
Dermal Drivers of Injury-Induced Inflammation: Contribution of Adipocytes and Fibroblasts


Abstract
1. Introduction
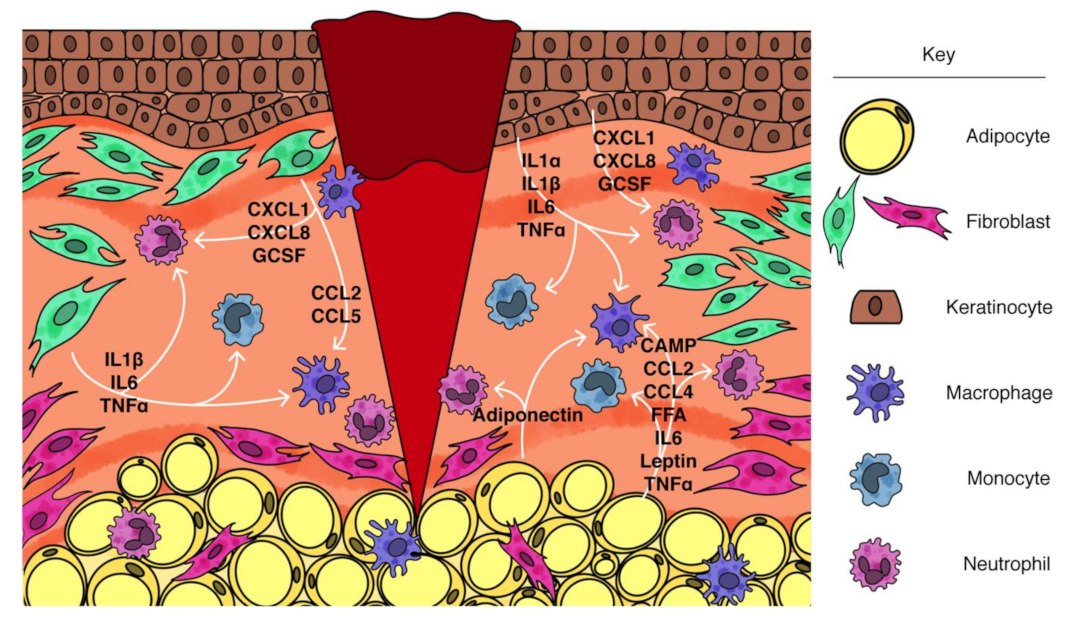
2. Contribution of Adipocytes to Inflammation
2.1. White Adipose Tissue
2.2. Dermal Adipocytes
2.3. WAT Inflammation
2.3.1. Neutrophil Recruitment
2.3.2. Macrophage Recruitment and Polarization
2.4. Adipocyte Response to Injury
3. Contribution of Fibroblasts to Injury-Induced Inflammation
3.1. Contribution of Fibroblasts to Tissue Inflammation
3.2. Signaling Pathways Regulating Inflammatory Fibroblast Phenotype
3.2.1. IL1 Signaling
3.2.2. TNFα Signaling
3.2.3. TLR Signaling
3.3. Molecular Regulation of Fibroblast Polarization
3.4. Functional Diversity in Fibroblasts
3.5. Communication between Adipocytes and Fibroblasts
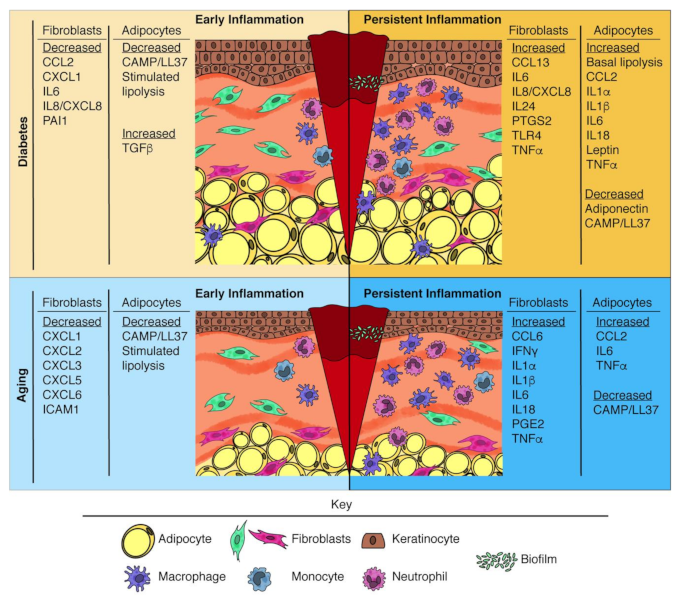
4. Altered Inflammatory Response during Impaired Wound Healing
4.1. Impaired Early Leukocyte Infiltration and Function
4.2. Persistence of Inflammation
5. Contribution of Adipocytes to Impaired Wound Healing
5.1. Diabetes-Associated Changes in Adipocyte Inflammatory Function
5.1.1. Impaired Early Leukocyte Infiltration and Function
5.1.2. Persistent Inflammation
5.2. Age-Associated Changes in Adipocyte Inflammatory Function
5.2.1. Impaired Early Leukocyte Infiltration and Function
5.2.2. Persistent Inflammation
6. Contribution of Fibroblasts to Impaired Wound Healing
6.1. Diabetes-Associated Changes in Fibroblast Inflammatory Function
6.1.1. Impaired Early Leukocyte Infiltration and Function
6.1.2. Persistent Inflammation
6.2. Age-Associated Changes in Fibroblast Inflammatory Function
6.2.1. Impaired Early Leukocyte Infiltration and Function
6.2.2. Persistent Inflammation
7. Methods
8. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACTA2 | actin alpha 2 |
| ATAC | Assay for Transposase-Accessible Chromatin |
| ATGL | adipose triglyceride lipase |
| BADGE | bisphenol A diglycidyl ether |
| CAMP | cathelicidin antimicrobial peptide |
| CCL | chemokine (C-C motif) ligand |
| CD | cluster of differentiation |
| COL | collagen |
| COX2 | cyclooxygenase-2 |
| CSF | colony stimulating factor |
| CTRP | C1q/TNF-receptor proteins |
| CXCL | chemokine (C-X-C motif) ligand |
| DPP4 | dipeptidyl peptidase 4 |
| DWAT | dermal white adipose tissue |
| EBF2 | early B-cell factor 2 |
| ECM | extracellular matrix |
| ERK | extracellular signal-related kinase |
| FAP | fibroblast activation protein |
| FFA | free fatty acid |
| GCSF | granulocyte colony stimulating factor |
| GPR84 | G protein-coupled receptor 84 |
| HMGB1 | high mobility group box 1 |
| IFNγ | interferon gamma |
| ICAM | intercellular adhesion molecule |
| IL | interleukin |
| LIF | leukemia inhibitory factor |
| LPS | lipopolysaccharide |
| LY6C | lymphocyte antigen 6 complex |
| MCP | monocyte chemoattractant protein |
| MFAP5 | microfibrillar associated protein 5 |
| MI | myocardial infarction |
| MIP | macrophage inflammatory protein |
| MMP | matrix metalloproteinase |
| NF-κB | nuclear factor kappa B |
| PDGF | platelet-derived growth factor |
| PDPN | podoplanin |
| PGE2 | prostaglandin E2 |
| PMN | polymorphonuclear leukocyte |
| PPARγ | peroxisome proliferator activated receptor gamma |
| PTGS2 | prostaglandin endoperoxide synthase 2 |
| ROS | reactive oxygen species |
| SARS-CoV2 | severe acute respiratory syndrome coronavirus 2 |
| SASP | senescence-associated secretory phenotype |
| SCA1 | stem cell antigen 1 |
| scRNA-seq | single-cell RNA sequencing |
| SERPINE1 | serine proteinase inhibitor 1 |
| SMA | smooth muscle actin |
| SWAT | subcutaneous white adipose tissue |
| TGFβ | transforming growth factor beta |
| THY1 | thy-1 cell surface antigen 1 |
| TLR | toll-like receptor |
| TNFα | tumor necrosis factor alpha |
| VEGF | vascular endothelial growth factor |
| VWAT | visceral white adipose tissue |
| WAT | white adipose tissue |
References
- Iwai, I.; Han, H.; den Hollander, L.; Svensson, S.; Öfverstedt, L.-G.; Anwar, J.; Brewer, J.; Bloksgaard, M.; Laloeuf, A.; Nosek, D.; et al. The Human Skin Barrier Is Organized as Stacked Bilayers of Fully Extended Ceramides with Cholesterol Molecules Associated with the Ceramide Sphingoid Moiety. J. Investig. Dermatol. 2012, 132, 2215–2225. [Google Scholar] [CrossRef]
- Janson, D.G.; Saintigny, G.; van Adrichem, A.; Mahé, C.; El Ghalbzouri, A. Different Gene Expression Patterns in Human Papillary and Reticular Fibroblasts. J. Investig. Dermatol. 2012, 132, 2565–2572. [Google Scholar] [CrossRef]
- Driskell, R.R.; Jahoda, C.A.B.; Chuong, C.-M.; Watt, F.M.; Horsley, V. Defining dermal adipose tissue. Exp. Dermatol. 2014, 23, 629–631. [Google Scholar] [CrossRef] [PubMed]
- Festa, E.; Fretz, J.; Berry, R.; Schmidt, B.; Rodeheffer, M.; Horowitz, M.; Horsley, V. Adipocyte Lineage Cells Contribute to the Skin Stem Cell Niche to Drive Hair Cycling. Cell 2011, 146, 761–771. [Google Scholar] [CrossRef]
- Plikus, M.V.; Mayer, J.A.; de la Cruz, D.; Baker, R.E.; Maini, P.K.; Maxson, R.; Chuong, C.-M. Cyclic dermal BMP signalling regulates stem cell activation during hair regeneration. Nature 2008, 451, 340–344. [Google Scholar] [CrossRef]
- Rahmani, W.; Abbasi, S.; Hagner, A.; Raharjo, E.; Kumar, R.; Hotta, A.; Magness, S.; Metzger, D.; Biernaskie, J. Hair follicle dermal stem cells regenerate the dermal sheath, repopulate the dermal papilla, and modulate hair type. Dev. Cell 2014, 31, 543–558. [Google Scholar] [CrossRef]
- Heitman, N.; Sennett, R.; Mok, K.W.; Saxena, N.; Srivastava, D.; Martino, P.; Grisanti, L.; Wang, Z.; Ma’ayan, A.; Rompolas, P.; et al. Dermal sheath contraction powers stem cell niche relocation during hair cycle regression. Science 2020, 367, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, B.A.; Horsley, V. Intradermal adipocytes mediate fibroblast recruitment during skin wound healing. Development 2013, 140, 1517–1527. [Google Scholar] [CrossRef] [PubMed]
- Shook, B.A.; Wasko, R.R.; Mano, O.; Rutenberg-Schoenberg, M.; Rudolph, M.C.; Zirak, B.; Rivera-Gonzalez, G.C.; López-Giráldez, F.; Zarini, S.; Rezza, A.; et al. Dermal Adipocyte Lipolysis and Myofibroblast Conversion Are Required for Efficient Skin Repair. Cell Stem Cell 2020, 26, 880–895.e6. [Google Scholar] [CrossRef] [PubMed]
- Shook, B.A.; Wasko, R.R.; Rivera-Gonzalez, G.C.; Salazar-Gatzimas, E.; López-Giráldez, F.; Dash, B.C.; Muñoz-Rojas, A.R.; Aultman, K.D.; Zwick, R.K.; Lei, V.; et al. Myofibroblast proliferation and heterogeneity are supported by macrophages during skin repair. Science 2018, 362, eaar2971. [Google Scholar] [CrossRef]
- Rinkevich, Y.; Walmsley, G.G.; Hu, M.S.; Maan, Z.N.; Newman, A.M.; Drukker, M.; Januszyk, M.; Krampitz, G.W.; Gurtner, G.C.; Lorenz, H.P.; et al. Identification and isolation of a dermal lineage with intrinsic fibrogenic potential. Science 2015, 348, aaa2151. [Google Scholar] [CrossRef] [PubMed]
- Plikus, M.V.; Guerrero-Juarez, C.F.; Ito, M.; Li, Y.R.; Dedhia, P.H.; Zheng, Y.; Shao, M.; Gay, D.L.; Ramos, R.; His, T.-C.; et al. Regeneration of fat cells from myofibroblasts during wound healing. Science 2017, aai8792. [Google Scholar] [CrossRef]
- Zhang, Z.; Shao, M.; Hepler, C.; Zi, Z.; Zhao, S.; An, Y.A.; Zhu, Y.; Ghaben, A.L.; Wang, M.-Y.; Li, N.; et al. Dermal adipose tissue has high plasticity and undergoes reversible dedifferentiation in mice. J. Clin. Investig. 2019, 129, 5327–5342. [Google Scholar] [CrossRef]
- Eming, S.A.; Martin, P.; Tomic-Canic, M. Wound repair and regeneration: Mechanisms, signaling, and translation. Sci. Transl. Med. 2014, 6, 265sr6. [Google Scholar] [CrossRef] [PubMed]
- Ridiandries, A.; Tan, J.; Bursill, C. The Role of Chemokines in Wound Healing. Int. J. Mol. Sci. 2018, 19, 3217. [Google Scholar] [CrossRef]
- Xiao, T.; Yan, Z.; Xiao, S.; Xia, Y. Proinflammatory cytokines regulate epidermal stem cells in wound epithelialization. Stem Cell Res. 2020, 11, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Klicznik, M.M.; Szenes-Nagy, A.B.; Campbell, D.J.; Gratz, I.K. Taking the lead—How keratinocytes orchestrate skin T cell immunity. Immunol. Lett. 2018, 200, 43–51. [Google Scholar] [CrossRef]
- Archer, N.K.; Jo, J.-H.; Lee, S.K.; Kim, D.; Smith, B.; Ortines, R.V.; Wang, Y.; Marchitto, M.C.; Ravipati, A.; Cai, S.S.; et al. Injury, dysbiosis, and filaggrin deficiency drive skin inflammation through keratinocyte IL-1α release. J. Allergy Clin. Immunol. 2019, 143, 1426–1443.e6. [Google Scholar] [CrossRef]
- Roupé, K.M.; Nybo, M.; Sjöbring, U.; Alberius, P.; Schmidtchen, A.; Sørensen, O.E. Injury Is a Major Inducer of Epidermal Innate Immune Responses during Wound Healing. J. Investig. Dermatol. 2010, 130, 1167–1177. [Google Scholar] [PubMed]
- Eming, S.A.; Krieg, T.; Davidson, J.M. Inflammation in Wound Repair: Molecular and Cellular Mechanisms. J. Investig. Dermatol. 2007, 127, 514–525. [Google Scholar] [CrossRef]
- de Oliveira, S.; Rosowski, E.E.; Huttenlocher, A. Neutrophil migration in infection and wound repair: Going forward in reverse. Nat. Rev. Immunol. 2016, 16, 378–391. [Google Scholar] [CrossRef] [PubMed]
- Daley, J.M.; Brancato, S.K.; Thomay, A.A.; Reichner, J.S.; Albina, J.E. The phenotype of murine wound macrophages. J. Leukoc. Biol. 2010, 87, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Pinhal-Enfield, G.; Ramanathan, M.; Hasko, G.; Vogel, S.N.; Salzman, A.L.; Boons, G.-J.; Leibovich, S.J. An Angiogenic Switch in Macrophages Involving Synergy between Toll-Like Receptors 2, 4, 7, and 9 and Adenosine A2A Receptors. Am. J. Phys. Anthropol. 2010, 163, 711–721. [Google Scholar] [CrossRef]
- Willenborg, S.; Lucas, T.; van Loo, G.; Knipper, J.A.; Krieg, T.; Haase, I.; Brachvogel, B.; Hammerschmidt, M.; Nagy, A.; Ferrara, N.; et al. CCR2 recruits an inflammatory macrophage subpopulation critical for angiogenesis in tissue repair. Blood 2012, 120, 613–625. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.S.; Borrelli, M.R.; Lorenz, H.P.; Longaker, M.T.; Wan, D.C. Mesenchymal Stromal Cells and Cutaneous Wound Healing: A Comprehensive Review of the Background, Role, and Therapeutic Potential. Stem Cells Int. 2018, 2018, 6901983. [Google Scholar] [CrossRef]
- Hu, M.S.; Walmsley, G.G.; Barnes, L.A.; Weiskopf, K.; Rennert, R.C.; Duscher, D.; Januszyk, M.; Maan, Z.N.; Hong, W.X.; Cheung, A.T.; et al. Delivery of monocyte lineage cells in a biomimetic scaffold enhances tissue repair. JCI Insight 2017, 2. [Google Scholar] [CrossRef]
- Nassiri, S.; Zakeri, I.; Weingarten, M.S.; Spiller, K.L. Relative Expression of Proinflammatory and Antiinflammatory Genes Reveals Differences between Healing and Nonhealing Human Chronic DiabeticFoot Ulcers. J. Investig. Dermatol. 2015, 135, 1700–1703. [Google Scholar] [CrossRef] [PubMed]
- Mirza, R.; Koh, T.J. Dysregulation of monocyte/macrophage phenotype in wounds of diabetic mice. Cytokine 2011, 56, 256–264. [Google Scholar] [CrossRef]
- Januszyk, M.; Chen, K.; Henn, D.; Foster, D.S.; Borrelli, M.R.; Bonham, C.A.; Sivaraj, D.; Wagh, D.; Longaker, M.T.; Wan, D.C.; et al. Characterization of Diabetic and Non-Diabetic Foot Ulcers Using Single-Cell RNA-Sequencing. Micromachines 2020, 11, 815. [Google Scholar] [CrossRef]
- Ansell, D.M.; Holden, K.A.; Hardman, M.J. Animal models of wound repair: Are they cutting it? Exp. Dermatol. 2012, 21, 581–585. [Google Scholar] [CrossRef] [PubMed]
- Nunan, R.; Harding, K.G.; Martin, P. Clinical challenges of chronic wounds: Searching for an optimal animal model to recapitulate their complexity. Dis. Models Mech. 2014, 7, 1205–1213. [Google Scholar] [CrossRef]
- Theocharidis, G.; Baltzis, D.; Roustit, M.; Tellechea, A.; Dangwal, S.; Khetani, R.S.; Shu, B.; Zhao, W.; Fu, J.; Bhasin, S.; et al. Integrated Skin Transcriptomics and Serum Multiplex Assays Reveal Novel Mechanisms of Wound Healing in Diabetic Foot Ulcers. Diabetes 2020, 69, 2157–2169. [Google Scholar] [CrossRef]
- Joshi, N.; Pohlmeier, L.; Ben Yehuda Greenwald, M.; Haertel, E.; Hiebert, P.; Kopf, M.; Werner, S. Comprehensive characterization of myeloid cells during wound healing in healthy and healing-impaired diabetic mice. Eur. J. Immunol. 2020, 50, 1335–1349. [Google Scholar] [CrossRef]
- Sawaya, A.P.; Stone, R.C.; Brooks, S.R.; Pastar, I.; Jozic, I.; Hasneen, K.; O’Neill, K.; Mehdizadeh, S.; Head, C.R.; Strbo, N.; et al. Deregulated immune cell recruitment orchestrated by FOXM1 impairs human diabetic wound healing. Nat. Commun. 2020, 11, 4678. [Google Scholar] [CrossRef] [PubMed]
- Shook, B.; Rivera-Gonzalez, G.; Ebmeier, S.; Grisotti, G.; Zwick, R.; Horsley, V. The Role of Adipocytes in Tissue Regeneration and Stem Cell Niches. Annu. Rev. Cell Dev. Biol. 2016, 32, 609–631. [Google Scholar] [PubMed]
- Zwick, R.K.; Guerrero-Juarez, C.F.; Horsley, V.; Plikus, M.V. Anatomical, Physiological, and Functional Diversity of Adipose Tissue. Cell Metab. 2018, 27, 68–83. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-J.; Wu, Y.; Fried, S.K. Adipose tissue heterogeneity: Implication of depot differences in adipose tissue for obesity complications. Mol. Asp. Med. 2013, 34, 1–11. [Google Scholar] [CrossRef]
- Dodson, M.V.; Du, M.; Wang, S.; Bergen, W.G.; Fernyhough-Culver, M.; Basu, U.; Poulos, S.P.; Hausman, G.J. Adipose depots differ in cellularity, adipokines produced, gene expression, and cell systems. Adipocyte 2014, 3, 236–241. [Google Scholar] [PubMed]
- Baglioni, S.; Cantini, G.; Poli, G.; Francalanci, M.; Squecco, R.; Di Franco, A.; Borgogni, E.; Frontera, S.; Nesi, G.; Liotta, F.; et al. Functional Differences in Visceral and Subcutaneous Fat Pads Originate from Differences in the Adipose Stem Cell. PLoS ONE 2012, 7, e36569. [Google Scholar] [CrossRef]
- Bulcão, C.; Ferreira, S.R.G.; Giuffrida, F.M.A.; Ribeiro-Filho, F.F. The new adipose tissue and adipocytokines. Curr. Diabetes Rev. 2006, 2, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Trayhurn, P.; Wood, I.S. Adipokines: Inflammation and the pleiotropic role of white adipose tissue. Br. J. Nutr. 2004, 92, 347–355. [Google Scholar] [CrossRef]
- Kershaw, E.E.; Flier, J.S. Adipose Tissue as an Endocrine Organ. J. Clin. Endocrinol. Metab. 2004, 89, 2548–2556. [Google Scholar] [CrossRef] [PubMed]
- Reilly, S.M.; Saltiel, A.R. Adapting to obesity with adipose tissue inflammation. Nat. Rev. Endocrinol. 2017, 13, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Francisco, V.; Pino, J.; Campos-Cabaleiro, V.; Ruiz-Fernández, C.; Mera, A.; Gonzalez-Gay, M.A.; Gómez, R.; Gualillo, O. Obesity, Fat Mass and Immune System: Role for Leptin. Front. Physiol. 2018, 9, 473. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Liu, M. Adiponectin: A versatile player of innate immunity. J. Mol. Cell Biol. 2016, 8, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Chiang, H.-C.; Sun, X.; Yuan, B.; Mitra, P.; Hu, Y.; Curiel, T.J.; Li, R. Genetic ablation of adipocyte PD-L1 reduces tumor growth but accentuates obesity-associated inflammation. J. Immunother. Cancer 2020, 8, e000964. [Google Scholar] [CrossRef]
- Gerhardt, C.C.; Romero, I.A.; Cancello, R.; Camoin, L.; Strosberg, A.D. Chemokines control fat accumulation and leptin secretion by cultured human adipocytes. Mol. Cell. Endocrinol. 2001, 175, 81–92. [Google Scholar] [PubMed]
- Kim, E.J.; Kim, Y.K.; Kim, S.; Kim, J.E.; Tian, Y.D.; Doh, E.J.; Lee, D.H.; Chung, J.H. Adipochemokines induced by ultraviolet irradiation contribute to impaired fat metabolism in subcutaneous fat cells. Br. J. Dermatol. 2018, 178, 492–501. [Google Scholar]
- Fischer, J.; Gutièrrez, S.; Ganesan, R.; Calabrese, C.; Ranjan, R.; Cildir, G.; Hos, N.J.; Rybniker, J.; Wolke, M.; Fries, J.W.U.; et al. Leptin signaling impairs macrophage defenses against Salmonella Typhimurium. Proc. Natl. Acad. Sci. USA 2019, 116, 16551–16560. [Google Scholar] [CrossRef]
- Teixeira, L.; Correia, A.; Oliveira, B.M.; Pinto, A.; Ferreira, P.G.; Vilanova, M. Modulation of Leptin and Leptin Receptor Expression in Mice Acutely Infected with Neospora caninum. Pathogens 2020, 9, 587. [Google Scholar] [CrossRef]
- Ohashi, K.; Parker, J.L.; Ouchi, N.; Higuchi, A.; Vita, J.A.; Gokce, N.; Pedersen, A.A.; Kalthoff, C.; Tullin, S.; Sams, A.; et al. Adiponectin promotes macrophage polarization toward an anti-inflammatory phenotype. J. Biol. Chem. 2010, 285, 6153–6160. [Google Scholar] [CrossRef]
- Yang, C.-C.; Sheu, H.-M.; Chung, P.-L.; Chang, C.-H.; Tsai, Y.-S.; Hughes, M.W.; Tuan, T.L.; Huang, L.L.H. Leptin of dermal adipose tissue is differentially expressed during the hair cycle and contributes to adipocyte-mediated growth inhibition of anagen-phase vibrissa hair. Exp. Dermatol. 2014, 24, 57–60. [Google Scholar] [CrossRef]
- Zhang, L.-J.; Guerrero-Juarez, C.F.; Hata, T.; Bapat, S.P.; Ramos, R.; Plikus, M.V.; Gallo, R.L. Dermal adipocytes protect against invasive Staphylococcus aureus skin infection. Science 2015, 347, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Cappellano, G.; Morandi, E.; Rainer, J.; Grubwieser, P.; Heinz, K.; Wolfram, D.; Bernhard, D.; Lobenwein, S.; Pierer, G.; Ploner, C. Human Macrophages Preferentially Infiltrate the Superficial Adipose Tissue. Int. J. Mol. Sci. 2018, 19, 1404–1414. [Google Scholar]
- Vitseva, O.I.; Tanriverdi, K.; Tchkonia, T.T.; Kirkland, J.L.; McDonnell, M.E.; Apovian, C.M.; Freedman, J.; Gokce, N. Inducible Toll-like receptor and NF-kappaB regulatory pathway expression in human adipose tissue. Obesity 2008, 16, 932–937. [Google Scholar] [CrossRef]
- Meijer, K.; de Vries, M.; Al-Lahham, S.; Bruinenberg, M.; Weening, D.; Dijkstra, M.; Kloosterhuis, N.; van der Leij, R.J.; van der Want, H.; Kroesen, B.-J.; et al. Human Primary Adipocytes Exhibit Immune Cell Function: Adipocytes Prime Inflammation Independent of Macrophages. PLoS ONE 2011, 6, e17154. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Lee, H.; Berg, A.H.; Lisanti, M.P.; Shapiro, L.; Scherer, P.E. The lipopolysaccharide-activated toll-like receptor (TLR)-4 induces synthesis of the closely related receptor TLR-2 in adipocytes. J. Biol. Chem. 2000, 275, 24255–24263. [Google Scholar] [CrossRef] [PubMed]
- Sethi, J.K.; Xu, H.; Uysal, K.T.; Wiesbrock, S.M.; Scheja, L.; Hotamisligil, G.S. Characterisation of receptor-specific TNFalpha functions in adipocyte cell lines lacking type 1 and 2 TNF receptors. Febs. Lett. 2000, 469, 77–82. [Google Scholar] [CrossRef]
- Cawthorn, W.P.; Sethi, J.K. TNF-α and adipocyte biology. Febs. Lett. 2007, 582, 117–131. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, J.; Zhang, Y.; Lau, W.B.; Jiao, L.-Y.; Liu, B.; Yuan, Y.; Wang, X.; Tao, L.; Gao, E.; et al. Differential regulation of TNF receptor 1 and receptor 2 in adiponectin expression following myocardial ischemia. Int. J. Cardiol. 2013, 168, 2201–2206. [Google Scholar] [CrossRef][Green Version]
- Doerrler, W.; Feingold, K.R.; Grünfeld, C. Cytokines induce catabolic effects in cultured adipocytes by multiple mechanisms. Cytokine 1994, 6, 478–484. [Google Scholar] [CrossRef]
- Nov, O.; Kohl, A.; Lewis, E.C.; Bashan, N.; Dvir, I.; Ben-Shlomo, S.; Fishman, S.; Wueest, S.; Konrad, D.; Rudich, A. Interleukin-1beta may mediate insulin resistance in liver-derived cells in response to adipocyte inflammation. Endocrinology 2010, 151, 4247–4256. [Google Scholar] [CrossRef]
- Lagathu, C.; Yvan-Charvet, L.; Bastard, J.P.; Maachi, M.; Quignard-Boulangé, A.; Capeau, J.; Caron, M. Long-term treatment with interleukin-1beta induces insulin resistance in murine and human adipocytes. Diabetologia 2006, 49, 2162–2173. [Google Scholar] [CrossRef]
- Feingold, K.R.; Doerrler, W.; Dinarello, C.A.; Fiers, W.; Grünfeld, C. Stimulation of lipolysis in cultured fat cells by tumor necrosis factor, interleukin-1, and the interferons is blocked by inhibition of prostaglandin synthesis. Endocrinology 1992, 130, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Nagai, Y.; Honda, H.; Okamoto, N.; Yanagibashi, T.; Ogasawara, M.; Yamamoto, S.; Imamura, R.; Takasaki, I.; Hara, H.; et al. Bidirectional crosstalk between neutrophils and adipocytes promotes adipose tissue inflammation. Faseb J. 2019, 33, 11821–11835. [Google Scholar] [CrossRef]
- Masoodi, M.; Kuda, O.; Rossmeisl, M.; Flachs, P.; Kopecky, J. Lipid signaling in adipose tissue: Connecting inflammation & metabolism. Bba Mol. Cell Biol. Lipids 2015, 1851, 503–518. [Google Scholar]
- Vered, E.-C.; Assaf, R.; Nurit, H.; Rachel, L. Neutrophils transiently infiltrate intra-abdominal fat early in the course of high-fat feeding. J. Lipid Res. 2008, 49, 1894–1903. [Google Scholar]
- Talukdar, S.; Da Young, O.; Bandyopadhyay, G.; Li, D.; Xu, J.; McNelis, J.; Lu, M.; Li, P.; Yan, Q.; Zhu, Y.; et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat. Med. 2019, 18, 1–8. [Google Scholar] [CrossRef]
- Mansuy-Aubert, V.; Zhou, Q.L.; Xie, X.; Gong, Z.; Huang, J.-Y.; Khan, A.R.; Aubert, G.; Candelaria, K.; Thomas, S.; Shin, D.-J.; et al. Imbalance between Neutrophil Elastase and its Inhibitor α1-Antitrypsin in Obesity Alters Insulin Sensitivity, Inflammation, and Energy Expenditure. Cell Metab. 2013, 17, 534–548. [Google Scholar] [CrossRef]
- Baumann, H.; Morella, K.K.; White, D.W.; Dembski, M.; Bailon, P.S.; Kim, H.; Lai, C.F.; Tartaglia, L.A. The full-length leptin receptor has signaling capabilities of interleukin 6-type cytokine receptors. Proc. Natl. Acad. Sci. USA 1996, 93, 8374–8378. [Google Scholar] [CrossRef]
- Caldefie-Chezet, F.; Poulin, A.; Vasson, M.P. Leptin Regulates Functional Capacities of Polymorphonuclear Neutrophils. Free Radic. Res. 2012, 37, 809–814. [Google Scholar] [CrossRef] [PubMed]
- Sundqvist, M.; Christenson, K.; Holdfeldt, A.; Gabl, M.; Mårtensson, J.; Björkman, L.; Dieckmann, R.; Dahlgren, C.; Forsman, H. Similarities and differences between the responses induced in human phagocytes through activation of the medium chain fatty acid receptor GPR84 and the short chain fatty acid receptor FFA2R. Bba Mol. Cell Res. 2018, 1865, 695–708. [Google Scholar] [CrossRef] [PubMed]
- Metzemaekers, M.; Gouwy, M.; Proost, P. Neutrophil chemoattractant receptors in health and disease: Double-edged swords. Cell Mol. Immunol. 2020, 17, 433–450. [Google Scholar] [CrossRef]
- Tynan, G.A.; Hearnden, C.H.; Oleszycka, E.; Lyons, C.L.; Coutts, G.; O’Connell, J.; Corrigan, M.A.; Lynch, L.; Campbell, M.; Callanan, J.J.; et al. Endogenous Oils Derived From Human Adipocytes Are Potent Adjuvants That Promote IL-1 -Dependent Inflammation. Diabetes 2014, 63, 2037–2050. [Google Scholar] [CrossRef]
- Staiger, H.; Staiger, K.; Stefan, N.; Wahl, H.G.; Machicao, F.; Kellerer, M.; Häring, H.-U. Palmitate-induced interleukin-6 expression in human coronary artery endothelial cells. Diabetes 2004, 53, 3209–3216. [Google Scholar] [CrossRef] [PubMed]
- Freigang, S.; Ampenberger, F.; Weiss, A.; Kanneganti, T.-D.; Iwakura, Y.; Hersberger, M.; Kopf, M. Fatty acid–induced mitochondrial uncoupling elicits inflammasome-independent IL-1α and sterile vascular inflammation in atherosclerosis. Nat. Immunol. 2013, 14, 1045–1053. [Google Scholar] [CrossRef]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef]
- Cinti, S.; Mitchell, G.; Barbatelli, G.; Murano, I.; Ceresi, E.; Faloia, E.; Wang, S.; Fortier, M.; Greenberg, A.S.; Obin, M.S. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J. Lipid Res. 2005, 46, 2347–2355. [Google Scholar] [CrossRef] [PubMed]
- den Brok, M.H.; Raaijmakers, T.K.; Collado-Camps, E.; Adema, G.J. Lipid Droplets as Immune Modulators in Myeloid Cells. Trends Immunol. 2018, 39, 380–392. [Google Scholar] [CrossRef] [PubMed]
- Nomura, M.; Liu, J.; Rovira, I.I.; Gonzalez-Hurtado, E.; Lee, J.; Wolfgang, M.J.; Finkel, T. Fatty acid oxidation in macrophage polarization. Nat. Publ. Group 2016, 17, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Suganami, T.; Nishida, J.; Ogawa, Y. A Paracrine Loop Between Adipocytes and Macrophages Aggravates Inflammatory Changes. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2062–2068. [Google Scholar] [CrossRef]
- Recio, C.; Lucy, D.; Purvis, G.S.D.; Iveson, P.; Zeboudj, L.; Iqbal, A.J.; Lin, D.; O’Callaghan, C.; Davison, L.; Griesbach, E.; et al. Activation of the Immune-Metabolic Receptor GPR84 Enhances Inflammation and Phagocytosis in Macrophages. Front. Immunol. 2018, 9, 81. [Google Scholar] [CrossRef]
- Vieira, W.A.; Gijsen, H.S.-V.; Ferris, W.F. Free fatty acid G-protein coupled receptor signaling in M1 skewed white adipose tissue macrophages. Cell Mol. Life Sci. 2016, 73, 3665–3676. [Google Scholar] [CrossRef]
- Alvarez-Curto, E.; Milligan, G. Metabolism meets immunity: The role of free fatty acid receptors in the immune system. Biochem. Pharmacol. 2016, 114, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Kasza, I.; Suh, Y.; Wollny, D.; Clark, R.J.; Roopra, A.; Colman, R.J.; MacDougald, O.A.; Shedd, T.A.; Nelson, D.W.; Yen, M.-I.; et al. Syndecan-1 Is Required to Maintain Intradermal Fat and Prevent Cold Stress. PLoS Genet. 2014, 10, e1004514. [Google Scholar] [CrossRef]
- Marangoni, R.G.; Korman, B.D.; Wei, J.; Wood, T.A.; Graham, L.V.; Whitfield, M.L.; Scherer, P.E.; Tourtellotte, W.G.; Varga, J. Myofibroblasts in murine cutaneous fibrosis originate from adiponectin-positive intradermal progenitors. Arthritis Rheumatol. 2015, 67, 1062–1073. [Google Scholar] [CrossRef] [PubMed]
- Franz, A.; Wood, W.; Martin, P. Fat Body Cells Are Motile and Actively Migrate to Wounds to Drive Repair and Prevent Infection. Dev. Cell 2018, 44, 460–470.e3. [Google Scholar] [CrossRef] [PubMed]
- Frank, S.; Stallmeyer, B.; Kämpfer, H.; Kolb, N.; Pfeilschifter, J. Leptin enhances wound re-epithelialization and constitutes a direct function of leptin in skin repair. J. Clin. Investig. 2000, 106, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Shibata, S.; Tada, Y.; Asano, Y.; Hau, C.S.; Kato, T.; Saeki, H.; Yamauchi, T.; Kubota, N.; Kadowaki, T.; Sato, S. Adiponectin regulates cutaneous wound healing by promoting keratinocyte proliferation and migration via the ERK signaling pathway. J. Immunol. 2012, 189, 3231–3241. [Google Scholar] [CrossRef]
- El-Hattab, M.Y.; Nagumo, Y.; Gourronc, F.A.; Klingelhutz, A.J.; Ankrum, J.A.; Sander, E.A. Human Adipocyte Conditioned Medium Promotes In Vitro Fibroblast Conversion to Myofibroblasts. Sci. Rep. 2020, 10, 10286. [Google Scholar] [CrossRef]
- Reitman, M.L.; Gavrilova, O. A-ZIP/F-1 mice lacking white fat: A model for understanding lipoatrophic diabetes. Int J. Obes Relat Metab Disord 2000, 24 (Suppl. 4), S11. [Google Scholar] [CrossRef][Green Version]
- Wang, Y.; Qian, Y.; Fang, Q.; Zhong, P.; Li, W.; Wang, L.; Fu, W.; Zhang, Y.; Xu, Z.; Li, X.; et al. Saturated palmitic acid induces myocardialinflammatory injuries through direct bindingto TLR4 accessory protein MD2. Nat. Commun. 2016, 8, 1–13. [Google Scholar]
- Van De Water, L.; Varney, S.; Tomasek, J.J. Mechanoregulation of the Myofibroblast in Wound Contraction, Scarring, and Fibrosis: Opportunities for New Therapeutic Intervention. Adv. Wound Care 2013, 2, 122–141. [Google Scholar] [CrossRef] [PubMed]
- Krausgruber, T.; Fortelny, N.; Fife-Gernedl, V.; Senekowitsch, M.; Schuster, L.C.; Lercher, A.; Nemc, A.; Schmidl, C.; Rendeiro, A.F.; Bergthaler, A.; et al. Structural cells are key regulators of organ-specific immune responses. Nature 2020, 583, 1–29. [Google Scholar] [CrossRef]
- Hughes, T.K.; Wadsworth, M.H.; Gierahn, T.M.; Do, T.; Weiss, D.; Andrade, P.R.; Ma, F.; de Andrade Silva, B.J.; Shao, S.; Tsoi, L.C.; et al. Second-Strand Synthesis-Based Massively Parallel scRNA-Seq Reveals Cellular States and Molecular Features of Human Inflammatory Skin Pathologies. Immunity 2020, 53, 878–894.e7. [Google Scholar] [CrossRef]
- Jolly, A.L.; Rau, S.; Chadha, A.K.; Abdulraheem, E.A.; Dean, D. Stromal Fibroblasts Drive Host Inflammatory Responses That Are Dependent on Chlamydia trachomatisStrain Type and Likely Influence Disease Outcomes. mBio 2019, 10, 143. [Google Scholar] [CrossRef]
- Shochet, G.E.; Brook, E.; Israeli-Shani, L.; Edelstein, E.; Shitrit, D. Fibroblast paracrine TNF-α signaling elevates integrin A5 expression in idiopathic pulmonary fibrosis (IPF). Respir Res. 2017, 18, 1–12. [Google Scholar]
- Mouton, A.J.; Ma, Y.; Gonzalez, O.J.R.; Daseke, M.J.; Flynn, E.R.; Freeman, T.C.; Garrett, M.R.; DeLeon-Pennell, K.Y.; Lindsey, M.L. Fibroblast polarization over the myocardial infarction time continuum shifts roles from inflammation to angiogenesis. Basic Res. Cardiol. 2019, 114, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Gęgotek, A.; Domingues, P.; Wroński, A.; Skrzydlewska, E. Changes in Proteome of Fibroblasts Isolated from Psoriatic Skin Lesions. Int. J. Mol. Sci. 2020, 21, 5363. [Google Scholar] [CrossRef]
- Arasa, J.; Terencio, M.C.; Andrés, R.M.; Marín-Castejón, A.; Valcuende-Cavero, F.; Payá, M.; Montesinos, M.C. Defective Induction of COX-2 Expression by Psoriatic Fibroblasts Promotes Pro-inflammatory Activation of Macrophages. Front. Immunol. 2019, 10, 536. [Google Scholar] [CrossRef]
- Löwa, A.; Graff, P.; Kaessmeyer, S.; Hedtrich, S. Fibroblasts from atopic dermatitis patients trigger inflammatory processes and hyperproliferation in human skin equivalents. J. Eur. Acad. Derm. Venereol. 2020, 34, e262–e265. [Google Scholar] [CrossRef] [PubMed]
- Ploeger, D.T.; Hosper, N.A.; Schipper, M.; Koerts, J.A.; de Rond, S.; Bank, R.A. Cell plasticity in wound healing: Paracrine factors of M1/ M2 polarized macrophages influence the phenotypical state of dermal fibroblasts. Cell Commun. Signal. 2013, 11, 29. [Google Scholar] [CrossRef] [PubMed]
- Sandstedt, J.; Sandstedt, M.; Lundqvist, A.; Jansson, M.; Sopasakis, V.R.; Jeppsson, A.; Hultén, L.M. Human cardiac fibroblasts isolated from patients with severe heart failure are immune-competent cells mediating an inflammatory response. Cytokine 2019, 113, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Daseke, M.J.; Tenkorang, M.A.A.; Chalise, U.; Konfrst, S.R.; Lindsey, M.L. Cardiac fibroblast activation during myocardial infarction wound healing: Fibroblast polarization after MI. Matrix Biol. 2020, 91–92, 109–116. [Google Scholar] [CrossRef]
- Richardson, R.J. Parallels between vertebrate cardiac and cutaneous wound healing and regeneration. npj Regen. Med. 2018, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Al-Rikabi, A.H.A.; Tobin, D.J.; Riches-Suman, K.; Thornton, M.J. Dermal fibroblasts cultured from donors with type 2 diabetes mellitus retain an epigenetic memory associated with poor wound healing responses. Sci. Rep. 2021, 11, 1–13. [Google Scholar] [CrossRef]
- Di Domizio, J.; Belkhodja, C.; Chenuet, P.; Fries, A.; Murray, T.; Mondéjar, P.M.; Demaria, O.; Conrad, C.; Homey, B.; Werner, S.; et al. The commensal skin microbiota triggers type I IFN–dependent innate repair responses in injured skin. Nat. Immunol. 2020, 21, 1–29. [Google Scholar] [CrossRef]
- Park, S.-Y.; Byun, E.; Lee, J.; Kim, S.; Kim, H. Air Pollution, Autophagy, and Skin Aging: Impact of Particulate Matter (PM10) on Human Dermal Fibroblasts Int. J. Mol. Sci. 2018, 19, 2727. [Google Scholar] [CrossRef]
- Eleftheriadis, T.; Liakopoulos, V.; Lawson, B.; Antoniadi, G.; Stefanidis, I.; Galaktidou, G. Lipopolysaccharide and hypoxia significantly alters interleukin-8 and macrophage chemoattractant protein-1 production by human fibroblasts but not fibrosis related factors. Hippokratia 2011, 15, 238–243. [Google Scholar]
- Suwara, M.I.; Green, N.J.; Borthwick, L.A.; Mann, J.; Mayer-Barber, K.D.; Barron, L.; Corris, P.A.; Farrow, S.N.; Wynn, T.A.; Fisher, A.J.; et al. IL-1α released from damaged epithelial cells is sufficient and essential to trigger inflammatory responses in human lung fibroblasts. Mucosal Immunol. 2013, 7, 684–693. [Google Scholar] [CrossRef]
- Paish, H.L.; Kalson, N.S.; Smith, G.R.; del Carpio Pons, A.; Baldock, T.E.; Smith, N.; Swist-Szulik, K.; Weir, D.J.; Bardgett, M.; Deehan, D.J.; et al. Fibroblasts Promote Inflammation and Pain via IL-1α Induction of the Monocyte Chemoattractant Chemokine (C-C Motif) Ligand 2. Am. J. Phys. Anthropol. 2018, 188, 696–714. [Google Scholar] [CrossRef]
- Spiekstra, S.W.; Breetveld, M.; Rustemeyer, T.; Scheper, R.J.; Gibbs, S. Wound-healing factors secreted by epidermal keratinocytes and dermal fibroblasts in skin substitutes. Wound Repair Regen. 2007, 15, 708–717. [Google Scholar] [CrossRef] [PubMed]
- Witowski, J.; Tayama, H.; Książek, K.; Wanic-Kossowska, M.; Bender, T.O.; Jörres, A. Human peritoneal fibroblasts are a potent source of neutrophil-targeting cytokines: A key role of IL-1beta stimulation. Lab. Investig. 2009, 89, 414–424. [Google Scholar] [CrossRef]
- Ng, S.B.; Tan, Y.H.; Guy, G.R. Differential induction of the interleukin-6 gene by tumor necrosis factor and interleukin-1. J. Biol. Chem. 1994, 269, 19021–19027. [Google Scholar] [CrossRef]
- Kitanaka, N.; Nakano, R.; Sugiura, K.; Kitanaka, T.; Namba, S.; Konno, T.; Nakayama, T.; Sugiya, H. Interleukin-1β promotes interleulin-6 expression via ERK1/2 signaling pathway in canine dermal fibroblasts. PLoS ONE 2019, 14, e0220262. [Google Scholar] [CrossRef] [PubMed]
- Akbar, M.; McLean, M.; Garcia-Melchor, E.; Crowe, L.A.; McMillan, P.; Fazzi, U.G.; Martin, D.; Arthur, A.; Reilly, J.H.; McInnes, I.B.; et al. Fibroblast activation and inflammation in frozen shoulder. PLoS ONE 2019, 14, e0215301. [Google Scholar] [CrossRef] [PubMed]
- Ritsu, M.; Kawakami, K.; Kanno, E.; Tanno, H.; Ishii, K.; Imai, Y.; Maruyama, R.; Tachi, M. Critical role of tumor necrosis factor-α in the early process of wound healing in skin. J. Dermatol. Dermatol. Surg. 2017, 21, 14–19. [Google Scholar] [CrossRef]
- Bae, S.; Jung, Y.; Choi, Y.M.; Li, S. Effects of er-miao-san extracts on TNF-alpha-induced MMP-1 expression in human dermal fibroblasts. Biol. Res. 2015, 48, 8. [Google Scholar] [CrossRef][Green Version]
- Goldberg, M.T.; Han, Y.-P.; Yan, C.; Shaw, M.C.; Garner, W.L. TNF-α Suppresses α-Smooth Muscle Actin Expression in Human Dermal Fibroblasts: An Implication for Abnormal Wound Healing. J. Investig. Dermatol. 2007, 127, 2645–2655. [Google Scholar] [CrossRef]
- Han, Y.P.; Tuan, T.L.; Wu, H.; Hughes, M.; Garner, W.L. TNF-alpha stimulates activation of pro-MMP2 in human skin through NF-(kappa)B mediated induction of MT1-MMP. J. Cell Biol. 2001, 114, 131–139. [Google Scholar]
- Fries, K.M.; Sempowski, G.D.; Gaspari, A.A.; Blieden, T.; Looney, R.J.; Phipps, R.P. CD40 expression by human fibroblasts. Clin. Immunol. Immunopathol. 1995, 77, 42–51. [Google Scholar] [CrossRef]
- Sempowski, G.D.; Chess, P.R.; Moretti, A.J.; Padilla, J.; Phipps, R.P.; Blieden, T.M. CD40 mediated activation of gingival and periodontal ligament fibroblasts. J. Periodontol. 1997, 68, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cao, H.J.; Graf, B.; Meekins, H.; Smith, T.J.; Phipps, R.P. CD40 engagement up-regulates cyclooxygenase-2 expression and prostaglandin E2 production in human lung fibroblasts. J. Immun. J. 1998, 160, 1053–1057. [Google Scholar]
- Kawai, M.; Masuda, A.; Kuwana, M. A CD40-CD154 interaction in tissue fibrosis. Arthritis Rheum. 2008, 58, 3562–3573. [Google Scholar] [CrossRef] [PubMed]
- Buckley, C.D. Defining a role for fibroblasts in the persistence of chronic inflammatory joint disease. Ann. Rheum. Dis. 2004, 63, ii92–ii95. [Google Scholar] [CrossRef]
- Ferrer, R.A.; Saalbach, A.; Grünwedel, M.; Lohmann, N.; Forstreuter, I.; Saupe, S.; Wandel, E.; Simon, J.C.; Franz, S. Dermal Fibroblasts Promote Alternative Macrophage Activation Improving Impaired Wound Healing. J. Investig. Dermatol. 2017, 137, 941–950. [Google Scholar] [CrossRef]
- Jang, S.; Park, J.-S.; Won, Y.-H.; Yun, S.-J.; Kim, S.-J. The Expression of Toll-Like Receptors (TLRs) in Cultured Human Skin Fibroblast is Modulated by Histamine. Chonnam Med. J. 2012, 48, 7–14. [Google Scholar] [CrossRef][Green Version]
- Yao, C.; Oh, J.-H.; Lee, D.H.; Bae, J.-S.; Jin, C.L.; Park, C.-H.; Chung, J.H. Toll-like receptor family members in skin fibroblasts are functional and have a higher expression compared to skin keratinocytes. Int. J. Mol. Med. 2015, 35, 1443–1450. [Google Scholar] [CrossRef]
- Wohlfahrt, T.; Rauber, S.; Uebe, S.; Luber, M.; Soare, A.; Ekici, A.; Weber, S.; Matei, A.-E.; Chen, C.-W.; Maier, C.; et al. PU.1 controls fibroblast polarization and tissue fibrosis. Nature 2019, 1–27. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, C.; Liu, L.; A, X.; Chen, B.; Li, Y.; Du, J. Macrophage-Derived mir-155-Containing Exosomes Suppress Fibroblast Proliferation and Promote Fibroblast Inflammation during Cardiac Injury. Mol. Ther. 2017, 25, 192–204. [Google Scholar] [CrossRef]
- Mahmoudi, S.; Mancini, E.; Xu, L.; Moore, A.; Jahanbani, F.; Hebestreit, K.; Srinivasan, R.; Li, X.; Devarajan, K.; Prélot, L.; et al. Heterogeneity in old fibroblasts is linked to variability in reprogramming and wound healing. Nature 2019, 574, 1–41. [Google Scholar] [CrossRef] [PubMed]
- Driskell, R.R.; Lichtenberger, B.M.; Hoste, E.; Kretzschmar, K.; Simons, B.D.; Charalambous, M.; Ferrón, S.R.; Herault, Y.; Pavlovic, G.; Ferguson-Smith, A.C.; et al. Distinct fibroblast lineages determine dermal architecture in skin development and repair. Nature 2013, 504, 277–281. [Google Scholar] [PubMed]
- Phan, Q.M.; Sinha, S.; Biernaskie, J.; Driskell, R.R. Single-cell transcriptomic analysis of small and large wounds reveals the distinct spatial organization of regenerative fibroblasts. Exp. Dermatol. 2020, 30, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Correa-Gallegos, D.; Jiang, D.; Christ, S.; Ramesh, P.; Ye, H.; Wannemacher, J.; Gopal, S.K.; Yu, Q.; Aichler, M.; Walch, A.; et al. Patch repair of deep wounds by mobilized fascia. Nature 2019, 576, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Philippeos, C.; Telerman, S.B.; Oulès, B.; Pisco, A.O.; Shaw, T.J.; Elgueta, R.; Lombardi, G.; Driskell, R.R.; Soldin, M.; Lynch, M.D.; et al. Spatial and Single-Cell Transcriptional Profiling Identifies Functionally Distinct Human Dermal Fibroblast Subpopulations. J. Investig. Dermatol. 2018, 138, 811–825. [Google Scholar] [CrossRef] [PubMed]
- Hepler, C.; Shan, B.; Zhang, Q.; Henry, G.H.; Shao, M.; Vishvanath, L.; Ghaben, A.L.; Mobley, A.B.; Strand, D.; Hon, G.C.; et al. Identification of functionally distinct fibro-inflammatory and adipogenic stromal subpopulations in visceral adipose tissue of adult mice. Elife 2018, 7, 771. [Google Scholar] [CrossRef]
- Mizoguchi, F.; Slowikowski, K.; Wei, K.; Marshall, J.L.; Rao, D.A.; Chang, S.K.; Nguyen, H.N.; Noss, E.H.; Turner, J.D.; Earp, B.E.; et al. Functionally distinct disease-associated fibroblast subsets in rheumatoid arthritis. Nat. Commun. 2018, 9, 789. [Google Scholar] [CrossRef]
- Croft, A.P.; Campos, J.; Jansen, K.; Turner, J.D.; Marshall, J.; Attar, M.; Savary, L.; Wehmeyer, C.; Naylor, A.J.; Kemble, S.; et al. Distinct fibroblast subsets drive inflammation and damage in arthritis. Nature 2019, 570, 1–24. [Google Scholar] [CrossRef]
- Ezure, T.; Amano, S. Adiponectin and leptin up-regulate extracellular matrix production by dermal fibroblasts. Biofactors 2007, 31, 229–236. [Google Scholar] [CrossRef]
- Marangoni, R.G.; Masui, Y.; Fang, F.; Korman, B.; Lord, G.; Lee, J.; Lakota, K.; Wei, J.; Scherer, P.E.; Otvos, L.; et al. Adiponectin is an endogenous anti-fibrotic mediator and therapeutic target. Sci. Rep. 2017, 7, 4397. [Google Scholar]
- Kim, E.J.; Kim, Y.K.; Kim, M.-K.; Kim, S.; Kim, J.Y.; Lee, D.H.; Chung, J.H. UV-induced inhibition of adipokine production in subcutaneous fat aggravates dermal matrix degradation in human skin. Sci. Rep. 2016, 6, 25616. [Google Scholar] [CrossRef] [PubMed]
- Gosain, A.; Di Pietro, L.A. Aging and Wound Healing. World J. Surg. 2004, 28, 321–326. [Google Scholar] [CrossRef]
- Brem, H.; Tomic-Canic, M. Cellular and molecular basis of wound healing in diabetes. J. Clin. Investig. 2007, 117, 1219–1222. [Google Scholar] [CrossRef] [PubMed]
- Dinh, T.; Elder, S.; Veves, A. Delayed wound healing in diabetes: Considering future treatments. Diabetes Manag. 2011, 1, 509–519. [Google Scholar] [CrossRef]
- Schnider, S.L.; Kohn, R.R. Effects of age and diabetes mellitus on the solubility and nonenzymatic glucosylation of human skin collagen. J. Clin. Investig. 1981, 67, 1630–1635. [Google Scholar] [CrossRef]
- Argyropoulos, A.J.; Robichaud, P.; Balimunkwe, R.M.; Fisher, G.J.; Hammerberg, C.; Yan, Y.; Quan, T. Alterations of Dermal Connective Tissue Collagen in Diabetes: Molecular Basis of Aged-Appearing Skin. PLoS ONE 2016, 11, e0153806. [Google Scholar] [CrossRef]
- Zou, Z.; Long, X.; Zhao, Q.; Zheng, Y.; Song, M.; Ma, S.; Jing, Y.; Wang, S.; He, Y.; Esteban, C.R.; et al. A Single-Cell Transcriptomic Atlas of Human Skin Aging. Dev. Cell 2020, 492, 438. [Google Scholar]
- Varani, J.; Dame, M.K.; Rittié, L.; Fligiel, S.E.G.; Kang, S.; Fisher, G.J.; Voorhees, J.J. Decreased collagen production in chronologically aged skin: Roles of age-dependent alteration in fibroblast function and defective mechanical stimulation. Am. J. Phys. Anthropol. 2006, 168, 1861–1868. [Google Scholar]
- Goulding, V. The effects of diabetes on collagen within wound healing. Diabet. Foot J. 2015, 18, 75–80. [Google Scholar]
- Nishiguchi, M.A.; Spencer, C.A.; Leung, D.H.; Leung, T.H. Aging Suppresses Skin-Derived Circulating SDF1 to Promote Full-Thickness Tissue Regeneration. Cell Rep. 2018, 24, 3383–3392.e5. [Google Scholar] [CrossRef]
- Grice, E.A.; Snitkin, E.S.; Yockey, L.J.; Bermudez, D.M.; NISC Comparative Sequencing Program; Liechty, K.W.; Segre, J.A. Longitudinal shift in diabetic wound microbiota correlates with prolonged skin defense response. Proc. Natl. Acad. Sci. USA 2010, 107, 14799–14804. [Google Scholar] [CrossRef]
- Roche, E.D.; Renick, P.J.; Tetens, S.P.; Ramsay, S.J.; Daniels, E.Q.; Carson, D.L. Increasing the presence of biofilm and healing delay in a porcine model of MRSA-infected wounds. Wound Repair Regen. 2012, 20, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Dowd, S.E.; Delton Hanson, J.; Rees, E.; Wolcott, R.D.; Zischau, A.M.; Sun, Y.; White, J.; Smith, D.M.; Kennedy, J.; Jones, C.E. Survey of fungi and yeast in polymicrobial infections in chronic wounds. J. Wound Care 2011, 20, 40–47. [Google Scholar] [CrossRef]
- McCarty, S.M.; Percival, S.L. Proteases and Delayed Wound Healing. Adv. Wound Care 2013, 2, 438–447. [Google Scholar] [CrossRef]
- Frykberg, R.G.; Banks, J. Challenges in the Treatment of Chronic Wounds. Adv. Wound Care 2015, 4, 560–582. [Google Scholar] [CrossRef]
- Motegi, S.-I.; Ishikawa, O. Mesenchymal stem cells: The roles and functions in cutaneous wound healing and tumor growth. J. Dermatol. Sci. 2017, 86, 83–89. [Google Scholar] [PubMed]
- Wood, S.; Jayaraman, V.; Huelsmann, E.J.; Bonish, B.; Burgad, D.; Sivaramakrishnan, G.; Qin, S.; DiPietro, L.A.; Zloza, A.; Zhang, C.; et al. Pro-Inflammatory Chemokine CCL2 (MCP-1) Promotes Healing in Diabetic Wounds by Restoring the Macrophage Response. PLoS ONE 2014, 9, e91574. [Google Scholar] [CrossRef] [PubMed]
- Phillipson, M.; Kubes, P. The Healing Power of Neutrophils. Trends Immunol. 2019, 40, 635–647. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Yang, P.; Gao, M.; Yu, T.; Shi, Y.; Zhang, M.; Yao, M.; Liu, Y.; Zhang, X. NLRP3 activation induced by neutrophil extracellular traps sustains inflammatory response in the diabetic wound. Clin. Sci. (Lond) 2019, 133, 565–582. [Google Scholar] [CrossRef]
- Yan, J.; Tie, G.; Wang, S.; Tutto, A.; DeMarco, N.; Khair, L.; Fazzio, T.G.; Messina, L.M. Diabetes impairs wound healing by Dnmt1-dependent dysregulation of hematopoietic stem cells differentiation towards macrophages. Nat. Commun. 2018, 9, 33. [Google Scholar] [CrossRef]
- Ogawa, T.; Kitagawa, M.; Hirokawa, K. Age-related changes of human bone marrow: A histometric estimation of proliferative cells, apoptotic cells, T cells, B cells and macrophages. Mech. Ageing Dev. 2000, 117, 57–68. [Google Scholar] [CrossRef]
- Mahbub, S.; Deburghgraeve, C.R.; Kovacs, E.J. Advanced age impairs macrophage polarization. J. Interferon Cytokine Res. 2012, 32, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Chelvarajan, R.L.; Collins, S.M.; Van Willigen, J.M.; Bondada, S. The unresponsiveness of aged mice to polysaccharide antigens is a result of a defect in macrophage function. J. Leukoc. Biol. 2005, 77, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, G.S.; Horan, M.A.; Ferguson, M.W. Aging alters the inflammatory and endothelial cell adhesion molecule profiles during human cutaneous wound healing. Lab. Investig. 1998, 78, 47–58. [Google Scholar] [PubMed]
- Swift, M.E.; Burns, A.L.; Gray, K.L.; DiPietro, L.A. Age-Related Alterations in the Inflammatory Response to Dermal Injury. J. Investig. Dermatol. 2001, 117, 1027–1035. [Google Scholar] [CrossRef]
- Kimball, A.; Schaller, M.; Joshi, A.; Davis, F.M.; denDekker, A.; Boniakowski, A.; Bermick, J.; Obi, A.; Moore, B.; Henke, P.K.; et al. Ly6CHi Blood Monocyte/Macrophage Drive Chronic Inflammation and Impair Wound Healing in Diabetes Mellitus. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1102–1114. [Google Scholar] [CrossRef] [PubMed]
- Tsalamandris, S.; Antonopoulos, A.S.; Oikonomou, E.; Papamikroulis, G.-A.; Vogiatzi, G.; Papaioannou, S.; Deftereos, S.; Tousoulis, D. The Role of Inflammation in Diabetes: Current Concepts and Future Perspectives. Eur. Cardiol. 2019, 14, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune-metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Investig. 2007, 117, 175–184. [Google Scholar] [CrossRef]
- Orliaguet, L.; Dalmas, E.; Drareni, K.; Venteclef, N.; Alzaid, F. Mechanisms of Macrophage Polarization in Insulin Signaling and Sensitivity. Front. Endocrinol. 2020, 11, 62. [Google Scholar] [CrossRef]
- Gallagher, K.A.; Joshi, A.; Carson, W.F.; Schaller, M.; Allen, R.; Mukerjee, S.; Kittan, N.; Feldman, E.L.; Henke, P.K.; Hogaboam, C.; et al. Epigenetic changes in bone marrow progenitor cells influence the inflammatory phenotype and alter wound healing in type 2 diabetes. Diabetes 2015, 64, 1420–1430. [Google Scholar] [CrossRef]
- Xuan, W.; Qu, Q.; Zheng, B.; Xiong, S.; Fan, G.-H. The chemotaxis of M1 and M2 macrophages is regulated by different chemokines. J. Leukoc. Biol. 2015, 97, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Fivenson, D.P.; Faria, D.T.; Nickoloff, B.J.; Poverini, P.J.; Kunkel, S.; Burdick, M.; Strieter, R.M. Chemokine and inflammatory cytokine changes during chronic wound healing. Wound Repair Regen. 1997, 5, 310–322. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.T.; Nguyen, X.-M.T.; Lane, J.; Wang, P. Relationship between obesity and diabetes in a US adult population: Findings from the National Health and Nutrition Examination Survey, 1999–2006. Obes. Surg. 2011, 21, 351–355. [Google Scholar] [CrossRef] [PubMed]
- Morigny, P.; Houssier, M.; Mouisel, E.; Langin, D. Adipocyte lipolysis and insulin resistance. Biochimie 2016, 125, 259–266. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Saltiel, A.R. Inflammatory links between obesity and metabolic disease. J. Clin. Investig. 2011, 121, 2111–2117. [Google Scholar] [PubMed]
- Zhu, Q.; An, Y.A.; Kim, M.; Zhang, Z.; Zhao, S.; Zhu, Y.; Asterholm, I.W.; Kusminski, C.M.; Scherer, P.E. Suppressing adipocyte inflammation promotes insulin resistance in mice. Mol. Metab. 2020, 39, 101010. [Google Scholar] [CrossRef] [PubMed]
- Asterholm, I.W.; Tao, C.; Morley, T.S.; Wang, Q.A.; Delgado-Lopez, F.; Wang, Z.V.; Scherer, P.E. Adipocyte Inflammation Is Essentialfor Healthy Adipose Tissue Expansion and Remodeling. Cell Metab. 2014, 20, 103–118. [Google Scholar]
- Zhou, H.; Zhang, Z.; Qian, G.; Zhou, J. Omentin-1 attenuates adipose tissue inflammation via restoration of TXNIP/NLRP3 signaling in high-fat diet-induced obese mice. Fundam. Clin. Pharm. 2020, 34, 721–735. [Google Scholar] [CrossRef] [PubMed]
- Fain, J.N.; Madan, A.K.; Hiler, M.L.; Cheema, P.; Bahouth, S.W. Comparison of the release of adipokines by adipose tissue, adipose tissue matrix, and adipocytes from visceral and subcutaneous abdominal adipose tissues of obese humans. Endocrinology 2004, 145, 2273–2282. [Google Scholar] [CrossRef]
- Kim, W.K.; Bae, K.-H.; Lee, S.C.; Oh, K.-J. The Latest Insights into Adipokines in Diabetes. J. Clin. Med. 2019, 8, 1874. [Google Scholar] [CrossRef]
- Gucalp, A.; Iyengar, N.M.; Zhou, X.K.; Giri, D.D.; Falcone, D.J.; Wang, H.; Williams, S.; Krasne, M.D.; Yaghnam, I.; Kunzel, B.; et al. Periprostatic adipose inflammation is associated with high-grade prostate cancer. Nat. Publ. Group 2017, 20, 1–6. [Google Scholar] [CrossRef]
- Pellegrinelli, V.; Rouault, C.; Rodriguez-Cuenca, S.; Albert, V.; Edom-Vovard, F.; Vidal-Puig, A.; Clement, K.; Butler-Browne, G.S.; Lacasa, D. Human Adipocytes Induce Inflammation and Atrophy in Muscle Cells During Obesity. Diabetes 2015, 64, 3121–3134. [Google Scholar] [CrossRef] [PubMed]
- Verboven, K.; Wouters, K.; Gaens, K.; Hansen, D.; Bijnen, M.; Wetzels, S.; Stehouwer, C.D.; Goossens, G.H.; Schalkwijk, C.G.; Blaak, E.E.; et al. Abdominal subcutaneous and visceral adipocyte size, lipolysis and inflammation relate to insulin resistance in male obese humans. Sci. Rep. 2018, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Czech, M.P. Cellular basis of insulin insensitivity in large rat adipocytes. J. Clin. Investig. 1976, 57, 1523–1532. [Google Scholar] [CrossRef] [PubMed]
- Jocken, J.W.E.; Langin, D.; Smit, E.; Saris, W.H.M.; Valle, C.; Hul, G.B.; Holm, C.; Arner, P.; Blaak, E.E. Adipose triglyceride lipase and hormone-sensitive lipase protein expression is decreased in the obese insulin-resistant state. J. Clin. Endocrinol. Metab. 2007, 92, 2292–2299. [Google Scholar] [CrossRef] [PubMed]
- Eichmann, T.O.; Kumari, M.; Haas, J.T.; Farese, R.V.; Zimmermann, R.; Lass, A.; Zechner, R. Studies on the substrate and stereo/regioselectivity of adipose triglyceride lipase, hormone-sensitive lipase, and diacylglycerol-O-acyltransferases. J. Biol. Chem. 2012, 287, 41446–41457. [Google Scholar] [CrossRef]
- Abrass, C.K.; Hori, M. Alterations in Fc receptor function of macrophages from streptozotocin-induced diabetic rats. J. Immun. J. 1984, 133, 1307–1312. [Google Scholar]
- Miao, M.; Niu, Y.; Xie, T.; Yuan, B.; Qing, C.; Lu, S. Diabetes-impaired wound healing and altered macrophage activation: A possible pathophysiologic correlation. Wound Repair Regen. 2012, 20, 203–213. [Google Scholar] [CrossRef]
- van Harten, R.M.; van Woudenbergh, E.; van Dijk, A.; Haagsman, H.P. Cathelicidins: Immunomodulatory Antimicrobials. Vaccines 2018, 6, 63. [Google Scholar] [CrossRef]
- Lishko, V.K.; Moreno, B.; Podolnikova, N.P.; Ugarova, T.P. Identification of Human Cathelicidin Peptide LL-37 as a Ligand for Macrophage Integrin αMβ2 (Mac-1, CD11b/CD18) that Promotes Phagocytosis by Opsonizing Bacteria. Res. Rep. Biochem. 2016, 2016, 39–55. [Google Scholar]
- van der Does, A.M.; Beekhuizen, H.; Ravensbergen, B.; Vos, T.; Ottenhoff, T.H.M.; van Dissel, J.T.; Drijfhout, J.W.; Hiemstra, P.S.; Nibbering, P.H. LL-37 directs macrophage differentiation toward macrophages with a proinflammatory signature. J. Immunol. 2010, 185, 1442–1449. [Google Scholar] [CrossRef]
- Hoang-Yen Tran, D.; Hoang-Ngoc Tran, D.; Mattai, S.A.; Sallam, T.; Ortiz, C.; Lee, E.C.; Robbins, L.; Ho, S.; Lee, J.E.; Fisseha, E.; et al. Cathelicidin suppresses lipid accumulation and hepatic steatosis by inhibition of the CD36 receptor. Int. J. Obes. 2016, 40, 1424–1434. [Google Scholar] [CrossRef]
- Rivas-Santiago, B.; Trujillo, V.; Montoya, A.; Gonzalez-Curiel, I.; Castañeda-Delgado, J.; Cardenas, A.; Rincon, K.; Hernandez, M.L.; Hernández-Pando, R. Expression of antimicrobial peptides in diabetic foot ulcer. J. Dermatol. Sci. 2012, 65, 19–26. [Google Scholar] [CrossRef]
- Zhang, L.-J.; Guerrero-Juarez, C.F.; Chen, S.X.; Zhang, X.; Yin, M.; Li, F.; Wu, S.; Chen, J.; Li, M.; Liu, Y.; et al. Diet-induced obesity promotes infection by impairment of the innate antimicrobial defense function of dermal adipocyte progenitors. Sci. Transl. Med. 2021, 13, eabb5280. [Google Scholar] [PubMed]
- Berndt, J.; Kralisch, S.; Klöting, N.; Ruschke, K.; Kern, M.; Fasshauer, M.; Schön, M.; Stumvoll, M.; Blüher, M. Adipose Triglyceride Lipase Gene Expression in Human Visceral Obesity. Exp. Clin. Endocrinol. Diabetes 2008, 116, 203–210. [Google Scholar] [CrossRef]
- Bialesova, L.; Kulyte, A.; Petrus, P.; Sinha, I.; Laurencikiene, J.; Zhao, C.; Wright, K.D.; Arner, P.; Dahlman, I. Epigenetic Regulation of PLIN1 in Obese Women and its Relation to Lipolysis. Sci. Rep. 2017, 1–11. [Google Scholar]
- Schoiswohl, G.; Stefanovic-Racic, M.; Menke, M.N.; Wills, R.C.; Surlow, B.A.; Basantani, M.K.; Sitnick, M.T.; Cai, L.; Yazbeck, C.F.; Stolz, D.B.; et al. Impact of Reduced ATGL-Mediated Adipocyte Lipolysis on Obesity-Associated Insulin Resistance and Inflammation in Male Mice. Endocrinology 2015, 156, 3610–3624. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-W.; Lee, M.; Oh, K.-J. Adipose Tissue-Derived Signatures for Obesity and Type 2 Diabetes: Adipokines, Batokines and MicroRNAs. J. Clin. Med. 2019, 8, 854. [Google Scholar] [CrossRef]
- Huber, J.; Kiefer, F.W.; Zeyda, M.; Ludvik, B.; Silberhumer, G.R.; Prager, G.; Zlabinger, G.J.; Stulnig, T.M. CC chemokine and CC chemokine receptor profiles in visceral and subcutaneous adipose tissue are altered in human obesity. J. Clin. Endocrinol. Metab. 2008, 93, 3215–3221. [Google Scholar] [CrossRef]
- Liu, W.; Zhou, X.; Li, Y.; Zhang, S.; Cai, X.; Zhang, R.; Gong, S.; Han, X.; Ji, L. Serum leptin, resistin, and adiponectin levels in obese and non-obese patients with newly diagnosed type 2 diabetes mellitus: A population-based study. Med. (Baltim.) 2020, 99, e19052. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, K.; Shibata, R.; Murohara, T.; Ouchi, N. Role of anti-inflammatory adipokines in obesity-related diseases. Trends Endocrinol. Metab. 2014, 25, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Pouget, C.; Dunyach-Remy, C.; Pantel, A.; Schuldiner, S.; Sotto, A.; Lavigne, J.-P. Biofilms in Diabetic Foot Ulcers: Significance and Clinical Relevance. Microorganisms 2020, 8, 1580. [Google Scholar] [CrossRef] [PubMed]
- Mishra, B.; Golla, R.M.; Lau, K.; Lushnikova, T.; Wang, G. Anti-Staphylococcal Biofilm Effects of Human Cathelicidin Peptides. Acs Med. Chem. Lett. 2016, 7, 117–121. [Google Scholar] [CrossRef]
- Schwartz, R.S.; Shuman, W.P.; Bradbury, V.L.; Cain, K.C.; Fellingham, G.W.; Beard, J.C.; Kahn, S.E.; Stratton, J.R.; Cerqueira, M.D.; Abrass, I.B. Body fat distribution in healthy young and older men. J. Gerontol. 1990, 45, M181. [Google Scholar] [CrossRef]
- Kruglikov, I.L.; Scherer, P.E. Skin aging: Are adipocytes the next target? Aging 2016, 8, 1457–1469. [Google Scholar] [CrossRef]
- Tyner, S.D.; Venkatachalam, S.; Choi, J.; Jones, S.; Ghebranious, N.; Igelmann, H.; Lu, X.; Soron, G.; Cooper, B.; Brayton, C.; et al. p53 mutant mice that display early ageing-associated phenotypes. Nature 2002, 415, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Gonzalez, G.C.; Shook, B.A.; Andrae, J.; Holtrup, B.; Bollag, K.; Betsholtz, C.; Rodeheffer, M.S.; Horsley, V. Skin Adipocyte Stem Cell Self-Renewal Is Regulated by a PDGFA/AKT-Signaling Axis. Cell Stem Cell. 2016, 19, 1–25. [Google Scholar] [CrossRef]
- Zhang, L.-J.; Chen, S.X.; Guerrero-Juarez, C.F.; Li, F.; Tong, Y.; Liang, Y.; Liggins, M.; Chen, X.; Chen, H.; Li, M.; et al. Age-Related Loss of Innate Immune Antimicrobial Function of Dermal Fat Is Mediated by Transforming Growth Factor Beta. Immunity 2019, 50, 121–136. [Google Scholar] [CrossRef]
- Kuk, J.L.; Saunders, T.J.; Davidson, L.E.; Ross, R. Age-related changes in total and regional fat distribution. Ageing Res. Rev. 2009, 8, 339–348. [Google Scholar] [CrossRef]
- Arai, Y.; Kamide, K.; Hirose, N. Adipokines and Aging: Findings From Centenarians and the Very Old. Front. Endocrinol. 2019, 10, 142. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, P.; Bouchard, B. The Impact of Aging on Adipose Function and Adipokine Synthesis. Front. Endocrinol. 2019, 10, 137. [Google Scholar] [CrossRef]
- Wan, M.; van der Does, A.M.; Tang, X.; Lindbom, L.; Agerberth, B.; Haeggström, J.Z. Antimicrobial peptide LL-37 promotes bacterial phagocytosis by human macrophages. J. Leukoc. Biol. 2014, 95, 971–981. [Google Scholar] [CrossRef] [PubMed]
- Schipper, B.M.; Marra, K.G.; Zhang, W.; Donnenberg, A.D.; Rubin, J.P. Regional anatomic and age effects on cell function of human adipose-derived stem cells. Ann. Plast. Surg. 2008, 60, 538–544. [Google Scholar]
- Caso, G.; McNurlan, M.A.; Mileva, I.; Zemlyak, A.; Mynarcik, D.C.; Gelato, M.C. Peripheral fat loss and decline in adipogenesis in older humans. Metabolism 2013, 62, 337–340. [Google Scholar] [CrossRef]
- Kirkland, J.L.; Hollenberg, C.H.; Gillon, W.S. Age, anatomic site, and the replication and differentiation of adipocyte precursors. Am. J. Physiol. 1990, 258, C206. [Google Scholar] [CrossRef]
- Zoico, E.; Di Francesco, V.; Olioso, D.; Fratta Pasini, A.M.; Sepe, A.; Bosello, O.; Cinti, S.; Cominacini, L.; Zamboni, M. In vitro aging of 3T3-L1 mouse adipocytes leads to altered metabolism and response to inflammation. Biogerontology 2009, 11, 111–122. [Google Scholar] [PubMed]
- Goldstein, S.; Moerman, E.J.; Soeldner, J.S.; Gleason, R.E.; Barnett, D.M. Diabetes mellitus and genetic prediabetes. Decreased replicative capacity of cultured skin fibroblasts. J. Clin. Investig. 1979, 63, 358–370. [Google Scholar] [CrossRef] [PubMed]
- Haydont, V.; Bernard, B.A.; Fortunel, N.O. Age-related evolutions of the dermis: Clinical signs, fibroblast and extracellular matrix dynamics. Mech. Ageing Dev. 2019, 177, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Stone, R.C.; Stojadinovic, O.; Ramirez, H.; Pastar, I.; Maione, A.G.; Smith, A.; Yanez, V.; Veves, A.; Kirsner, R.S.; et al. Integrative analysis of miRNA and mRNA paired expression profiling of primary fibroblast derived from diabetic foot ulcers reveals multiple impaired cellular functions. Wound Repair Regen. 2016, 24, 943–953. [Google Scholar] [CrossRef]
- Maione, A.G.; Brudno, Y.; Stojadinovic, O.; Park, L.K.; Smith, A.; Tellechea, A.; Leal, E.C.; Kearney, C.J.; Veves, A.; Tomic-Canic, M.; et al. Three-Dimensional Human Tissue Models That Incorporate Diabetic Foot Ulcer-Derived Fibroblasts Mimic In VivoFeatures of Chronic Wounds. Tissue Eng. Part. C Methods 2015, 21, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Freund, A.; Orjalo, A.V.; Desprez, P.-Y.; Campisi, J. Inflammatory networks during cellular senescence: Causes and consequences. Trends Mol. Med. 2010, 16, 238–246. [Google Scholar] [CrossRef]
- Wilkinson, H.N.; Clowes, C.; Banyard, K.L.; Matteuci, P.; Mace, K.A.; Hardman, M.J. Elevated Local Senescence in Diabetic Wound Healing Is Linked to Pathological Repair via CXCR2. J. Investig. Dermatol. 2019, 139, 1171–1181.e6. [Google Scholar] [CrossRef] [PubMed]
- Mavrogonatou, E.; Konstantinou, A.; Kletsas, D. Long-term exposure to TNF-α leads human skin fibroblasts to a p38 MAPK- and ROS-mediated premature senescence. Biogerontology 2018, 19, 237–249. [Google Scholar] [CrossRef]
- Xuan, Y.H.; Huang, B.B.; Tian, H.S.; Chi, L.S.; Duan, Y.M.; Wang, X.; Zhu, Z.X.; Cai, W.H.; Zhu, Y.T.; Wei, T.M.; et al. High-Glucose Inhibits Human Fibroblast Cell Migration in Wound Healing via Repression of bFGF-Regulating JNK Phosphorylation. PLoS ONE 2014, 9, e108182. [Google Scholar] [CrossRef] [PubMed]
- Wall, S.J.; Sampson, M.J.; Levell, N.; Murphy, G. Elevated matrix metalloproteinase-2 and -3 production from human diabetic dermal fibroblasts. Br. J. Dermatol. 2003, 149, 13–16. [Google Scholar] [CrossRef]
- Pang, L.; Wang, Y.; Zheng, M.; Wang, Q.; Lin, H.; Zhang, L.; Wu, L. Transcriptomic study of high-glucose effects on human skin fibroblast cells. Mol. Med. Rep. 2016, 13, 2627–2634. [Google Scholar] [CrossRef]
- Portou, M.J.; Yu, R.; Baker, D.; Xu, S.; Abraham, D.; Tsui, J. Hyperglycaemia and Ischaemia Impair Wound Healing via Toll-like Receptor 4 Pathway Activation in vitro and in an Experimental Murine Model. Eur. J. Vasc. Endovasc. Surg. 2020, 59, 117–127. [Google Scholar] [CrossRef]
- Dasu, M.R.; Jialal, I. Amelioration in wound healing in diabetic toll-like receptor-4 knockout mice. J. Diabetes Its Complicat. 2013, 27, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Park, L.K.; Maione, A.G.; Smith, A.; Gerami-Naini, B.; Iyer, L.K.; Mooney, D.J.; Veves, A.; Garlick, J.A. Genome-wide DNA methylation analysis identifies a metabolic memory profile in patient-derived diabetic foot ulcer fibroblasts. Epigenetics 2014, 9, 1339–1349. [Google Scholar] [CrossRef]
- Solé-Boldo, L.; Raddatz, G.; Schütz, S.; Mallm, J.-P.; Rippe, K.; Lonsdorf, A.S.; Rodríguez-Paredes, M.; Lyko, F. Single-cell transcriptomes of the human skin reveal age-related loss of fibroblast priming. Commun. Biol. 2020, 3, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Salzer, M.C.; Lafzi, A.; Berenguer-Llergo, A.; Youssif, C.; Castellanos, A.; Solanas, G.; Peixoto, F.O.; Attolini, C.S.-O.; Prats, N.; Aguilera, M.; et al. Identity Noise and Adipogenic Traits Characterize Dermal Fibroblast Aging. Cell 2018, 175, 1–39. [Google Scholar] [CrossRef]
- Li, Y.; Lei, D.; Swindell, W.R.; Xia, W.; Weng, S.; Fu, J.; Worthen, C.A.; Okubo, T.; Johnston, A.; Gudjonsson, J.E.; et al. Age-Associated Increase in Skin Fibroblast-Derived Prostaglandin E2 Contributes to Reduced Collagen Levels in Elderly Human Skin. J. Investig. Dermatol. 2015, 135, 2181–2188. [Google Scholar] [CrossRef] [PubMed]
- Wall, I.B.; Moseley, R.; Baird, D.M.; Kipling, D.; Giles, P.; Laffafian, I.; Price, P.E.; Thomas, D.W.; Stephens, P. Fibroblast dysfunction is a key factor in the non-healing of chronic venous leg ulcers. J. Investig. Dermatol. 2008, 128, 2526–2540. [Google Scholar] [CrossRef]
- Wolf, J.; Weinberger, B.; Arnold, C.R.; Maier, A.B.; Westendorp, R.G.J.; Grubeck-Loebenstein, B. The effect of chronological age on the inflammatory response of human fibroblasts. Exp. Gerontol. 2012, 47, 749–753. [Google Scholar] [CrossRef] [PubMed]
- Lago, J.C.; Puzzi, M.B. The effect of aging in primary human dermal fibroblasts. PLoS ONE 2019, 14, e0219165. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Inflammatory Defect during Wound Healing | Mesenchymal Cell-Based Approach to Treat Inflammatory Defect | |
|---|---|---|
| Adipocytes | Fibroblasts | |
| Impaired early myeloid cell recruitment | ↑ Stimulated lipolysis ↑ Cathelicidin (CAMP/LL37) | ↑ Chemokine expression (CCL2, CXCL1, CXCL2, IL8/CXCL8) |
| Persistent inflammation | ↓ Basal lipolysis ↑ Cathelicidin (CAMP/LL37) ↑ Anti-inflammatory adipokine expression (Adiponectin) ↓ Pro-inflammatory adipokine expression (CCL2, IL1, IL6, IL18, Leptin, TNFα) | ↓ Pro-inflammatory cytokine expression (IL1, IL6, IL8/CXCL8, TNFα) ↓ SASP |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cooper, P.O.; Haas, M.R.; Noonepalle, S.k.R.; Shook, B.A. Dermal Drivers of Injury-Induced Inflammation: Contribution of Adipocytes and Fibroblasts. Int. J. Mol. Sci. 2021, 22, 1933. https://doi.org/10.3390/ijms22041933
Cooper PO, Haas MR, Noonepalle SkR, Shook BA. Dermal Drivers of Injury-Induced Inflammation: Contribution of Adipocytes and Fibroblasts. International Journal of Molecular Sciences. 2021; 22(4):1933. https://doi.org/10.3390/ijms22041933
Chicago/Turabian StyleCooper, Paula O., MaryEllen R. Haas, Satish kumar R. Noonepalle, and Brett A. Shook. 2021. "Dermal Drivers of Injury-Induced Inflammation: Contribution of Adipocytes and Fibroblasts" International Journal of Molecular Sciences 22, no. 4: 1933. https://doi.org/10.3390/ijms22041933
APA StyleCooper, P. O., Haas, M. R., Noonepalle, S. k. R., & Shook, B. A. (2021). Dermal Drivers of Injury-Induced Inflammation: Contribution of Adipocytes and Fibroblasts. International Journal of Molecular Sciences, 22(4), 1933. https://doi.org/10.3390/ijms22041933