA 2011, 108, 11332–11337. [Google Scholar] [CrossRef] [PubMed]
- Tu, C.-F.; Wu, M.-Y.; Lin, Y.-C.; Kannagi, R.; Yang, R.-B. FUT8 Promotes Breast Cancer Cell Invasiveness by Remodeling TGF-β Receptor Core Fucosylation. Breast Cancer Res. 2017, 19, 111. [Google Scholar] [CrossRef] [PubMed]
- Brooks, S.A.; Leathem, A.J. Expression of the CD15 Antigen (Lewis x) in Breast Cancer. Histochem. J. 1995, 27, 689–693. [Google Scholar] [CrossRef]
- Blanas, A.; Sahasrabudhe, N.M.; Rodríguez, E.; van Kooyk, Y.; van Vliet, S.J. Fucosylated Antigens in Cancer: An Alliance toward Tumor Progression, Metastasis, and Resistance to Chemotherapy. Front. Oncol. 2018, 8, 39. [Google Scholar] [CrossRef] [PubMed]
- Macauley, M.S.; Crocker, P.R.; Paulson, J.C. Siglec-Mediated Regulation of Immune Cell Function in Disease. Nat. Rev. Immunol. 2014, 14, 653–666. [Google Scholar] [CrossRef]
- Van de Wall, S.; Santegoets, K.C.M.; van Houtum, E.J.H.; Büll, C.; Adema, G.J. Sialoglycans and Siglecs Can Shape the Tumor Immune Microenvironment. Trends Immunol. 2020, 41, 274–285. [Google Scholar] [CrossRef]
- Läubli, H.; Borsig, L. Altered Cell Adhesion and Glycosylation Promote Cancer Immune Suppression and Metastasis. Front. Immunol. 2019, 10, 2120. [Google Scholar] [CrossRef] [PubMed]
- Beatson, R.; Graham, R.; Grundland Freile, F.; Cozzetto, D.; Kannambath, S.; Pfeifer, E.; Woodman, N.; Owen, J.; Nuamah, R.; Mandel, U.; et al. Cancer-Associated Hypersialylated MUC1 Drives the Differentiation of Human Monocytes into Macrophages with a Pathogenic Phenotype. Commun. Biol. 2020, 3, 644. [Google Scholar] [CrossRef]
- Barkal, A.A.; Brewer, R.E.; Markovic, M.; Kowarsky, M.; Barkal, S.A.; Zaro, B.W.; Krishnan, V.; Hatakeyama, J.; Dorigo, O.; Barkal, L.J.; et al. CD24 Signalling through Macrophage Siglec-10 Is a Target for Cancer Immunotherapy. Nature 2019, 572, 392–396. [Google Scholar] [CrossRef]
- Takamiya, R.; Ohtsubo, K.; Takamatsu, S.; Taniguchi, N.; Angata, T. The Interaction between Siglec-15 and Tumor-Associated Sialyl-Tn Antigen Enhances TGF- Secretion from Monocytes/Macrophages through the DAP12-Syk Pathway. Glycobiology 2013, 23, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sun, J.; Liu, L.N.; Flies, D.B.; Nie, X.; Toki, M.; Zhang, J.; Song, C.; Zarr, M.; Zhou, X.; et al. Siglec-15 as an Immune Suppressor and Potential Target for Normalization Cancer Immunotherapy. Nat. Med. 2019, 25, 656–666. [Google Scholar] [CrossRef]
- Murugesan, G.; Correia, V.G.; Palma, A.S.; Chai, W.; Li, C.; Feizi, T.; Martin, E.; Laux, B.; Franz, A.; Fuchs, K.; et al. Siglec-15 Recognition of Sialoglycans on Tumor Cell Lines Can Occur Independently of Sialyl Tn Antigen Expression. Glycobiology 2020, cwaa048. [Google Scholar] [CrossRef] [PubMed]
- Zizzari, I.G.; Napoletano, C.; Battisti, F.; Rahimi, H.; Caponnetto, S.; Pierelli, L.; Nuti, M.; Rughetti, A. MGL Receptor and Immunity: When the Ligand Can Make the Difference. J. Immunol. Res. 2015, 2015, 450695. [Google Scholar] [CrossRef] [PubMed]
- Merlotti, A.; Malizia, A.L.; Michea, P.; Bonte, P.-E.; Goudot, C.; Carregal, M.S.; Nuñez, N.; Sedlik, C.; Ceballos, A.; Soumelis, V.; et al. Aberrant Fucosylation Enables Breast Cancer Clusterin to Interact with Dendritic Cell-Specific ICAM-Grabbing Non-Integrin (DC-SIGN). OncoImmunology 2019, 8, e1629257. [Google Scholar] [CrossRef]
- Rohne, P.; Prochnow, H.; Koch-Brandt, C. The CLU-Files: Disentanglement of a Mystery. Biomol. Concepts 2016, 7, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.B.; Yeh, E.S.; Soloff, A.C. Tumor-Associated Macrophages: Unwitting Accomplices in Breast Cancer Malignancy. Npj Breast Cancer 2016, 2, 15025. [Google Scholar] [CrossRef] [PubMed]
- Ngambenjawong, C.; Gustafson, H.H.; Pun, S.H. Progress in Tumor-Associated Macrophage (TAM)-Targeted Therapeutics. Adv. Drug Deliv. Rev. 2017, 114, 206–221. [Google Scholar] [CrossRef]
- Mereiter, S.; Balmaña, M.; Campos, D.; Gomes, J.; Reis, C.A. Glycosylation in the Era of Cancer-Targeted Therapy: Where Are We Heading? Cancer Cell 2019, 36, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Wielgat, P.; Rogowski, K.; Niemirowicz-Laskowska, K.; Car, H. Sialic Acid-Siglec Axis as Molecular Checkpoints Targeting of Immune System: Smart Players in Pathology and Conventional Therapy. Int. J. Mol. Sci. 2020, 21, 4361. [Google Scholar] [CrossRef]
- Xiao, H.; Woods, E.C.; Vukojicic, P.; Bertozzi, C.R. Precision Glycocalyx Editing as a Strategy for Cancer Immunotherapy. Proc. Natl. Acad. Sci. USA 2016, 113, 10304–10309. [Google Scholar] [CrossRef]
- Gray, M.A.; Stanczak, M.A.; Mantuano, N.R.; Xiao, H.; Pijnenborg, J.F.A.; Malaker, S.A.; Miller, C.L.; Weidenbacher, P.A.; Tanzo, J.T.; Ahn, G.; et al. Targeted Glycan Degradation Potentiates the Anticancer Immune Response in Vivo. Nat. Chem. Biol. 2020, 16, 1376–1384. [Google Scholar] [CrossRef]
- Zitvogel, L.; Pitt, J.M.; Daillère, R.; Smyth, M.J.; Kroemer, G. Mouse Models in Oncoimmunology. Nat. Rev. Cancer 2016, 16, 759–773. [Google Scholar] [CrossRef]
- Bissell, M.J.; Radisky, D. Putting Tumours in Context. Nat. Rev. Cancer 2001, 1, 46–54. [Google Scholar] [CrossRef]
- Hickman, J.A.; Graeser, R.; de Hoogt, R.; Vidic, S.; Brito, C.; Gutekunst, M.; van der Kuip, H.; IMI PREDECT Consortium. Three-Dimensional Models of Cancer for Pharmacology and Cancer Cell Biology: Capturing Tumor Complexity in Vitro/Ex Vivo. Biotechnol. J. 2014, 9, 1115–1128. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Wang, J.; Liu, W.N.; Zhao, Y. Cancer Immunotherapies and Humanized Mouse Drug Testing Platforms. Transl. Oncol. 2019, 12, 987–995. [Google Scholar] [CrossRef] [PubMed]
- Santo, V.E.; Estrada, M.F.; Rebelo, S.P.; Abreu, S.; Silva, I.; Pinto, C.; Veloso, S.C.; Serra, A.T.; Boghaert, E.; Alves, P.M.; et al. Adaptable Stirred-Tank Culture Strategies for Large Scale Production of Multicellular Spheroid-Based Tumor Cell Models. J. Biotechnol. 2016, 221, 118–129. [Google Scholar] [CrossRef]
- Cartaxo, A.L.; Estrada, M.F.; Domenici, G.; Roque, R.; Silva, F.; Gualda, E.J.; Loza-Alvarez, P.; Sflomos, G.; Brisken, C.; Alves, P.M.; et al. A Novel Culture Method That Sustains ERα Signaling in Human Breast Cancer Tissue Microstructures. J. Exp. Clin. Cancer Res. 2020, 39, 161. [Google Scholar] [CrossRef] [PubMed]
- Bahcecioglu, G.; Basara, G.; Ellis, B.W.; Ren, X.; Zorlutuna, P. Breast Cancer Models: Engineering the Tumor Microenvironment. Acta Biomater. 2020, 106, 1–21. [Google Scholar] [CrossRef]
- Rodrigues, J.; Heinrich, M.A.; Teixeira, L.M.; Prakash, J. 3D In Vitro Model (R)Evolution: Unveiling Tumor–Stroma Interactions. Trends Cancer 2020, S2405-8033(20)30283-1. [Google Scholar] [CrossRef]
- Estrada, M.F.; Rebelo, S.P.; Davies, E.J.; Pinto, M.T.; Pereira, H.; Santo, V.E.; Smalley, M.J.; Barry, S.T.; Gualda, E.J.; Alves, P.M.; et al. Modelling the Tumour Microenvironment in Long-Term Microencapsulated 3D Co-Cultures Recapitulates Phenotypic Features of Disease Progression. Biomaterials 2016, 78, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Rebelo, S.P.; Pinto, C.; Martins, T.R.; Harrer, N.; Estrada, M.F.; Loza-Alvarez, P.; Cabeçadas, J.; Alves, P.M.; Gualda, E.J.; Sommergruber, W.; et al. 3D-3-Culture: A Tool to Unveil Macrophage Plasticity in the Tumour Microenvironment. Biomaterials 2018, 163, 185–197. [Google Scholar] [CrossRef]
- Lopes, N.; Cartaxo, A.L.; Pinto, C.; Rebelo, S.; Brito, C. Exploiting 3D Co-Culture Models to Depict the Phenotype of Tumor Associated Macrophages in Breast Cancer. In Proceedings of the 8th International Conference on Tumor Microenvironment Progression, Therapy & Prevention, Lisbon, Portugal, 10–14 June 2018. O50. [Google Scholar] [CrossRef]
- Balmaña, M.; Mereiter, S.; Diniz, F.; Feijão, T.; Barrias, C.; Reis, C. Multicellular Human Gastric Cancer Spheroids Mimic the Glycosylation Phenotype of Gastric Carcinomas. Molecules 2018, 23, 2815. [Google Scholar] [CrossRef]
- Coelho, R.; Marcos-Silva, L.; Mendes, N.; Pereira, D.; Brito, C.; Jacob, F.; Steentoft, C.; Mandel, U.; Clausen, H.; David, L.; et al. Mucins and Truncated O-Glycans Unveil Phenotypic Discrepancies between Serous Ovarian Cancer Cell Lines and Primary Tumours. Int. J. Mol. Sci. 2018, 19, 2045. [Google Scholar] [CrossRef]
- Mao, Y.; Zhao, Y.; Zhang, Y.; Yang, H. In-Depth Characterization and Comparison of the N-Glycosylated Proteome of Two-Dimensional- and Three-Dimensional-Cultured Breast Cancer Cells and Xenografted Tumors. PLoS ONE 2020, 15, e0243789. [Google Scholar] [CrossRef]
- Lübbers, J.; Rodríguez, E.; van Kooyk, Y. Modulation of Immune Tolerance via Siglec-Sialic Acid Interactions. Front. Immunol. 2018, 9, 2807. [Google Scholar] [CrossRef] [PubMed]
- Solinas, G.; Germano, G.; Mantovani, A.; Allavena, P. Tumor-Associated Macrophages (TAM) as Major Players of the Cancer-Related Inflammation. J. Leukoc. Biol. 2009, 86, 1065–1073. [Google Scholar] [CrossRef] [PubMed]
- De Haan, N.; Wuhrer, M.; Ruhaak, L.R. Mass Spectrometry in Clinical Glycomics: The Path from Biomarker Identification to Clinical Implementation. Clin. Mass Spectrom. 2020, 18, 1–12. [Google Scholar] [CrossRef]
- Kailemia, M.J.; Xu, G.; Wong, M.; Li, Q.; Goonatilleke, E.; Leon, F.; Lebrilla, C.B. Recent Advances in the Mass Spectrometry Methods for Glycomics and Cancer. Anal. Chem. 2018, 90, 208–224. [Google Scholar] [CrossRef] [PubMed]
- Palma, A.S.; Feizi, T.; Childs, R.A.; Chai, W.; Liu, Y. The Neoglycolipid (NGL)-Based Oligosaccharide Microarray System Poised to Decipher the Meta-Glycome. Curr. Opin. Chem. Biol. 2014, 18, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Gao, C.; Zhang, Y.; Palma, A.S.; Childs, R.A.; Silva, L.M.; Liu, Y.; Jiang, X.; Liu, Y.; Chai, W.; et al. O-Glycome Beam Search Arrays for Carbohydrate Ligand Discovery. Mol. Cell. Proteom. 2018, 17, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, Q.; Ashline, D.; Zhu, Y.; Lasanajak, Y.; Chernova, T.; Reinhold, V.; Cummings, R.D.; Wang, P.G.; Ju, T.; et al. Amplification and Preparation of Cellular O-Glycomes for Functional Glycomics. Anal. Chem. 2020, 92, 10390–10401. [Google Scholar] [CrossRef] [PubMed]
- Gimeno, A.; Valverde, P.; Ardá, A.; Jiménez-Barbero, J. Glycan Structures and Their Interactions with Proteins. A NMR View. Curr. Opin. Struct. Biol. 2020, 62, 22–30. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lopes, N.; Correia, V.G.; Palma, A.S.; Brito, C. Cracking the Breast Cancer Glyco-Code through Glycan-Lectin Interactions: Targeting Immunosuppressive Macrophages. Int. J. Mol. Sci. 2021, 22, 1972. https://doi.org/10.3390/ijms22041972
Lopes N, Correia VG, Palma AS, Brito C. Cracking the Breast Cancer Glyco-Code through Glycan-Lectin Interactions: Targeting Immunosuppressive Macrophages. International Journal of Molecular Sciences. 2021; 22(4):1972. https://doi.org/10.3390/ijms22041972
Chicago/Turabian StyleLopes, Nuno, Viviana G. Correia, Angelina S. Palma, and Catarina Brito. 2021. "Cracking the Breast Cancer Glyco-Code through Glycan-Lectin Interactions: Targeting Immunosuppressive Macrophages" International Journal of Molecular Sciences 22, no. 4: 1972. https://doi.org/10.3390/ijms22041972
APA StyleLopes, N., Correia, V. G., Palma, A. S., & Brito, C. (2021). Cracking the Breast Cancer Glyco-Code through Glycan-Lectin Interactions: Targeting Immunosuppressive Macrophages. International Journal of Molecular Sciences, 22(4), 1972. https://doi.org/10.3390/ijms22041972