
Allosteric Modulation of GPCRs of Class A by Cholesterol


Abstract
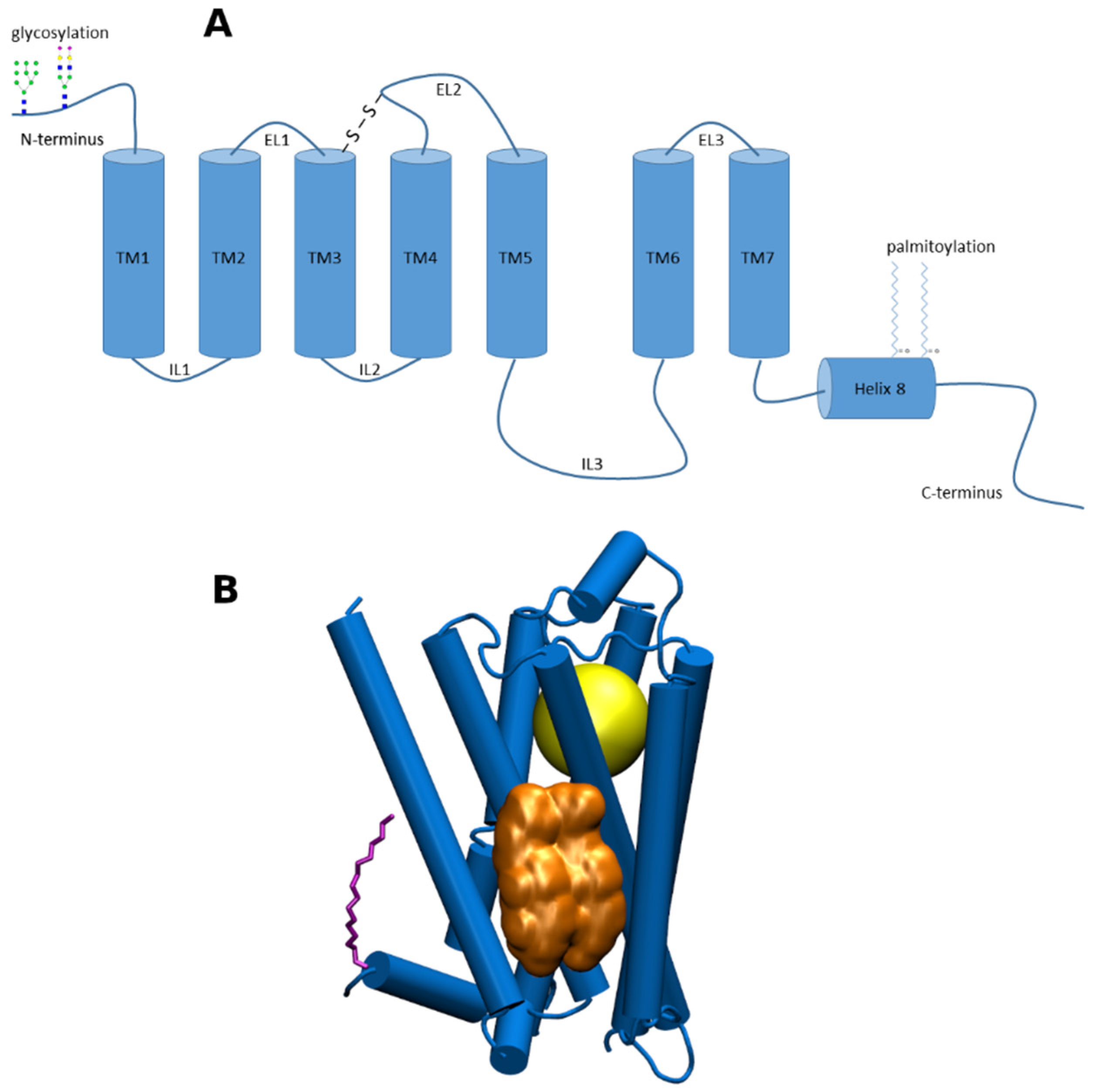
1. Introduction
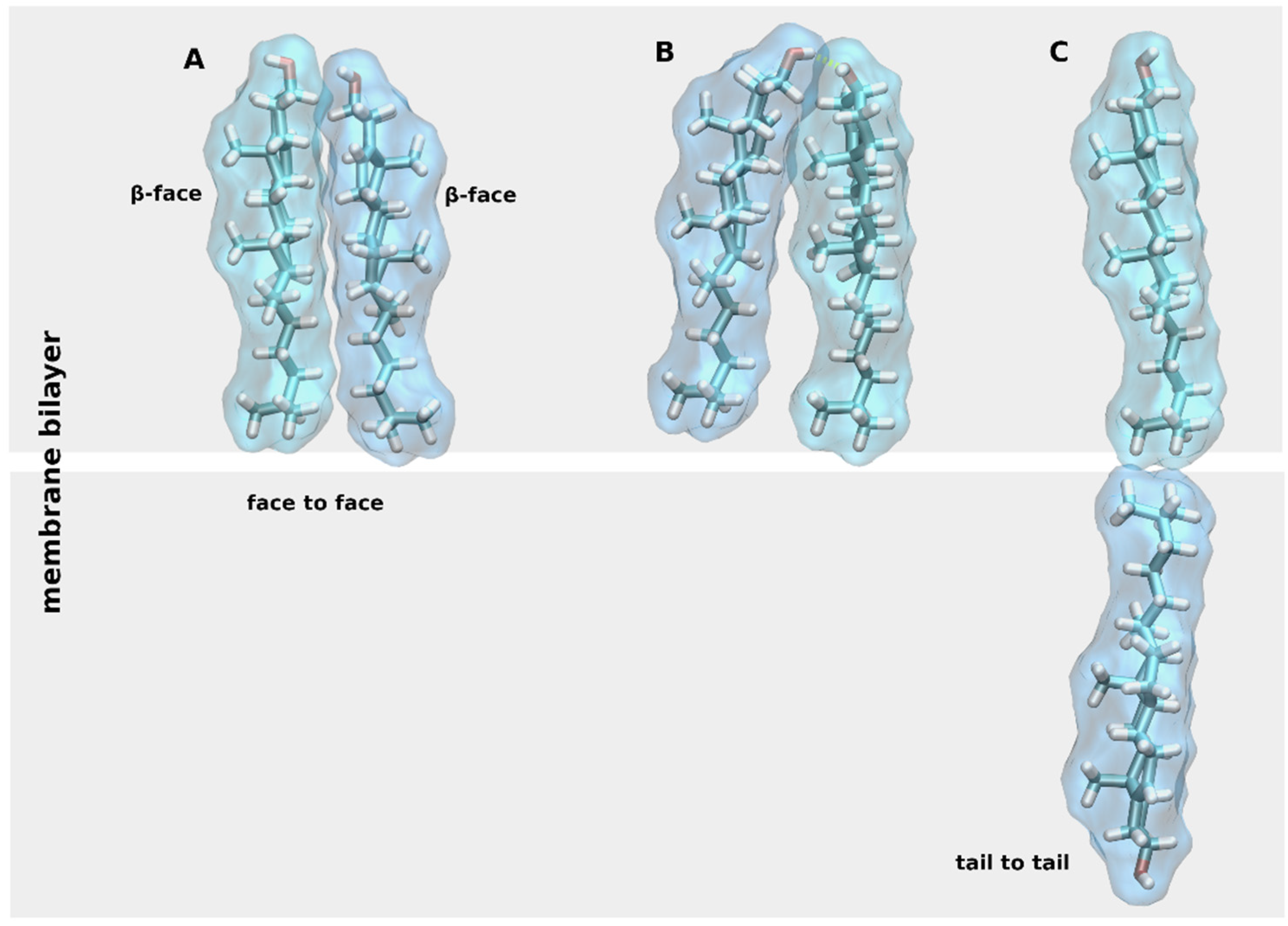
2. Chemical Properties of Membrane CLR
3. General Mechanisms of Cholesterol Action at GPCRs
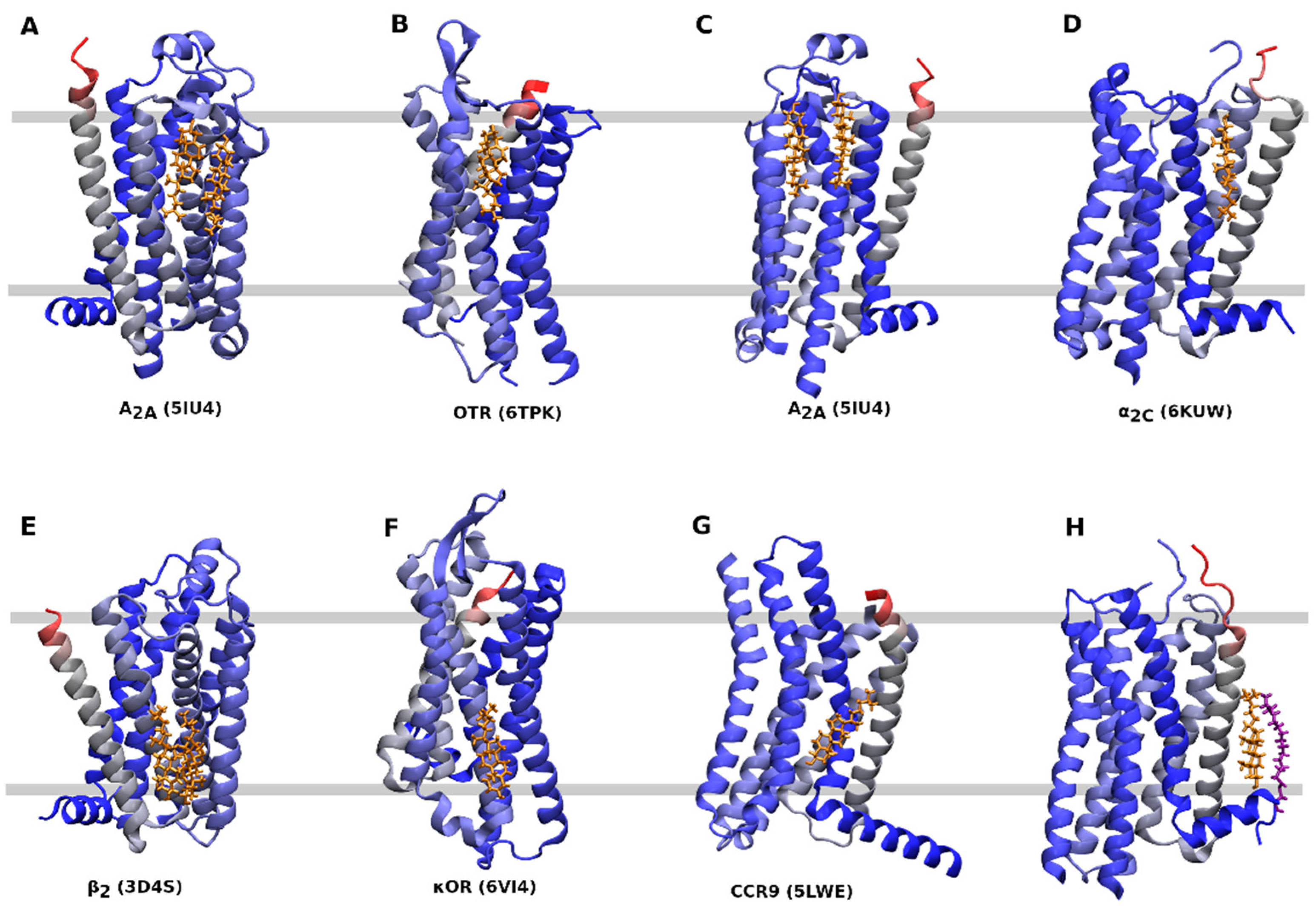
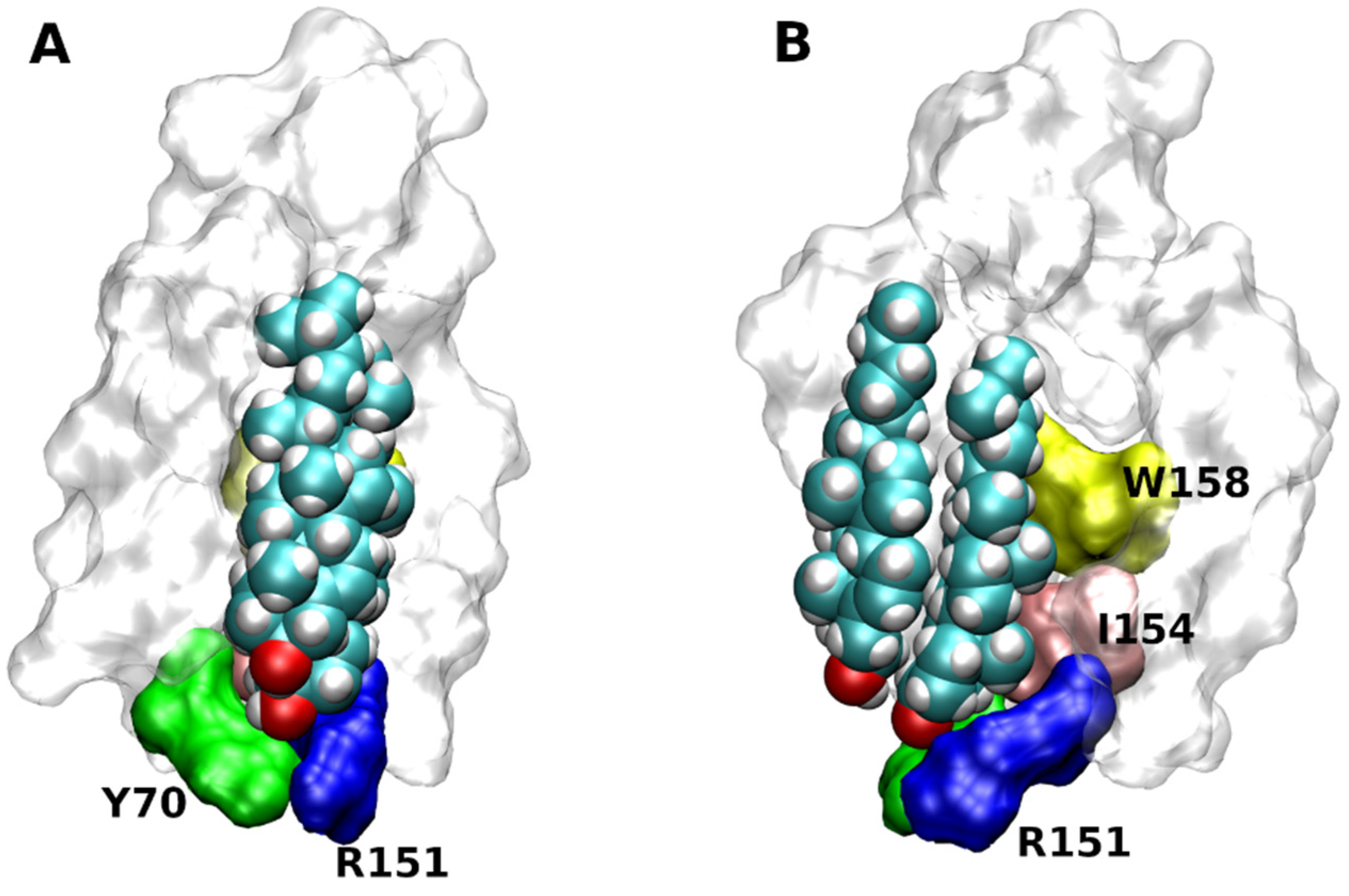
4. Binding of Cholesterol to GPCRs
5. Effects of CLR on Ligand Binding
6. Effects of CLR on the Functional Response of GPCRs
7. Effects of CLR on Oligomerization of GPCRs
8. Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schiöth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef]
- Zhang, Y.; Doruker, P.; Kaynak, B.; Zhang, S.; Krieger, J.; Li, H.; Bahar, I. Intrinsic dynamics is evolutionarily optimized to enable allosteric behavior. Curr. Opin. Struct. Biol. 2020, 62, 14–21. [Google Scholar] [CrossRef]
- Gimpl, G. Interaction of G protein coupled receptors and cholesterol. Chem. Phys. Lipids 2016, 199, 61–73. [Google Scholar] [CrossRef]
- Sarkar, P.; Chattopadhyay, A. Cholesterol interaction motifs in G protein-coupled receptors: Slippery hot spots? Wiley Interdiscip. Rev. Syst. Biol. Med. 2020, e1481. [Google Scholar] [CrossRef]
- Rose, I.A.; Hanson, K.R.; Wilkinson, K.D.; Wimmer, M.J. A suggestion for naming faces of ring compounds. Proc. Natl. Acad. Sci. USA 1980, 77, 2439–2441. [Google Scholar] [CrossRef]
- Bandara, A.; Panahi, A.; Pantelopulos, G.A.; Straub, J.E. Exploring the structure and stability of cholesterol dimer formation in multicomponent lipid bilayers. J. Comput. Chem. 2017, 38, 1479–1488. [Google Scholar] [CrossRef]
- Fantini, J.; Barrantes, F.J. How cholesterol interacts with membrane proteins: An exploration of cholesterol-binding sites including CRAC, CARC, and tilted domains. Front. Physiol. 2013, 4, 31. [Google Scholar] [CrossRef]
- Mukherjee, S.; Chattopadhyay, A. Monitoring cholesterol organization in membranes at low concentrations utilizing the wavelength-selective fluorescence approach. Chem. Phys. Lipids 2005, 134, 79–84. [Google Scholar] [CrossRef]
- Niemelä, P.S.; Ollila, S.; Hyvönen, M.T.; Karttunen, M.; Vattulainen, I. Assessing the nature of lipid raft membranes. PLoS Comput. Biol. 2007, 3, 0304–0312. [Google Scholar] [CrossRef]
- Simons, K.; Toomre, D. Lipid rafts and signal transduction. Nat. Rev. Mol. cell Biol. 2000, 1, 31–39. [Google Scholar] [CrossRef]
- Killian, J.A. Hydrophobic mismatch between proteins and lipids in membranes. Biochim. Biophys. Acta Rev. Biomembr. 1998, 1376, 401–416. [Google Scholar] [CrossRef]
- Lei, B.; Morris, D.P.; Smith, M.P.; Schwinn, D.A. Lipid rafts constrain basal α1A-adrenergic receptor signaling by maintaining receptor in an inactive conformation. Cell. Signal. 2009, 21, 1532–1539. [Google Scholar] [CrossRef]
- Ostrom, R.S.; Insel, P.A. The evolving role of lipid rafts and caveolae in G protein-coupled receptor signaling: Implications for molecular pharmacology. Br. J. Pharmacol. 2004, 143, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Legler, D.F.; Matti, C.; Laufer, J.M.; Jakobs, B.D.; Purvanov, V.; Uetz-von Allmen, E.; Thelen, M. Modulation of Chemokine Receptor Function by Cholesterol: New Prospects for Pharmacological Intervention. Mol. Pharmacol. 2017, 91, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Sviridov, D.; Mukhamedova, N.; Miller, Y.I. Lipid rafts as a therapeutic target. J. Lipid Res. 2020, 61, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Cherezov, V.; Rosenbaum, D.M.; Hanson, M.A.; Rasmussen, S.G.F.; Thian, F.S.; Kobilka, T.S.; Choi, H.-J.; Kuhn, P.; Weis, W.I.; Kobilka, B.K.; et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science 2007, 318, 1258–1265. [Google Scholar] [CrossRef]
- Hanson, M.A.; Cherezov, V.; Griffith, M.T.; Roth, C.B.; Jaakola, V.-P.; Chien, E.Y.T.; Velasquez, J.; Kuhn, P.; Stevens, R.C. A specific cholesterol binding site is established by the 2.8 A structure of the human beta2-adrenergic receptor. Structure 2008, 16, 897–905. [Google Scholar] [CrossRef]
- Ishchenko, A.; Stauch, B.; Han, G.W.; Batyuk, A.; Shiriaeva, A.; Li, C.; Zatsepin, N.; Weierstall, U.; Liu, W.; Nango, E.; et al. Toward G protein-coupled receptor structure-based drug design using X-ray lasers. IUCrJ 2019, 6, 1106–1119. [Google Scholar] [CrossRef]
- Claff, T.; Yu, J.; Blais, V.; Patel, N.; Martin, C.; Wu, L.; Han, G.W.; Holleran, B.J.; Van der Poorten, O.; White, K.L.; et al. Elucidating the active δ-opioid receptor crystal structure with peptide and small-molecule agonists. Sci. Adv. 2019, 5, eaax9115. [Google Scholar] [CrossRef]
- Che, T.; English, J.; Krumm, B.E.; Kim, K.; Pardon, E.; Olsen, R.H.J.; Wang, S.; Zhang, S.; Diberto, J.F.; Sciaky, N.; et al. Nanobody-enabled monitoring of kappa opioid receptor states. Nat. Commun. 2020, 11, 1145. [Google Scholar] [CrossRef]
- Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Mathiesen, J.M.; Sunahara, R.K.; Pardo, L.; Weis, W.I.; Kobilka, B.K.; Granier, S. Crystal structure of the µ-opioid receptor bound to a morphinan antagonist. Nature 2012, 485, 321–326. [Google Scholar] [CrossRef]
- Huang, W.; Manglik, A.; Venkatakrishnan, A.J.; Laeremans, T.; Feinberg, E.N.; Sanborn, A.L.; Kato, H.E.; Livingston, K.E.; Thorsen, T.S.; Kling, R.C.; et al. Structural insights into µ-opioid receptor activation. Nature 2015, 524, 315–321. [Google Scholar] [CrossRef]
- Wu, H.; Wacker, D.; Mileni, M.; Katritch, V.; Han, G.W.; Vardy, E.; Liu, W.; Thompson, A.A.; Huang, X.-P.; Carroll, F.I.; et al. Structure of the human κ-opioid receptor in complex with JDTic. Nature 2012, 485, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Wacker, D.; Wang, S.; McCorvy, J.D.; Betz, R.M.; Venkatakrishnan, A.J.; Levit, A.; Lansu, K.; Schools, Z.L.; Che, T.; Nichols, D.E.; et al. Crystal Structure of an LSD-Bound Human Serotonin Receptor. Cell 2017, 168, 377–389. [Google Scholar] [CrossRef]
- McCorvy, J.D.; Wacker, D.; Wang, S.; Agegnehu, B.; Liu, J.; Lansu, K.; Tribo, A.R.; Olsen, R.H.J.; Che, T.; Jin, J.; et al. Structural determinants of 5-HT2B receptor activation and biased agonism. Nat. Struct. Mol. Biol. 2018, 25, 787–796. [Google Scholar] [CrossRef]
- Liu, W.; Chun, E.; Thompson, A.A.; Chubukov, P.; Xu, F.; Katritch, V.; Han, G.W.; Roth, C.B.; Heitman, L.H.; IJzerman, A.P.; et al. Structural basis for allosteric regulation of GPCRs by sodium ions. Science 2012, 337, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Segala, E.; Guo, D.; Cheng, R.K.Y.; Bortolato, A.; Deflorian, F.; Doré, A.S.; Errey, J.C.; Heitman, L.H.; IJzerman, A.P.; Marshall, F.H.; et al. Controlling the Dissociation of Ligands from the Adenosine A2A Receptor through Modulation of Salt Bridge Strength. J. Med. Chem. 2016, 59, 6470–6479. [Google Scholar] [CrossRef] [PubMed]
- Wingler, L.M.; Skiba, M.A.; McMahon, C.; Staus, D.P.; Kleinhenz, A.L.W.W.; Suomivuori, C.-M.M.; Latorraca, N.R.; Dror, R.O.; Lefkowitz, R.J.; Kruse, A.C. Angiotensin and biased analogs induce structurally distinct active conformations within a GPCR. Science 2020, 367, 888–892. [Google Scholar] [CrossRef]
- Krishna Kumar, K.; Shalev-Benami, M.; Robertson, M.J.; Hu, H.; Banister, S.D.; Hollingsworth, S.A.; Latorraca, N.R.; Kato, H.E.; Hilger, D.; Maeda, S.; et al. Structure of a Signaling Cannabinoid Receptor 1-G Protein Complex. Cell 2019, 176, 448–458.e12. [Google Scholar] [CrossRef] [PubMed]
- Xing, C.; Zhuang, Y.; Xu, T.H.; Feng, Z.; Zhou, X.E.; Chen, M.; Wang, L.; Meng, X.; Xue, Y.; Wang, J.; et al. Cryo-EM Structure of the Human Cannabinoid Receptor CB2-Gi Signaling Complex. Cell 2020, 180, 645–654.e13. [Google Scholar] [CrossRef]
- Oswald, C.; Rappas, M.; Kean, J.; Doré, A.S.; Errey, J.C.; Bennett, K.; Deflorian, F.; Christopher, J.A.; Jazayeri, A.; Mason, J.S.; et al. Intracellular allosteric antagonism of the CCR9 receptor. Nature 2016, 540, 462–465. [Google Scholar] [CrossRef] [PubMed]
- Gusach, A.; Luginina, A.; Marin, E.; Brouillette, R.L.; Besserer-Offroy, É.; Longpré, J.-M.; Ishchenko, A.; Popov, P.; Patel, N.; Fujimoto, T.; et al. Structural basis of ligand selectivity and disease mutations in cysteinyl leukotriene receptors. Nat. Commun. 2019, 10, 5573. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Wu, L.; Yuan, S.; Wu, M.; Xu, Y.; Sun, Q.; Li, S.; Zhao, S.; Hua, T.; Liu, Z.-J. Structural basis of CXC chemokine receptor 2 activation and signalling. Nature 2020, 585, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Miles, T.F.; Spiess, K.; Jude, K.M.; Tsutsumi, N.; Burg, J.S.; Ingram, J.R.; Waghray, D.; Hjorto, G.M.; Larsen, O.; Ploegh, H.L.; et al. Viral GPCR US28 can signal in response to chemokine agonists of nearly unlimited structural degeneracy. Elife 2018, 7. [Google Scholar] [CrossRef]
- Shihoya, W.; Nishizawa, T.; Yamashita, K.; Inoue, A.; Hirata, K.; Kadji, F.M.N.; Okuta, A.; Tani, K.; Aoki, J.; Fujiyoshi, Y.; et al. X-ray structures of endothelin ETB receptor bound to clinical antagonist bosentan and its analog. Nat. Struct. Mol. Biol. 2017, 24, 758–764. [Google Scholar] [CrossRef]
- Chen, T.; Xiong, M.; Zong, X.; Ge, Y.; Zhang, H.; Wang, M.; Won Han, G.; Yi, C.; Ma, L.; Ye, R.D.; et al. Structural basis of ligand binding modes at the human formyl peptide receptor 2. Nat. Commun. 2020, 11, 1208. [Google Scholar] [CrossRef]
- Zhuang, Y.; Liu, H.; Edward Zhou, X.; Kumar Verma, R.; de Waal, P.W.; Jang, W.; Xu, T.-H.; Wang, L.; Meng, X.; Zhao, G.; et al. Structure of formylpeptide receptor 2-Gi complex reveals insights into ligand recognition and signaling. Nat. Commun. 2020, 11, 885. [Google Scholar] [CrossRef]
- Waltenspühl, Y.; Schöppe, J.; Ehrenmann, J.; Kummer, L.; Plückthun, A. Crystal structure of the human oxytocin receptor. Sci. Adv. 2020, 6, eabb5419. [Google Scholar] [CrossRef]
- Zhang, K.; Zhang, J.; Gao, Z.-G.; Zhang, D.; Zhu, L.; Han, G.W.; Moss, S.M.; Paoletta, S.; Kiselev, E.; Lu, W.; et al. Structure of the human P2Y12 receptor in complex with an antithrombotic drug. Nature 2014, 509, 115–118. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, Z.-G.; Zhang, K.; Kiselev, E.; Crane, S.; Wang, J.; Paoletta, S.; Yi, C.; Ma, L.; Zhang, W.; et al. Two disparate ligand-binding sites in the human P2Y1 receptor. Nature 2015, 520, 317–321. [Google Scholar] [CrossRef]
- Li, H.; Papadopoulos, V. Peripheral-type benzodiazepine receptor function in cholesterol transport. Identification of a putative cholesterol recognition/interaction amino acid sequence and consensus pattern. Endocrinology 1998, 139, 4991–4997. [Google Scholar] [CrossRef] [PubMed]
- Jafurulla, M.; Tiwari, S.; Chattopadhyay, A. Identification of cholesterol recognition amino acid consensus (CRAC) motif in G-protein coupled receptors. Biochem. Biophys. Res. Commun. 2011, 404, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Thal, D.M.; Sun, B.; Feng, D.; Nawaratne, V.; Leach, K.; Felder, C.C.; Bures, M.G.; Evans, D.A.; Weis, W.I.; Bachhawat, P.; et al. Crystal structures of the M1 and M4 muscarinic acetylcholine receptors. Nature 2016, 531, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Xu, J.; Kadji, F.M.N.; Clark, M.J.; Zhao, J.; Tsutsumi, N.; Aoki, J.; Sunahara, R.K.; Inoue, A.; Garcia, K.C.; et al. Structure and selectivity engineering of the M1 muscarinic receptor toxin complex. Science 2020, 369, 161–167. [Google Scholar] [CrossRef]
- Sengupta, D.; Chattopadhyay, A. Molecular dynamics simulations of GPCR-cholesterol interaction: An emerging paradigm. Biochim. Biophys. Acta 2015, 1848, 1775–1782. [Google Scholar] [CrossRef]
- Lee, A.G. Interfacial Binding Sites for Cholesterol on G Protein-Coupled Receptors. Biophys. J. 2019, 116, 1586–1597. [Google Scholar] [CrossRef]
- Cang, X.; Du, Y.; Mao, Y.; Wang, Y.; Yang, H.; Jiang, H. Mapping the functional binding sites of cholesterol in β2-adrenergic receptor by long-time molecular dynamics simulations. J. Phys. Chem. B 2013, 117, 1085–1094. [Google Scholar] [CrossRef]
- McGraw, C.; Yang, L.; Levental, I.; Lyman, E.; Robinson, A.S. Membrane cholesterol depletion reduces downstream signaling activity of the adenosine A2A receptor. Biochim. Biophys. Acta Biomembr. 2019, 1861, 760–767. [Google Scholar] [CrossRef]
- Rouviere, E.; Arnarez, C.; Yang, L.; Lyman, E. Identification of Two New Cholesterol Interaction Sites on the A2A Adenosine Receptor. Biophys. J. 2017, 113, 2415–2424. [Google Scholar] [CrossRef] [PubMed]
- Randáková, A.; Dolejší, E.; Rudajev, V.; Zimčík, P.; Doležal, V.; El-Fakahany, E.E.; Jakubík, J. Role of membrane cholesterol in differential sensitivity of muscarinic receptor subtypes to persistently bound xanomeline. Neuropharmacology 2018, 133, 129–144. [Google Scholar] [CrossRef] [PubMed]
- Ballesteros, J.A.; Weinstein, H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. In Methods in Neurosciences; Academic Press: San Diego, CA, USA, 1995; Volume 25, pp. 366–428. ISBN 9780121852955. [Google Scholar]
- Gater, D.L.; Saurel, O.; Iordanov, I.; Liu, W.; Cherezov, V.; Milon, A. Two classes of cholesterol binding sites for the β2AR revealed by thermostability and NMR. Biophys. J. 2014, 107, 2305–2312. [Google Scholar] [CrossRef] [PubMed]
- Gimpl, G.; Klein, U.; Reiländer, H.; Fahrenholz, F. Expression of the human oxytocin receptor in baculovirus-infected insect cells: High-affinity binding is induced by a cholesterol-cyclodextrin complex. Biochemistry 1995, 34, 13794–13801. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, A.; Jafurulla, M.; Kalipatnapu, S.; Pucadyil, T.J.; Harikumar, K.G. Role of cholesterol in ligand binding and G-protein coupling of serotonin1A receptors solubilized from bovine hippocampus. Biochem. Biophys. Res. Commun. 2005, 327, 1036–1041. [Google Scholar] [CrossRef]
- Jafurulla, M.; Rao, B.D.; Sreedevi, S.; Ruysschaert, J.-M.; Covey, D.F.; Chattopadhyay, A. Stereospecific requirement of cholesterol in the function of the serotonin1A receptor. Biochim. Biophys. Acta 2014, 1838, 158–163. [Google Scholar] [CrossRef]
- Calmet, P.; Cullin, C.; Cortès, S.; Vang, M.; Caudy, N.; Baccouch, R.; Dessolin, J.; Maamar, N.T.; Lecomte, S.; Tillier, B.; et al. Cholesterol impacts chemokine CCR5 receptor ligand-binding activity. FEBS J. 2020, 287, 2367–2385. [Google Scholar] [CrossRef] [PubMed]
- Babcock, G.J.; Farzan, M.; Sodroski, J. Ligand-independent dimerization of CXCR4, a principal HIV-1 coreceptor. J. Biol. Chem. 2003, 278, 3378–3385. [Google Scholar] [CrossRef]
- Ruthirakuhan, M.; Herrmann, N.; Andreazza, A.C.; Verhoeff, N.P.L.G.; Gallagher, D.; Black, S.E.; Kiss, A.; Lanctôt, K.L. 24S-Hydroxycholesterol Is Associated with Agitation Severity in Patients with Moderate-to-Severe Alzheimer’s Disease: Analyses from a Clinical Trial with Nabilone. J. Alzheimer’s Dis. 2019, 71, 21–31. [Google Scholar] [CrossRef]
- Guixà-González, R.; Albasanz, J.L.; Rodriguez-Espigares, I.; Pastor, M.; Sanz, F.; Martí-Solano, M.; Manna, M.; Martinez-Seara, H.; Hildebrand, P.W.; Martín, M.; et al. Membrane cholesterol access into a G-protein-coupled receptor. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Michal, P.; Rudajev, V.; El-Fakahany, E.E.; Dolezal, V. Membrane cholesterol content influences binding properties of muscarinic M2 receptors and differentially impacts activation of second messenger pathways. Eur. J. Pharmacol. 2009, 606, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Michal, P.; El-Fakahany, E.E.; Doležal, V. Changes in Membrane Cholesterol Differentially Influence Preferential and Non-preferential Signaling of the M1 and M3 Muscarinic Acetylcholine Receptors. Neurochem. Res. 2015, 40, 2068–2077. [Google Scholar] [CrossRef]
- André, A.; Gaibelet, G.; Le Guyader, L.; Welby, M.; Lopez, A.; Lebrun, C. Membrane partitioning of various δ-opioid receptor forms before and after agonist activations: The effect of cholesterol. Biochim. Biophys. Acta Biomembr. 2008, 1778, 1483–1492. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Hu, Y.; Kaindl, J.; Risel, P.; Hübner, H.; Maeda, S.; Niu, X.; Li, H.; Gmeiner, P.; Jin, C.; et al. Conformational Complexity and Dynamics in a Muscarinic Receptor Revealed by NMR Spectroscopy. Mol. Cell 2019, 75, 53–65.e7. [Google Scholar] [CrossRef] [PubMed]
- Nygaard, R.; Zou, Y.; Dror, R.O.; Mildorf, T.J.; Arlow, D.H.; Manglik, A.; Pan, A.C.; Liu, C.W.; Fung, J.J.; Bokoch, M.P.; et al. The dynamic process of β2-adrenergic receptor activation. Cell 2013, 152, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Jakubík, J.; Randáková, A.; Chetverikov, N.; El-Fakahany, E.E.; Doležal, V. The operational model of allosteric modulation of pharmacological agonism. Sci. Rep. 2020, 10, 14421. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.G. Lipid-protein interactions in biological membranes: A structural perspective. Biochim. Biophys. Acta 2003, 1612, 1–40. [Google Scholar] [CrossRef]
- Trzaskowski, B.; Latek, D.; Yuan, S.; Ghoshdastider, U.; Debinski, A.; Filipek, S. Action of molecular switches in GPCRs—Theoretical and experimental studies. Curr. Med. Chem. 2012, 19, 1090–1109. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Yang, D.; Wu, M.; Guo, Y.; Guo, W.; Zhong, L.; Cai, X.; Dai, A.; Jang, W.; Shakhnovich, E.I.; et al. Common activation mechanism of class A GPCRs. Elife 2019, 8. [Google Scholar] [CrossRef]
- Hulme, E.C. GPCR activation: A mutagenic spotlight on crystal structures. Trends Pharmacol. Sci. 2013, 34, 67–84. [Google Scholar] [CrossRef]
- Gimpl, G.; Burger, K.; Fahrenholz, F. Cholesterol as modulator of receptor function. Biochemistry 1997, 36, 10959–10974. [Google Scholar] [CrossRef]
- Levitt, E.S.; Clark, M.J.; Jenkins, P.M.; Martens, J.R.; Traynor, J.R. Differential effect of membrane cholesterol removal on μ- and δ-opioid receptors. A parallel comparison of acute and chronic signaling to adenylyl cyclase. J. Biol. Chem. 2009, 284, 22108–22122. [Google Scholar] [CrossRef]
- Neale, C.; Herce, H.D.; Pomès, R.; García, A.E. Can Specific Protein-Lipid Interactions Stabilize an Active State of the Beta 2 Adrenergic Receptor? Biophys. J. 2015, 109, 1652–1662. [Google Scholar] [CrossRef]
- Pontier, S.M.; Percherancier, Y.; Galandrin, S.; Breit, A.; Galés, C.; Bouvier, M. Cholesterol-dependent separation of the beta2-adrenergic receptor from its partners determines signaling efficacy: Insight into nanoscale organization of signal transduction. J. Biol. Chem. 2008, 283, 24659–24672. [Google Scholar] [CrossRef]
- Paila, Y.D.; Jindal, E.; Goswami, S.K.; Chattopadhyay, A. Cholesterol depletion enhances adrenergic signaling in cardiac myocytes. Biochim. Biophys. Acta 2011, 1808, 461–465. [Google Scholar] [CrossRef]
- Manna, M.; Niemelä, M.; Tynkkynen, J.; Javanainen, M.; Kulig, W.; Müller, D.J.; Rog, T.; Vattulainen, I. Mechanism of allosteric regulation of β2-adrenergic receptor by cholesterol. Elife 2016, 5, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Ferré, S.; Casadó, V.; Devi, L.A.; Filizola, M.; Jockers, R.; Lohse, M.J.; Milligan, G.; Pin, J.-P.; Guitart, X. G protein-coupled receptor oligomerization revisited: Functional and pharmacological perspectives. Pharmacol. Rev. 2014, 66, 413–434. [Google Scholar] [CrossRef] [PubMed]
- Jordan, B.A.; Devi, L.A. G-protein-coupled receptor heterodimerization modulates receptor function. Nature 1999, 399, 697–700. [Google Scholar] [CrossRef]
- Gomes, I.; Gupta, A.; Filipovska, J.; Szeto, H.H.; Pintar, J.E.; Devi, L.A. A role for heterodimerization of mu and delta opiate receptors in enhancing morphine analgesia. Proc. Natl. Acad. Sci. USA 2004, 101, 5135–5139. [Google Scholar] [CrossRef]
- Tiwari, V.; He, S.-Q.; Huang, Q.; Liang, L.; Yang, F.; Chen, Z.; Tiwari, V.; Fujita, W.; Devi, L.A.; Dong, X.; et al. Activation of µ-δ opioid receptor heteromers inhibits neuropathic pain behavior in rodents. Pain 2020, 161, 842–855. [Google Scholar] [CrossRef] [PubMed]
- Kuszak, A.J.; Pitchiaya, S.; Anand, J.P.; Mosberg, H.I.; Walter, N.G.; Sunahara, R.K. Purification and functional reconstitution of monomeric mu-opioid receptors: Allosteric modulation of agonist binding by Gi2. J. Biol. Chem. 2009, 284, 26732–26741. [Google Scholar] [CrossRef]
- Meral, D.; Provasi, D.; Prada-Gracia, D.; Möller, J.; Marino, K.; Lohse, M.J.; Filizola, M. Molecular details of dimerization kinetics reveal negligible populations of transient µ-opioid receptor homodimers at physiological concentrations. Sci. Rep. 2018, 8, 7705. [Google Scholar] [CrossRef] [PubMed]
- Möller, J.; Isbilir, A.; Sungkaworn, T.; Osberg, B.; Karathanasis, C.; Sunkara, V.; Grushevskyi, E.O.; Bock, A.; Annibale, P.; Heilemann, M.; et al. Single-molecule analysis reveals agonist-specific dimer formation of µ-opioid receptors. Nat. Chem. Biol. 2020, 16, 946–954. [Google Scholar] [CrossRef]
- Fotiadis, D.; Liang, Y.; Filipek, S.; Saperstein, D.A.; Engel, A.; Palczewski, K. The G protein-coupled receptor rhodopsin in the native membrane. FEBS Lett. 2004, 564, 281–288. [Google Scholar] [CrossRef]
- Whorton, M.R.; Jastrzebska, B.; Park, P.S.H.; Fotiadis, D.; Engel, A.; Palczewski, K.; Sunahara, R.K. Efficient coupling of transducin to monomeric rhodopsin in a phospholipid bilayer. J. Biol. Chem. 2008, 283, 4387–4394. [Google Scholar] [CrossRef] [PubMed]
- Modzelewska, A.; Filipek, S.; Palczewski, K.; Park, P.S.H. Arrestin interaction with rhodopsin: Conceptual models. Cell Biochem. Biophys. 2006, 46, 1–15. [Google Scholar] [CrossRef]
- Bayburt, T.H.; Vishnivetskiy, S.A.; McLean, M.A.; Morizumi, T.; Huang, C.-C.; Tesmer, J.J.G.; Ernst, O.P.; Sligar, S.G.; Gurevich, V.V. Monomeric rhodopsin is sufficient for normal rhodopsin kinase (GRK1) phosphorylation and arrestin-1 binding. J. Biol. Chem. 2011, 286, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.Y.; Pöge, M.; Morizumi, T.; Gulati, S.; Van Eps, N.; Zhang, J.; Miszta, P.; Filipek, S.; Mahamid, J.; Plitzko, J.M.; et al. Cryo-EM structure of the native rhodopsin dimer in nanodiscs. J. Biol. Chem. 2019, 294, 14215–14230. [Google Scholar] [CrossRef]
- Milligan, G. The prevalence, maintenance, and relevance of G protein-coupled receptor oligomerization. Mol. Pharmacol. 2013, 84, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Dainese, E.; Oddi, S.; Maccarrone, M. Lipid-mediated dimerization of beta2-adrenergic receptor reveals important clues for cannabinoid receptors. Cell. Mol. Life Sci. 2008, 65, 2277–2279. [Google Scholar] [CrossRef]
- Wang, J.; He, L.; Combs, C.A.; Roderiquez, G.; Norcross, M.A. Dimerization of CXCR4 in living malignant cells: Control of cell migration by a synthetic peptide that reduces homologous CXCR4 interactions. Mol. Cancer Ther. 2006, 5, 2474–2483. [Google Scholar] [CrossRef]
- Angers, S.; Salahpour, A.; Joly, E.; Hilairet, S.; Chelsky, D.; Dennis, M.; Bouvier, M. Detection of beta 2-adrenergic receptor dimerization in living cells using bioluminescence resonance energy transfer (BRET). Proc. Natl. Acad. Sci. USA 2000, 97, 3684–3689. [Google Scholar] [CrossRef]
- Prasanna, X.; Chattopadhyay, A.; Sengupta, D. Cholesterol modulates the dimer interface of the β2- adrenergic receptor via cholesterol occupancy sites. Biophys. J. 2014, 106, 1290–1300. [Google Scholar] [CrossRef]
- Paila, Y.D.; Kombrabail, M.; Krishnamoorthy, G.; Chattopadhyay, A. Oligomerization of the serotonin1A receptor in live cells: A time-resolved fluorescence anisotropy approach. J. Phys. Chem. B 2011, 115, 11439–11447. [Google Scholar] [CrossRef]
- Ganguly, S.; Clayton, A.H.A.; Chattopadhyay, A. Organization of higher-order oligomers of the serotonin₁(A) receptor explored utilizing homo-FRET in live cells. Biophys. J. 2011, 100, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Ayoub, M.A.; Couturier, C.; Lucas-Meunier, E.; Angers, S.; Fossier, P.; Bouvier, M.; Jockers, R. Monitoring of ligand-independent dimerization and ligand-induced conformational changes of melatonin receptors in living cells by bioluminescence resonance energy transfer. J. Biol. Chem. 2002, 277, 21522–21528. [Google Scholar] [CrossRef]
- Gorinski, N.; Kowalsman, N.; Renner, U.; Wirth, A.; Reinartz, M.T.; Seifert, R.; Zeug, A.; Ponimaskin, E.; Niv, M.Y. Computational and experimental analysis of the transmembrane domain 4/5 dimerization interface of the serotonin 5-HT(1A) receptor. Mol. Pharmacol. 2012, 82, 448–463. [Google Scholar] [CrossRef]
- Massaccesi, L.; Laudadio, E.; Mobbili, G.; Minnelli, C.; Galeazzi, R. Cholesterol-mediated oligomerization pathways of serotonin G-coupled receptor 5-HT2C. Int. J. Biol. Macromol. 2020, 160, 1090–1100. [Google Scholar] [CrossRef] [PubMed]
- Park, P.S.H.; Wells, J.W. Oligomeric potential of the M2 muscarinic cholinergic receptor. J. Neurochem. 2004, 90, 537–548. [Google Scholar] [CrossRef]
- McMillin, S.M.; Heusel, M.; Liu, T.; Costanzi, S.; Wess, J. Structural basis of M3 muscarinic receptor dimer/oligomer formation. J. Biol. Chem. 2011, 286, 28584–28598. [Google Scholar] [CrossRef] [PubMed]
- Park, P.S.H.; Sum, C.S.; Pawagi, A.B.; Wells, J.W. Cooperativity and oligomeric status of cardiac muscarinic cholinergic receptors. Biochemistry 2002, 41, 5588–5604. [Google Scholar] [CrossRef]
- Redka, D.S.; Pisterzi, L.F.; Wells, J.W. Binding of orthosteric ligands to the allosteric site of the M(2) muscarinic cholinergic receptor. Mol. Pharmacol. 2008, 74, 834–843. [Google Scholar] [CrossRef]
- Redka, D.S.; Morizumi, T.; Elmslie, G.; Paranthaman, P.; Shivnaraine, R.V.; Ellis, J.; Ernst, O.P.; Wells, J.W. Coupling of G proteins to reconstituted monomers and tetramers of the M2 muscarinic receptor. J. Biol. Chem. 2014, 289, 24347–24365. [Google Scholar] [CrossRef] [PubMed]
- Colozo, A.T.; Park, P.S.-H.; Sum, C.S.; Pisterzi, L.F.; Wells, J.W. Cholesterol as a determinant of cooperativity in the M2 muscarinic cholinergic receptor. Biochem. Pharmacol. 2007, 74, 236–255. [Google Scholar] [CrossRef] [PubMed]
- Liste, M.J.V.; Caltabiano, G.; Ward, R.J.; Alvarez-Curto, E.; Marsango, S.; Milligan, G. The molecular basis of oligomeric organization of the human M3 muscarinic acetylcholine receptor. Mol. Pharmacol. 2015, 87, 936–953. [Google Scholar] [CrossRef]
- Nguyen, D.H.; Taub, D. Cholesterol is essential for macrophage inflammatory protein 1β binding and conformational integrity of CC chemokine receptor 5. Blood 2002, 99, 4298–4306. [Google Scholar] [CrossRef]
- Pluhackova, K.; Gahbauer, S.; Kranz, F.; Wassenaar, T.A.; Böckmann, R.A. Dynamic Cholesterol-Conditioned Dimerization of the G Protein Coupled Chemokine Receptor Type 4. PLoS Comput. Biol. 2016, 12, e1005169. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, M.; Siauw, M.; Horackova, M. Modulation of cardiac M2 muscarinic receptor binding by progesterone-related steroids. J. Mol. Cell. Cardiol. 1995, 27, 1831–1839. [Google Scholar] [CrossRef]
- Dolejší, E.; Szánti-Pintér, E.; Chetverikov, N.; Nelic, D.; Randáková, A.; Doležal, V.; Kudová, E.; Jakubík, J. Steroids as the novel class of high-affinity allosteric modulators of muscarinic. Res. Sq. 2021. [Google Scholar] [CrossRef]
- Robichaud, M.; Debonnel, G. Modulation of the firing activity of female dorsal raphe nucleus serotonergic neurons by neuroactive steroids. J. Endocrinol. 2004, 182, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-M.; Qi, Y.-J.; Wu, P.-Y.; Zhu, Y.; Dong, Y.-L.; Cheng, Z.-X.; Zhu, Y.-H.; Dong, Y.; Ma, L.; Zheng, P. Neuroactive steroid pregnenolone sulphate inhibits long-term potentiation via activation of alpha2-adrenoreceptors at excitatory synapses in rat medial prefrontal cortex. Int. J. Neuropsychopharmacol. 2008, 11, 611–624. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
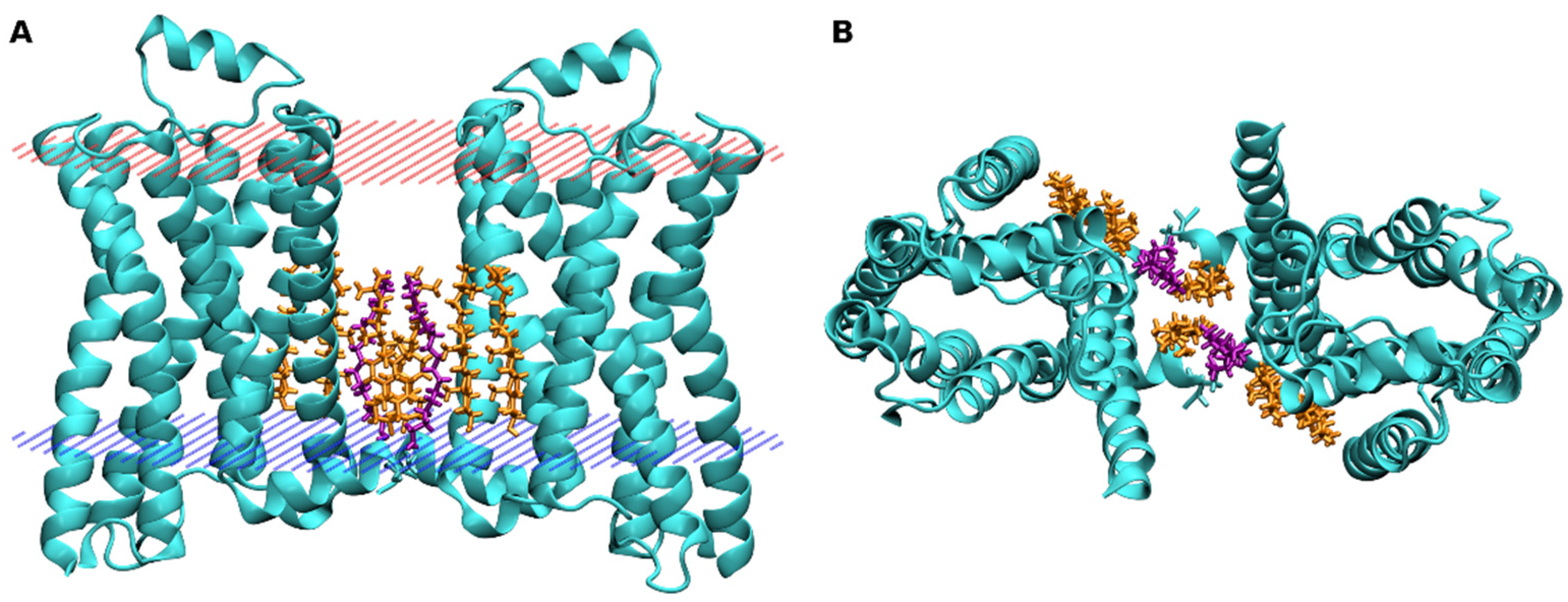{kind=link}
| Rec. | Code | G-prot. | Conf. | CLR | Leaflet | TM | Notes | Ref. |
|---|---|---|---|---|---|---|---|---|
| α2C | 6KUW | Gi | Inactive | Monomer | Out | 1, 7 | cholesterol is a part of the protomer-protomer interface | TBP |
| β2 | 2RH1 | Gs | Inactive | Dimer | In | 2, 3, 4 | [16] | |
| Monomer | In | 1, 8 | ||||||
| β2 | 3D4S | Gs | Inactive | Dimer | In | 2, 3, 4 | [17] | |
| β2 | 6PS0 | Gs | Inactive | Monomer | In | 2, 3, 4 | 6PS2, 6PS3, 6PS4, 6PS5, 6PS7 same | [18] |
| κOR | 6PT2 | Gi | Active | Monomer | Out | 6 | only with one protomer | [19] |
| κOR | 6B73 | Gi | Active | Monomer | Out | 6 | [20] | |
| κOR | 6VI4 | Gi | Inactive | Monomer | In | 4, 5 | only with one protomer | [20] |
| μOR | 4DKL | Gi | Inactive | Monomer | Out | 6 | [21] | |
| μOR | 5C1M | Gi | Active | Monomer | Out | 6 | [22] | |
| 5-HT2B | 4IB4 | Gq | Active | Monomer | In | 1, 8 | [23] | |
| 5-HT2B | 5TVN | Gq | Active | Monomer | In | 1, 8 | [24] | |
| 5-HT2B | 6DRX | Gq | Active | Monomer | In | 1, 8 | 6DRY, 6DRZ, 6DS0 same | [25] |
| A2A | 4EIY | Gs | Inactive | Dimer | Out | 6 | [26] | |
| Monomer | Out | 2, 3, 4 | ||||||
| A2A | 5IU4 | Gs | Inactive | Dimer | Out | 6 | 5UI7, 5UI8, 5UIA and 5UIB same | [27] |
| Dimer | Out | 2, 3, 4 | 5UIB only monomer. | |||||
| AT1 | 6OS1 | Gq | Active | Monomer | In | 1, 8 | 6OS0 and 6OD1 no cholesterol | [28] |
| CB1 | 6N4B | Gi | Active | Monomer | In | 3, 4 | [29] | |
| Monomer | In | 3, 4 | unexpected orientation with OH in the middle of the membrane | |||||
| CB2 | 6PT0 | Gi | Active | Dimer | Out | 6 | [30] | |
| Monomer | In | 5, 6 | parallel to membrane | |||||
| Monomer | In | 3, 4 | parallel to membrane | |||||
| CCR9 | 5LWE | Inactive | Monomer | In | 6 | [31] | ||
| CLT2 | 6RZ7 | Gq | Inactive | Monomer | In | 6 | 6RZ6, 6RZ9 dtto. | [32] |
| CXCR2 | 6LFM | Gi | Active | Monomer | In | 2, 3, 4 | 6LFO ditto, 6LFL no cholesterol | [33] |
| CXCR3 | 5WB2 | Gi | Active | Dimer | Out | 6 | [34] | |
| ETB | 5X93 | Gq | Inactive | Monomer | Out | 1, 7 | 5XPR no cholesterol | [35] |
| FPR2 | 6LW5 | Gi | Active | Monomer | In | 6 | [36] | |
| Monomer | In | 2, 3, 4 | ||||||
| FPR2 | 6OMM | Gi | Active | Dimer | Out | 1, 2 | [37] | |
| Monomer | Out | 6 | ||||||
| Monomer | In | 6 | ||||||
| Monomer | In | 3, 4, 5 | ||||||
| OTR | 6TPK | Gq | Inactive | Monomer | Out | 4, 5 | [38] | |
| P2Y12 | 4NTJ | Gi | Inactive | Monomer | Out | 1, 7 | [39] | |
| Monomer | In | 3, 4 | unexpected orientation, binding to Y in DRY motif | |||||
| P2Y12 | 4PXZ | Gi | Inactive | Monomer | In | 2, 3, 4 | [39] | |
| P2Y1 | 4XNV | Gq | Inactive | Monomer | Out | 2, 3, 4 | [40] |
| Rec. | Code | CLR | Leaflet | TM | Predicted | Confirmed? | CLR-Interacting Residues |
|---|---|---|---|---|---|---|---|
| α2C | 6KUW | Monomer | Out | 1, 7 | CCM | No | Q45, Y46, E112, K420 |
| β2 | 2RH1 | Dimer | In | 2, 3, 4 | CCM | Yes | Y70, T73, S74, R151, W158 |
| Monomer | In | 1, 8 | T56, C341_Plm | ||||
| β2 | 3D4S | Dimer | In | 2, 3, 4 | CCM | Yes | Y70, T73, S74, R151, W158 |
| β2 | 6PS0 | Monomer | In | 2, 3, 4 | CCM | Yes | Y70, T73, S74, R151, W158, F166 |
| κOR | 6PT2 | Monomer | Out | 6 | CCM | No | F280, D293 (o2) |
| κOR | 6B73 | Monomer | Out | 6 | CCM | No | T288, F293, T302, S303, S311 |
| κOR | 6VI4 | Monomer | In | 4, 5 | CCM | No | F147, T150, Y157, H162 |
| μOR | 4DKL | Monomer | Out | 6 | CCM Q | Yes | T294, Y299, F313, Q314, S317 |
| μOR | 5C1M | Monomer | Out | 6 | CCM Q | Yes | T294, Y299, F313, Q314, S317 |
| 5-HT2B | 4IB4 | Monomer | In | 1, 8 | CCM | No | T73, Y394, Y399, C397_Plm |
| 5-HT2B | 5TVN | Monomer | In | 1, 8 | CCM | No | T73, Y394, Y399 |
| 5-HT2B | 6DRX | Monomer | In | 1, 8 | CCM | No | T73, Y394, Y399 |
| A2A | 4EIY | Dimer | Out | 6 | F182, 183, 255, 258, S263, H264 | ||
| Monomer | Out | 2, 3, 4 | CCM | No | F70, F79, Q163 (o2) | ||
| A2A | 5IU4 | Dimer | Out | 6 | F182, 183, 255, 258, S263, H264 | ||
| Dimer | Out | 2, 3, 4 | CCM | No | F70, H75, F79, F133, Q163 | ||
| AT1 | 6OS1 | Monomer | In | 1, 8 | CCM | No | F39, F44, S47 |
| CB1 | 6N4B | Monomer | In | 3, 4 | CRAC | Partly | F208, K232 |
| Monomer | In | 3, 4 | F208, Y215, H219, R220 | ||||
| CB2 | 6PT0 | Dimer | Out | 6 | None | -- | Q276, K279, F283 |
| Monomer | In | 6 | Y207, H211, W214, H217, R238, D240 | ||||
| Monomer | In | 3, 4 | D130, Y137, T153, R149 | ||||
| CCR9 | 5LWE | Monomer | In | 6 | CCM | Yes | K254, T258, F263, F308, F319 |
| CLT2 | 6RZ7 | Monomer | In | 5 | CARC | Yes | S218, Y221, R226, F257 |
| CXCR2 | 6LFM | Monomer | In | 2, 3, 4 | CRAC W | Yes | N89, N129, K163, W170, L174 |
| CXCR3 | 5WB2 | Dimer | Out | 6 | CCM | Yes | T247, F265, S268, R271, T276 |
| ETB | 5X93 | Monomer | Out | 1 | None | -- | Y102, T105 |
| FPR2 | 6LW5 | Monomer | In | 6 | S215 | ||
| Monomer | In | 2, 3, 4 | CRAC | No | N66, W150 | ||
| FPR2 | 6OMM | Dimer | Out | 1, 2 | -- | F37, | |
| Monomer | Out | 6 | F206, F255, W267 | ||||
| Monomer | In | 6 | H229 | ||||
| Monomer | In | 3, 4, 5 | CRAC | No | F118, H129, W132 | ||
| OTR | 6TPK | Monomer | Out | 4, 5 | CCM | No | F191, W195, Y200, W203 |
| P2Y12 | 4NTJ | Monomer | Out | 1, 7 | F28, Y278, S282, W285 | ||
| Monomer | In | 3, 4 | CCM | No | Y123, Q124 | ||
| P2Y12 | 4PXZ | Monomer | In | 2, 3, 4 | CCM | Yes | F51, S55, K64, N65, F106, K142, W149, F153 |
| P2Y1 | 4XNV | Monomer | In | 2, 3, 4 | CCM | No | Y189, T221, Y217, S213 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jakubík, J.; El-Fakahany, E.E. Allosteric Modulation of GPCRs of Class A by Cholesterol. Int. J. Mol. Sci. 2021, 22, 1953. https://doi.org/10.3390/ijms22041953
Jakubík J, El-Fakahany EE. Allosteric Modulation of GPCRs of Class A by Cholesterol. International Journal of Molecular Sciences. 2021; 22(4):1953. https://doi.org/10.3390/ijms22041953
Chicago/Turabian StyleJakubík, Jan, and Esam E. El-Fakahany. 2021. "Allosteric Modulation of GPCRs of Class A by Cholesterol" International Journal of Molecular Sciences 22, no. 4: 1953. https://doi.org/10.3390/ijms22041953
APA StyleJakubík, J., & El-Fakahany, E. E. (2021). Allosteric Modulation of GPCRs of Class A by Cholesterol. International Journal of Molecular Sciences, 22(4), 1953. https://doi.org/10.3390/ijms22041953