Controlling the Heterodimerisation of the Phytosulfokine Receptor 1 (PSKR1) via Island Loop Modulation

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
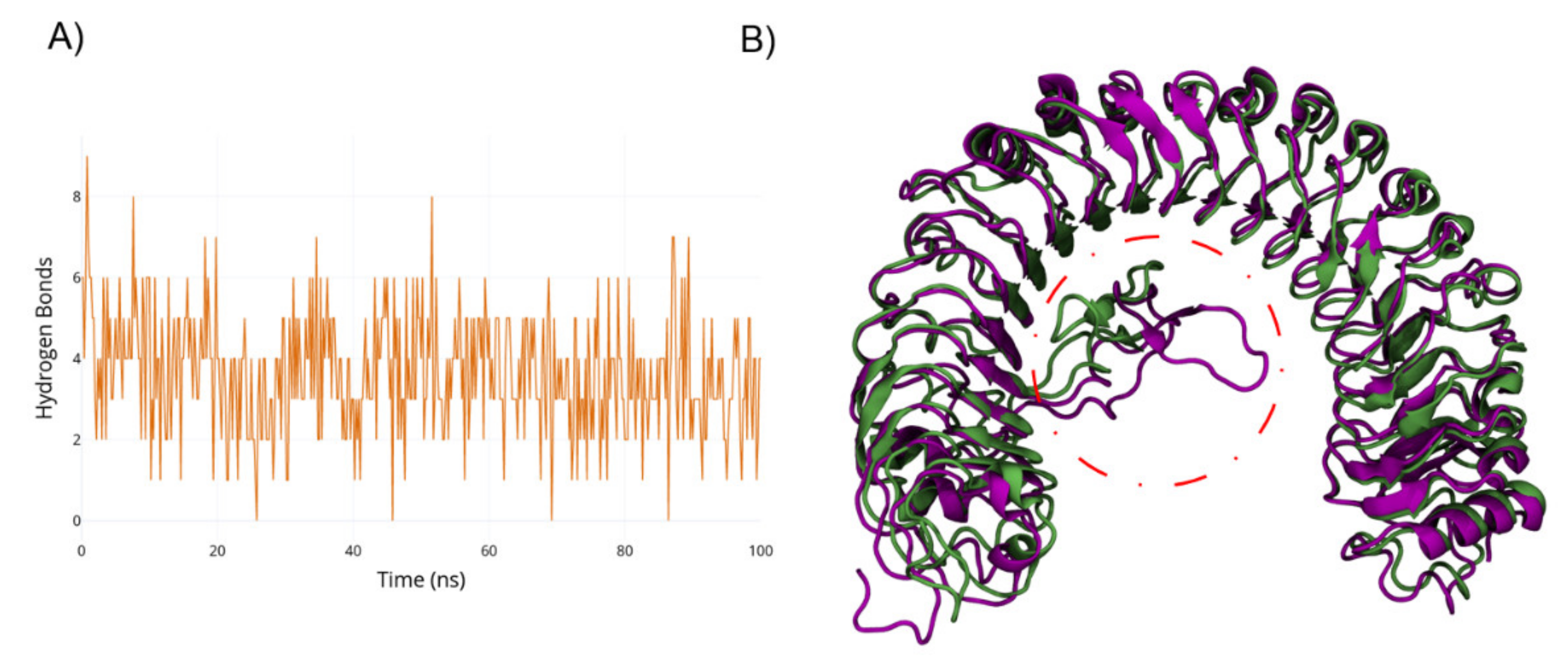
2.1. Metadynamics of Island Loop Transition
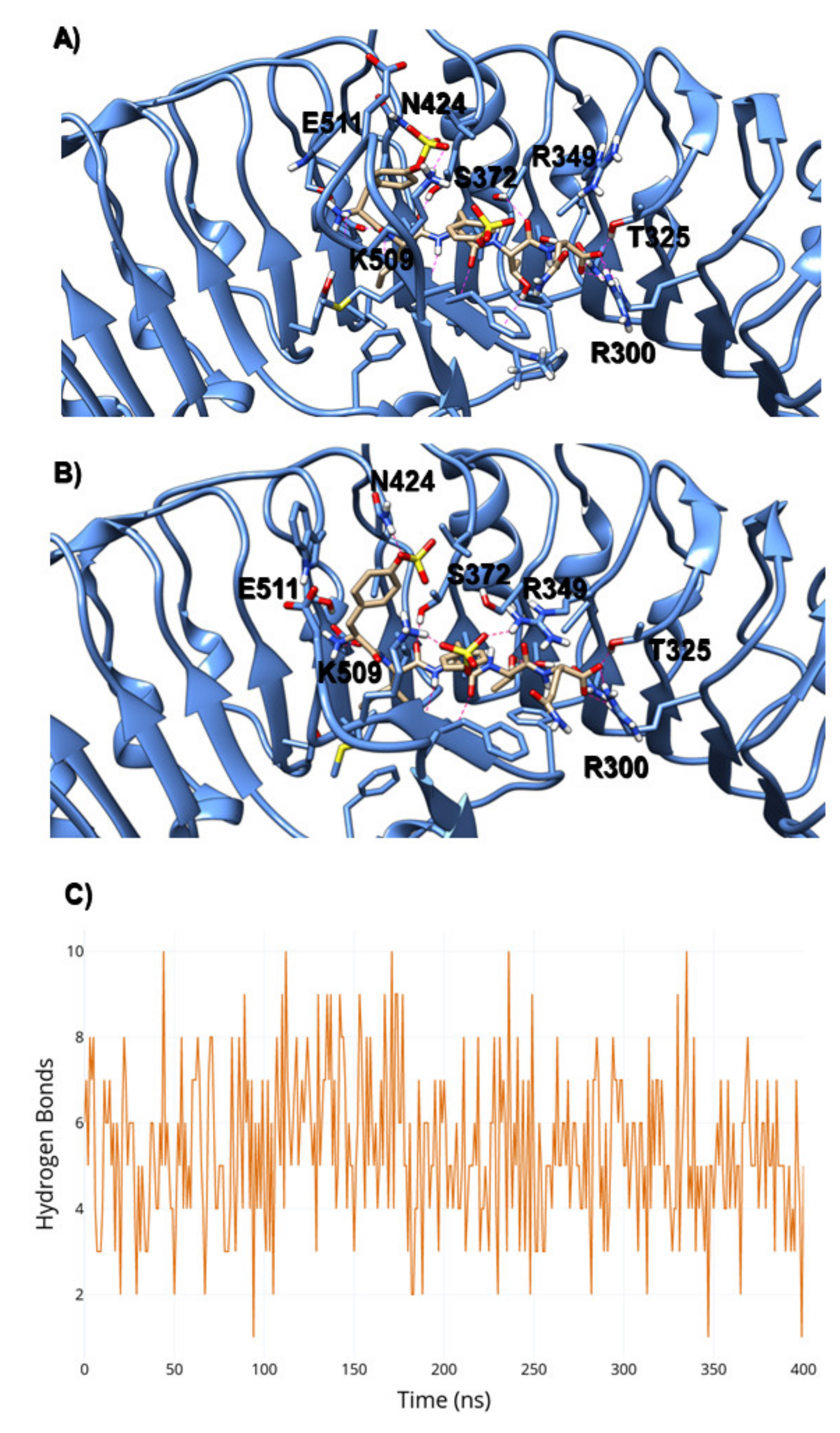
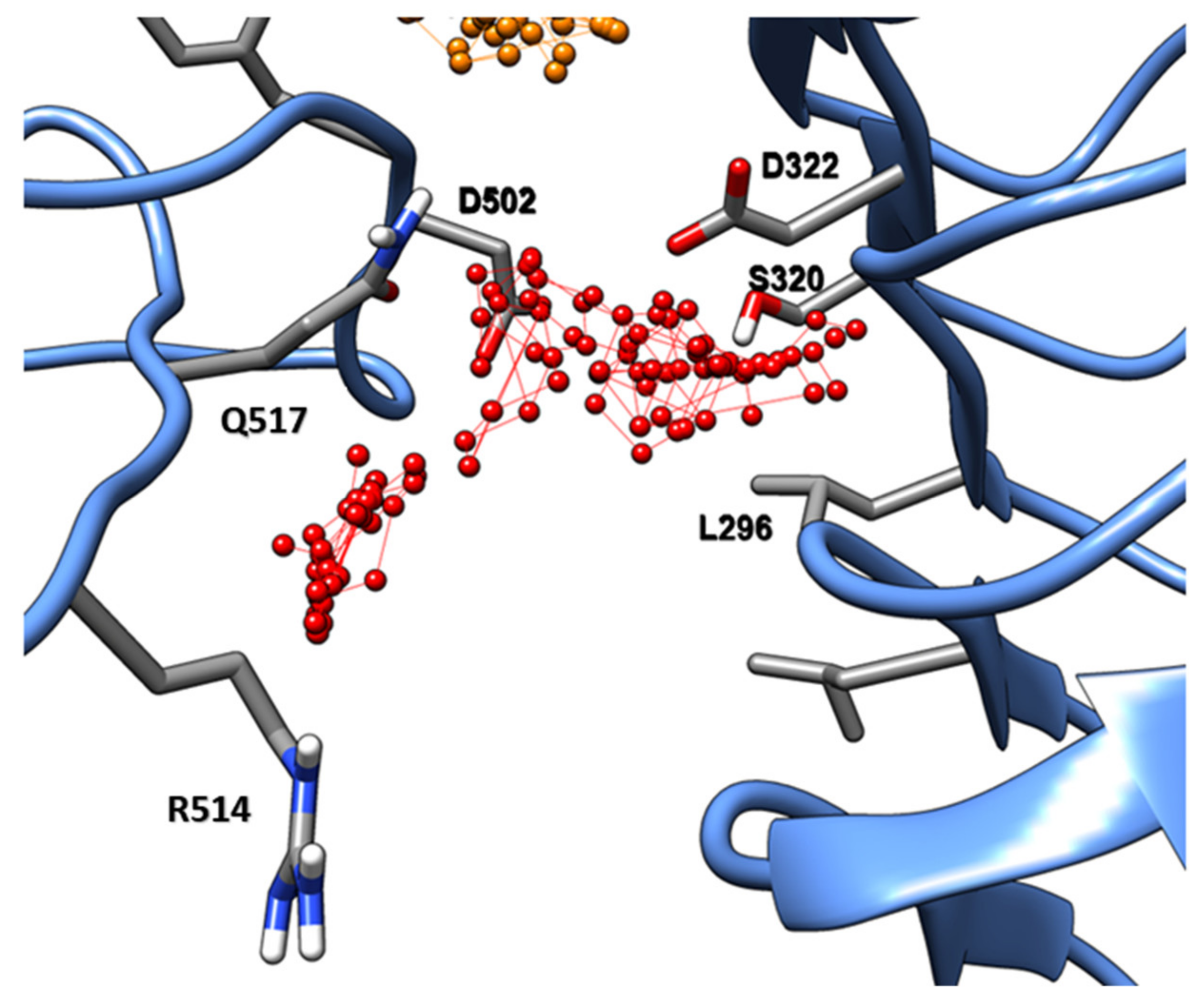
2.2. Evaluation of State B “Druggability”
3. Discussion
PSK Directly Affects the Folding Energy of the Island Loop
4. Materials and Methods
4.1. Molecular Modelling
4.2. Equilibrium Molecular Dynamics (MD) Simulations
4.3. Island Domain Loop Well-Tempered Metadynamics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PSK | Phytosulfokine |
| PSKR1 | Phytosulfokine Receptor 1 |
| ID | Island Loop |
| DcPSKR1 | Daucus Carota Phytosulfokine Receptor 1 |
| LRR | Leucine-rich repeat |
| LRR-RK | Leucine-rich repeat receptor Kinase |
| BRI1 | brassinosteroid-insensitive 1 |
| FLS2 | flagellin insensitive 2 |
| BAK1 | BRI1-associated kinase 1 |
| SERK1 | somatic embryogenesis receptor-like kinase 1 |
| RMSF | Root Mean Square Fluctuation |
| RMSD | Root Mean Square Deviation |
References
- Godel-Jędrychowska, K.; Maćkowska, K.; Kurczyńska, E.; Grzebelus, E. Composition of the reconstituted cell wall in protoplast-derived cells of daucus is affected by phytosulfokine (PSK). Int. J. Mol. Sci. 2019, 20, 5490. [Google Scholar] [CrossRef] [PubMed]
- Matsubayashi, Y. Posttranslationally modified small-peptide signals in plants. Annu. Rev. Plant Biol. 2014, 65, 385–413. [Google Scholar] [CrossRef] [PubMed]
- Matsubayashi, Y.; Sakagami, Y. Phytosulfokine, sulfated peptides that induce the proliferation of single mesophyll cells of Asparagus officinalis L. Proc. Natl. Acad. Sci. USA 1996, 93, 7623–7627. [Google Scholar] [CrossRef] [PubMed]
- Sauter, M. Phytosulfokine peptide signalling. J. Exp. Bot. 2015, 66, 5161–5169. [Google Scholar] [CrossRef]
- Stührwohldt, N.; Dahlke, R.I.; Kutschmar, A.; Peng, X.; Sun, M.X.; Sauter, M. Phytosulfokine peptide signaling controls pollen tube growth and funicular pollen tube guidance in Arabidopsis thaliana. Physiol. Plant. 2015, 153, 643–653. [Google Scholar] [CrossRef]
- Stührwohldt, N.; Dahlke, R.I.; Steffens, B.; Johnson, A.; Sauter, M. Phytosulfokine-α controls hypocotyl length and cell expansion in Arabidopsis thaliana through phytosulfokine receptor 1. PLoS ONE 2011, 6, e21054. [Google Scholar] [CrossRef]
- Kaufmann, C.; Sauter, M.; Kopriva, S. Sulfated plant peptide hormones. J. Exp. Bot. 2019, 70, 4267–4277. [Google Scholar] [CrossRef]
- Murphy, E.; Smith, S.; De Smeta, I. Small signaling peptides in Arabidopsis development: How cells communicate over a short distance. Plant Cell 2012, 24, 3198–3217. [Google Scholar] [CrossRef]
- Kutschmar, A.; Rzewuski, G.; Stührwohldt, N.; Beemster, G.T.S.; Inzé, D.; Sauter, M. PSK-α promotes root growth in Arabidopsis. New Phytol. 2009, 181, 820–831. [Google Scholar] [CrossRef]
- Hanai, H.; Matsuno, T.; Yamamoto, M.; Matsubayashi, Y.; Kobayashi, T.; Kamada, H.; Sakagami, Y. A secreted peptide growth factor, phytosulfokine, acting as a stimulatory factor of carrot somatic embryo formation. Plant Cell Physiol. 2000, 41, 27–32. [Google Scholar] [CrossRef]
- Srivastava, R.; Liu, J.X.; Howell, S.H. Proteolytic processing of a precursor protein for a growth-promoting peptide by a subtilisin serine protease in Arabidopsis. Plant J. 2008, 56, 219–227. [Google Scholar] [CrossRef]
- Mosher, S.; Seybold, H.; Rodriguez, P.; Stahl, M.; Davies, K.A.; Dayaratne, S.; Morillo, S.A.; Wierzba, M.; Favery, B.; Keller, H.; et al. The tyrosine-sulfated peptide receptors PSKR1 and PSY1R modify the immunity of Arabidopsis to biotrophic and necrotrophic pathogens in an antagonistic manner. Plant J. 2013, 73, 469–482. [Google Scholar] [CrossRef]
- Nagar, P.; Kumar, A.; Jain, M.; Kumari, S.; Mustafiz, A. Genome-wide analysis and transcript profiling of PSKR gene family members in Oryza sativa. PLoS ONE 2020, 15, e0236349. [Google Scholar] [CrossRef]
- Matsubayashi, Y.; Ogawa, M.; Kihara, H.; Niwa, M.; Sakagami, Y. Disruption and overexpression of Arabidopsis phytosulfokine receptor gene affects cellular longevity and potential for growth. Plant Physiol. 2006, 142, 45–53. [Google Scholar] [CrossRef]
- Amano, Y.; Tsubouchi, H.; Shinohara, H.; Ogawa, M.; Matsubayashi, Y. Tyrosine-sulfated glycopeptide involved in cellular proliferation and expansion in Arabidopsis. Proc. Natl. Acad. Sci. USA 2007, 104, 18333–18338. [Google Scholar] [CrossRef]
- Wang, J.; Li, H.; Han, Z.; Zhang, H.; Wang, T.; Lin, G.; Chang, J.; Yang, W.; Chai, J. Allosteric receptor activation by the plant peptide hormone phytosulfokine. Nature 2015, 525, 265–268. [Google Scholar] [CrossRef]
- She, J.; Han, Z.; Kim, T.W.; Wang, J.; Cheng, W.; Chang, J.; Shi, S.; Wang, J.; Yang, M.; Wang, Z.Y.; et al. Structural insight into brassinosteroid perception by BRI1. Nature 2011, 474, 472–477. [Google Scholar] [CrossRef]
- Wang, X.; Kota, U.; He, K.; Blackburn, K.; Li, J.; Goshe, M.B.; Huber, S.C.; Clouse, S.D. Sequential Transphosphorylation of the BRI1/BAK1 Receptor Kinase Complex Impacts Early Events in Brassinosteroid Signaling. Dev. Cell 2008, 15, 220–235. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Li, L.; Macho, A.P.; Han, Z.; Hu, Z.; Zipfel, C.; Zhou, J.M.; Chai, J. Structural basis for flg22-induced activation of the Arabidopsis FLS2-BAK1 immune complex. Science 2013, 342, 624–628. [Google Scholar] [CrossRef] [PubMed]
- Holzwart, E.; Huerta, A.I.; Glöckner, N.; Gómez, B.G.; Wanke, F.; Augustin, S.; Askani, J.C.; Schürholz, A.K.; Harter, K.; Wolf, S. BRI1 controls vascular cell fate in the arabidopsis root through RLP44 and phytosulfokine signaling. Proc. Natl. Acad. Sci. USA 2018, 115, 11838–11843. [Google Scholar] [CrossRef] [PubMed]
- Schwessinger, B.; Roux, M.; Kadota, Y.; Ntoukakis, V.; Sklenar, J.; Jones, A.; Zipfel, C. Phosphorylation-dependent differential regulation of plant growth, cell death, and innate immunity by the regulatory receptor-like kinase BAK1. PLoS Genet. 2011, 7, e1002046. [Google Scholar] [CrossRef]
- Kou, X.; Liu, Q.; Sun, Y.; Wang, P.; Zhang, S.; Wu, J. The Peptide PbrPSK2 From Phytosulfokine Family Induces Reactive Oxygen Species (ROS) Production to Regulate Pear Pollen Tube Growth. Front. Plant Sci. 2020, 11, 1799. [Google Scholar] [CrossRef]
- Zhang, H.; Hu, Z.; Lei, C.; Zheng, C.; Wang, J.; Shao, S.; Li, X.; Xia, X.; Cai, X.; Zhou, J.; et al. A Plant Phytosulfokine Peptide Initiates Auxin-Dependent Immunity through Cytosolic Ca2+ Signaling in Tomato. Plant Cell 2018, 30, 652–667. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, C.; Boutrot, F.; Segonzac, C.; Schwessinger, B.; Gimenez-Ibanez, S.; Chinchilla, D.; Rathjen, J.P.; De Vries, S.C.; Zipfel, C. Brassinosteroids inhibit pathogen-associated molecular pattern-triggered immune signaling independent of the receptor kinase BAK1. Proc. Natl. Acad. Sci. USA 2012, 109, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Aghdam, M.S.; Alikhani-Koupaei, M. Exogenous phytosulfokine α (PSKα) applying delays senescence and relief decay in strawberry fruits during cold storage by sufficient intracellular ATP and NADPH availability. Food Chem. 2021, 336, 127685. [Google Scholar] [CrossRef] [PubMed]
- Aghdam, M.S.; Sayyari, M.; Luo, Z. Exogenous application of phytosulfokine α (PSKα) delays yellowing and preserves nutritional quality of broccoli florets during cold storage. Food Chem. 2020, 333, 127481. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.; Sali, A. Comparative protein structure modeling using MODELLER. Curr. Protoc. Bioinforma. 2014, 47, 5.6.1–5.6.32. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef]
- Jo, S.; Song, K.C.; Desaire, H.; MacKerell, A.D.; Im, W. Glycan reader: Automated sugar identification and simulation preparation for carbohydrates and glycoproteins. J. Comput. Chem. 2011, 32, 3135–3141. [Google Scholar] [CrossRef]
- Park, S.J.; Lee, J.; Qi, Y.; Kern, N.R.; Lee, H.S.; Jo, S.; Joung, I.; Joo, K.; Lee, J.; Im, W. CHARMM-GUI Glycan Modeler for modeling and simulation of carbohydrates and glycoconjugates. Glycobiology 2019, 29, 320–331. [Google Scholar] [CrossRef]
- Park, S.J.; Lee, J.; Patel, D.S.; Ma, H.; Lee, H.S.; Jo, S.; Im, W. Glycan Reader is improved to recognize most sugar types and chemical modifications in the Protein Data Bank. Bioinformatics 2017, 33, 3051–3057. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N log( N ) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Hess, B. P-LINCS: A Parallel Linear Constraint Solver for Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 116–122. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Crystal structure and pair potentials: A molecular-dynamics study. Phys. Rev. Lett. 1980, 45, 1196–1199. [Google Scholar] [CrossRef]
- Barducci, A.; Bussi, G.; Parrinello, M. Well-tempered metadynamics: A smoothly converging and tunable free-energy method. Phys. Rev. Lett. 2008, 100, 020603. [Google Scholar] [CrossRef] [PubMed]
- Bonomi, M.; Bussi, G.; Camilloni, C.; Tribello, G.A.; Banáš, P.; Barducci, A.; Bernetti, M.; Bolhuis, P.G.; Bottaro, S.; Branduardi, D.; et al. Promoting transparency and reproducibility in enhanced molecular simulations. Nat. Methods 2019, 16, 670–673. [Google Scholar] [CrossRef]
- Tribello, G.A.; Bonomi, M.; Branduardi, D.; Camilloni, C.; Bussi, G. PLUMED 2: New feathers for an old bird. Comput. Phys. Commun. 2014, 185, 604–613. [Google Scholar] [CrossRef]
- Bonomi, M.; Branduardi, D.; Bussi, G.; Camilloni, C.; Provasi, D.; Raiteri, P.; Donadio, D.; Marinelli, F.; Pietrucci, F.; Broglia, R.A.; et al. PLUMED: A portable plugin for free-energy calculations with molecular dynamics. Comput. Phys. Commun. 2009, 180, 1961–1972. [Google Scholar] [CrossRef]
- Kozakov, D.; Grove, L.E.; Hall, D.R.; Bohnuud, T.; Mottarella, S.E.; Luo, L.; Xia, B.; Beglov, D.; Vajda, S. The FTMap family of web servers for determining and characterizing ligand-binding hot spots of proteins. Nat. Protoc. 2015, 10, 733–755. [Google Scholar] [CrossRef]
- BioSolveIT—SeeSAR. Available online: https://www.biosolveit.de/SeeSAR/ (accessed on 6 January 2020).
- Schneider, N.; Lange, G.; Hindle, S.; Klein, R.; Rarey, M. A consistent description of HYdrogen bond and DEhydration energies in protein-ligand complexes: Methods behind the HYDE scoring function. J. Comput. Aided. Mol. Des. 2013, 27, 15–29. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Souza, J.V.; Kondal, M.; Zaborniak, P.; Cairns, R.; Bronowska, A.K. Controlling the Heterodimerisation of the Phytosulfokine Receptor 1 (PSKR1) via Island Loop Modulation. Int. J. Mol. Sci. 2021, 22, 1806. https://doi.org/10.3390/ijms22041806
de Souza JV, Kondal M, Zaborniak P, Cairns R, Bronowska AK. Controlling the Heterodimerisation of the Phytosulfokine Receptor 1 (PSKR1) via Island Loop Modulation. International Journal of Molecular Sciences. 2021; 22(4):1806. https://doi.org/10.3390/ijms22041806
Chicago/Turabian Stylede Souza, João V., Matthew Kondal, Piotr Zaborniak, Ryland Cairns, and Agnieszka K. Bronowska. 2021. "Controlling the Heterodimerisation of the Phytosulfokine Receptor 1 (PSKR1) via Island Loop Modulation" International Journal of Molecular Sciences 22, no. 4: 1806. https://doi.org/10.3390/ijms22041806
APA Stylede Souza, J. V., Kondal, M., Zaborniak, P., Cairns, R., & Bronowska, A. K. (2021). Controlling the Heterodimerisation of the Phytosulfokine Receptor 1 (PSKR1) via Island Loop Modulation. International Journal of Molecular Sciences, 22(4), 1806. https://doi.org/10.3390/ijms22041806