
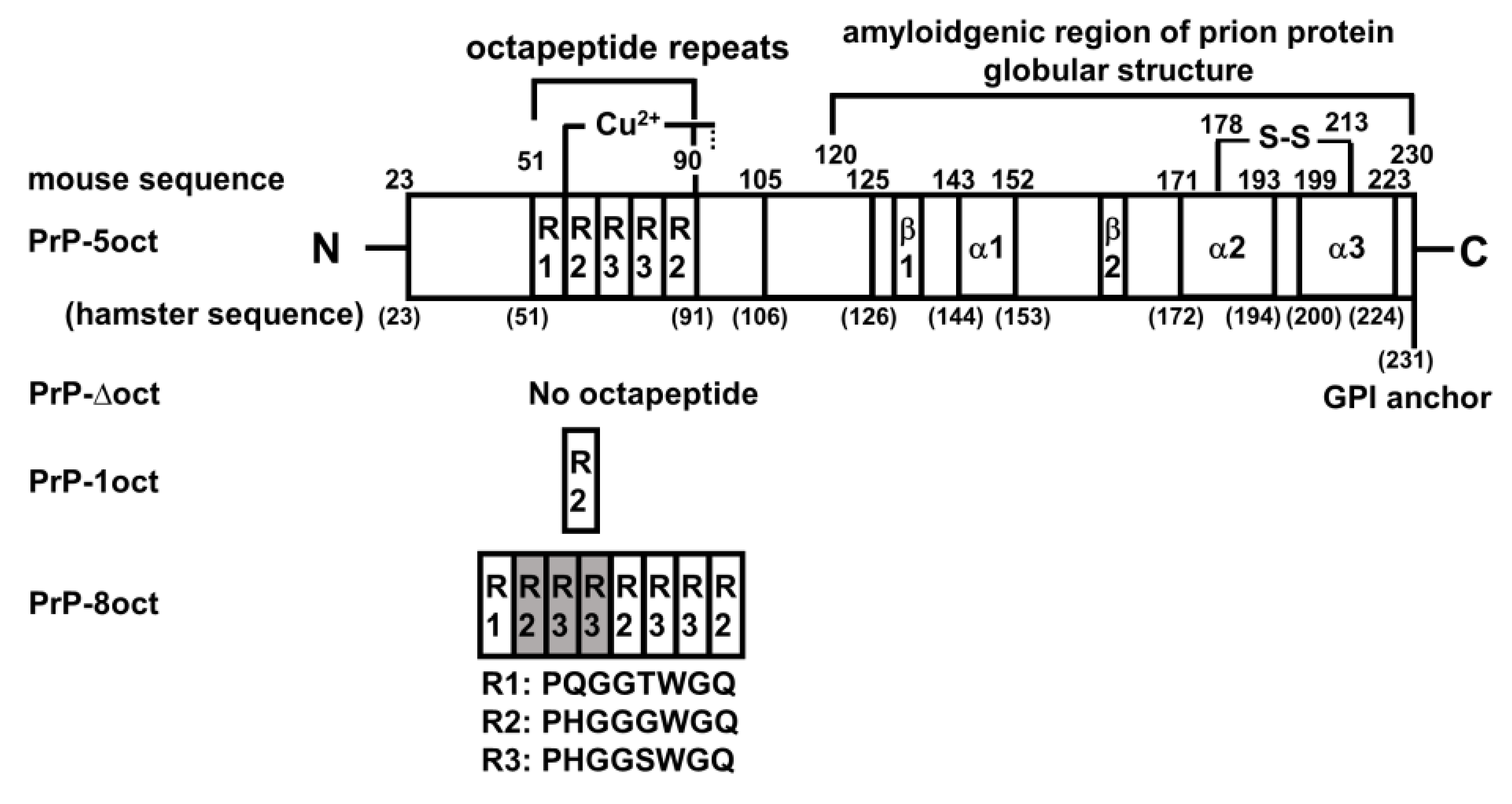
The Effect of Octapeptide Repeats on Prion Folding and Misfolding


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
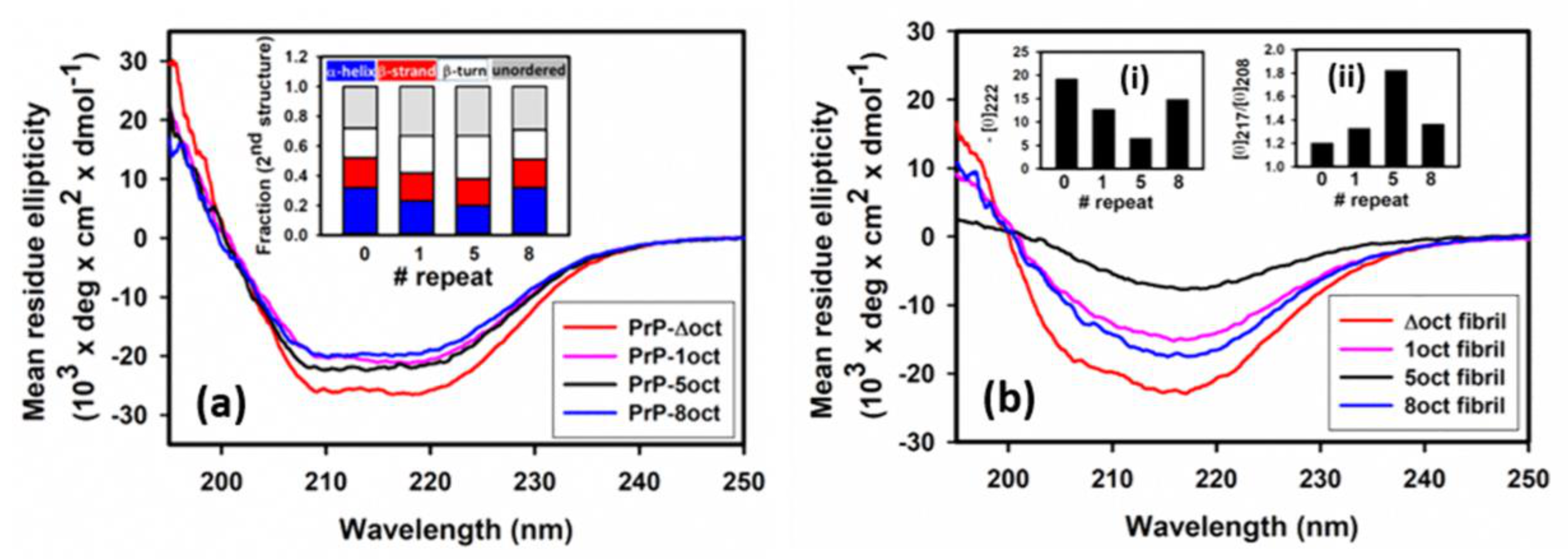
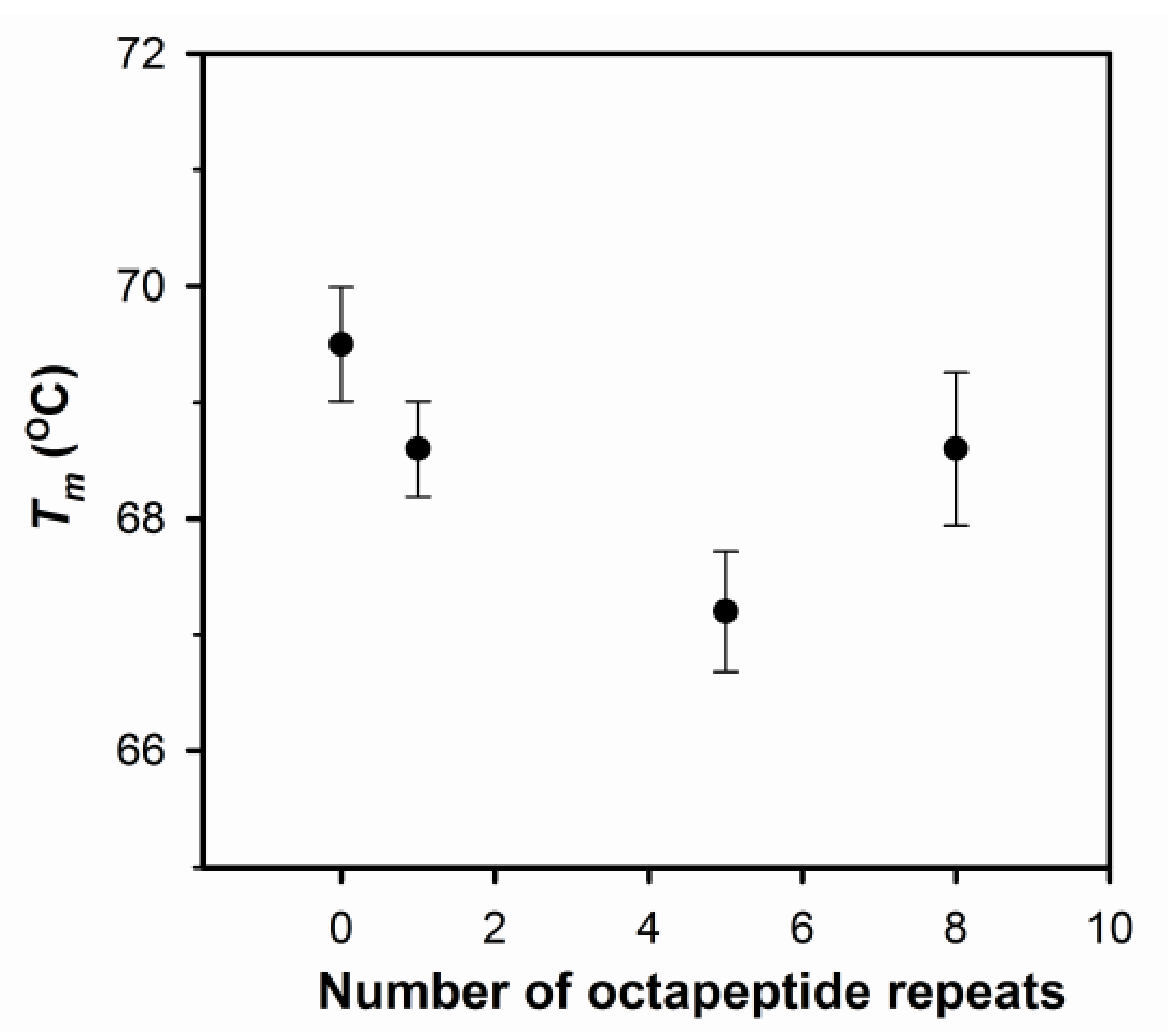
2.1. The Number of Octapeptide Repeats Affects the Structure and the Thermal Stability of Prion Proteins
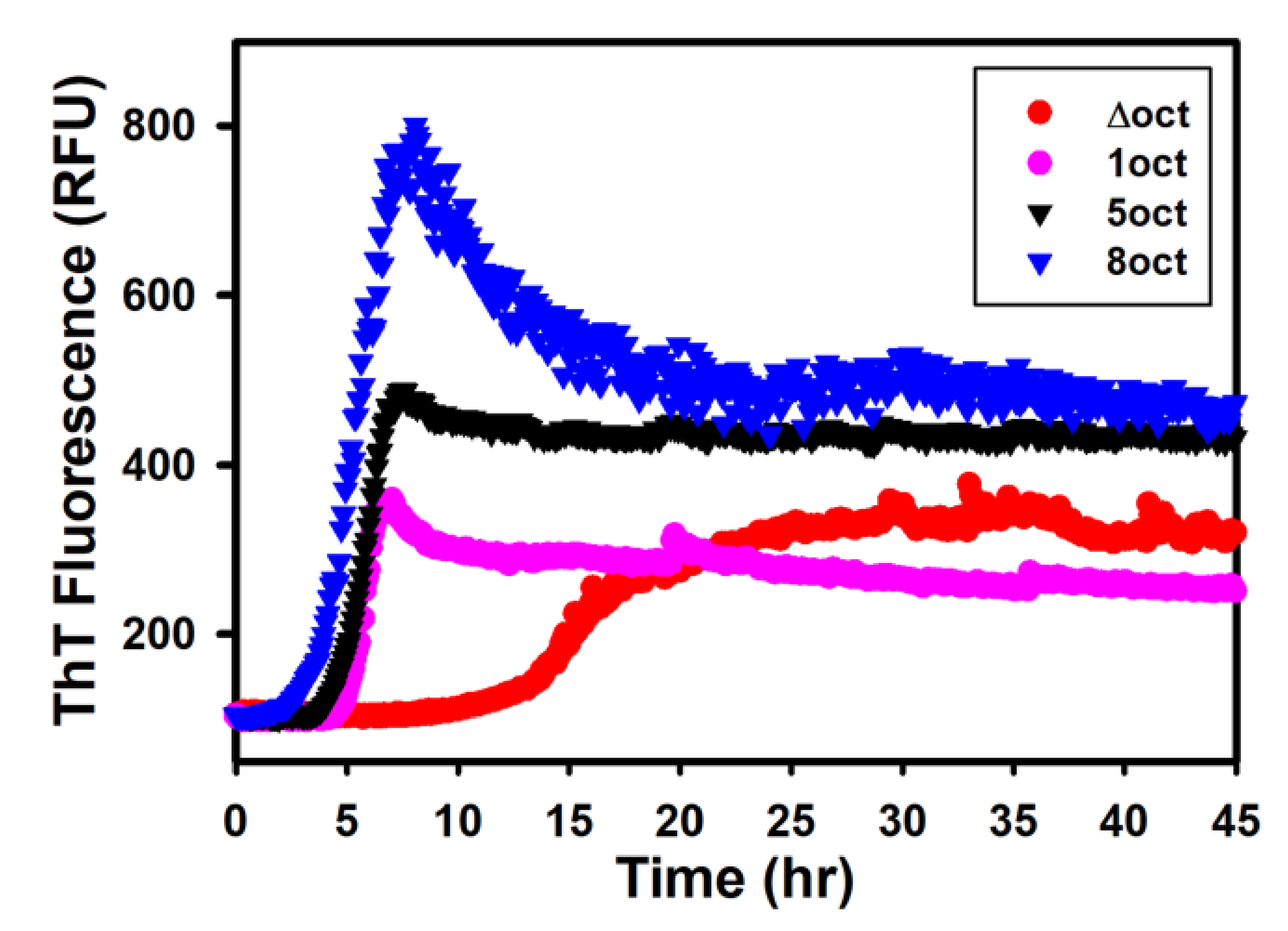
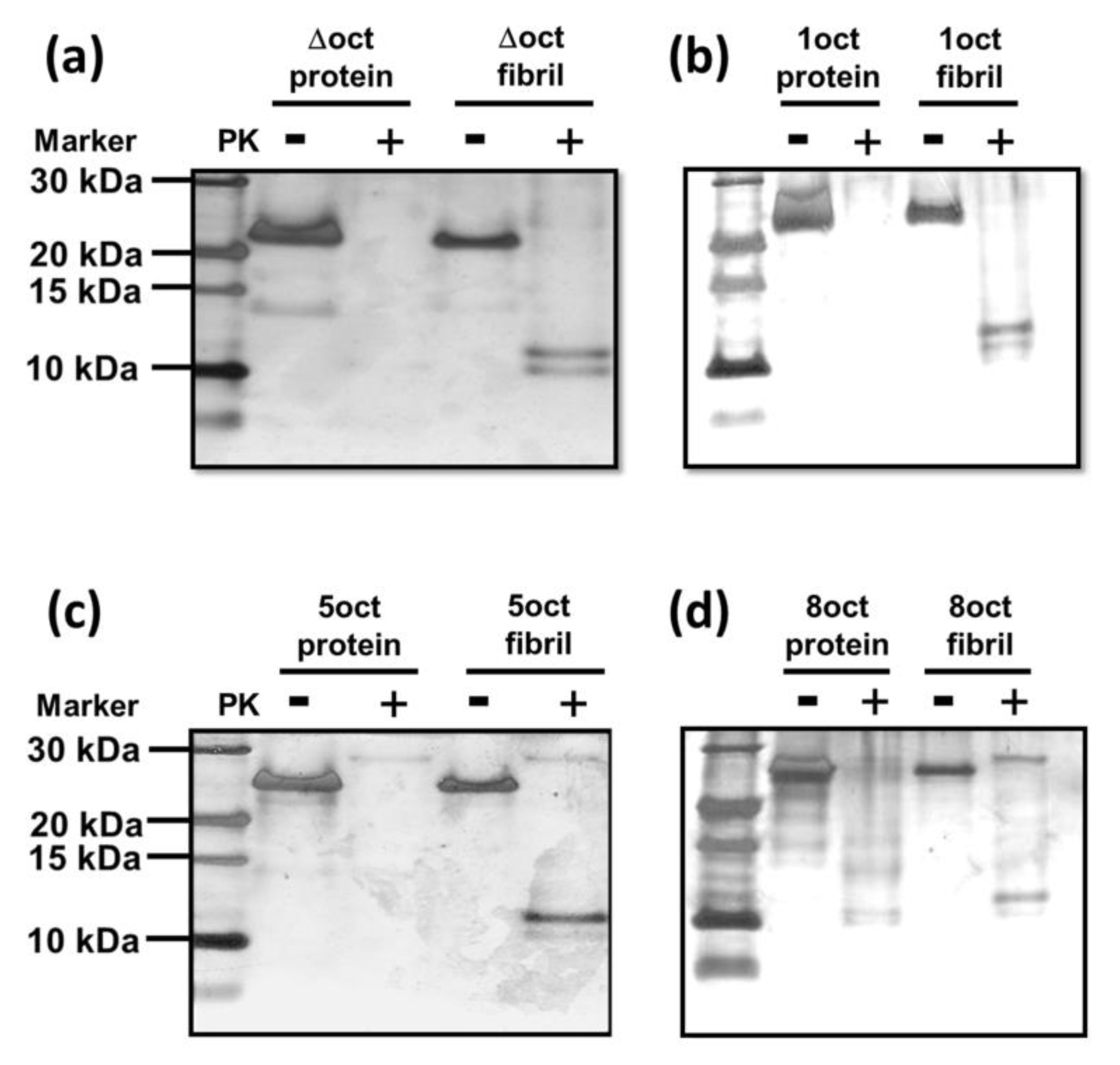
2.2. The Expansion Rather Than the Deletion of Octapeptides Enhances the Kinetics of Fibril Conversion
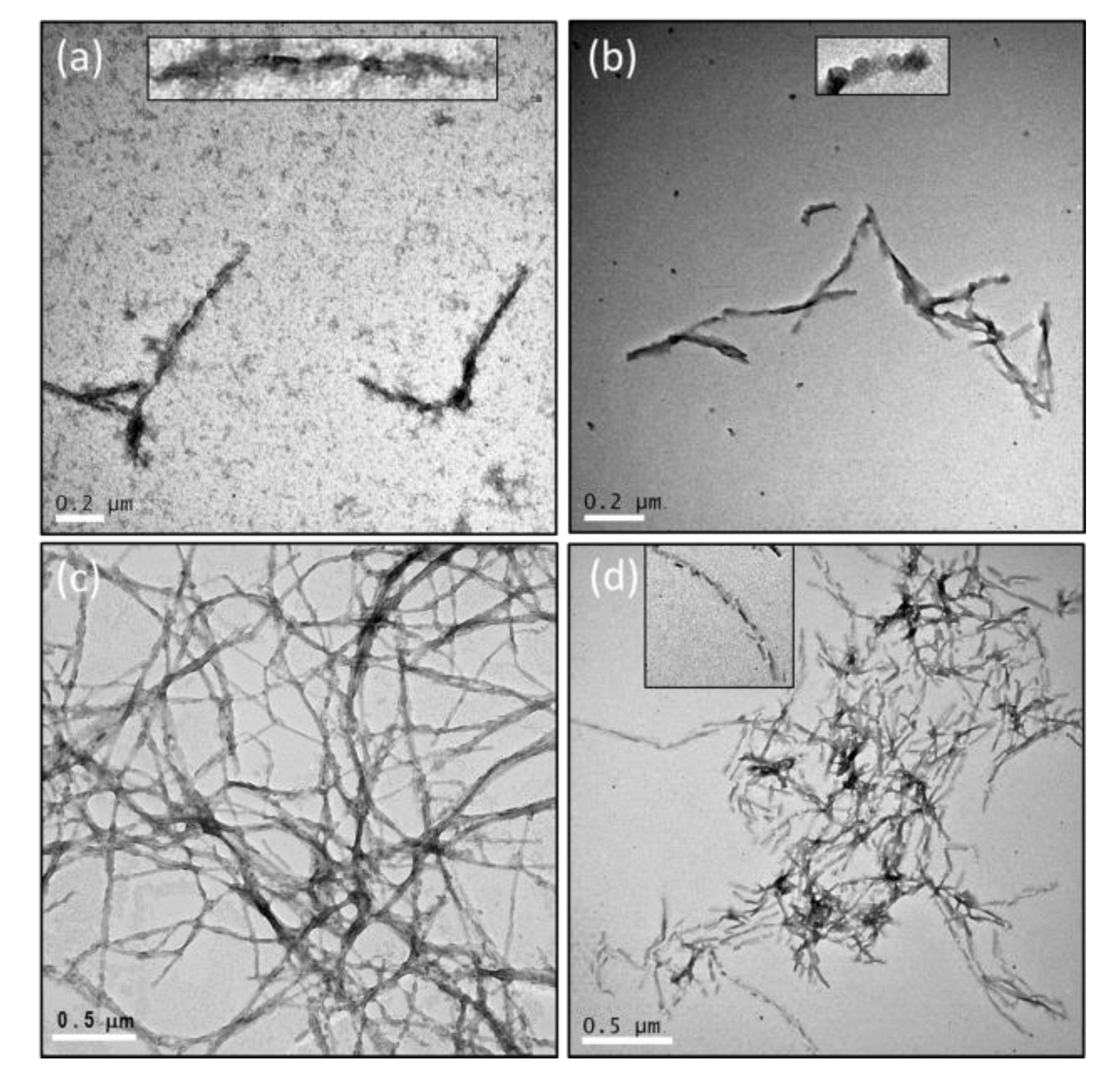
2.3. The Number of Octapeptide Repeats Affects the Structures of Fibrils
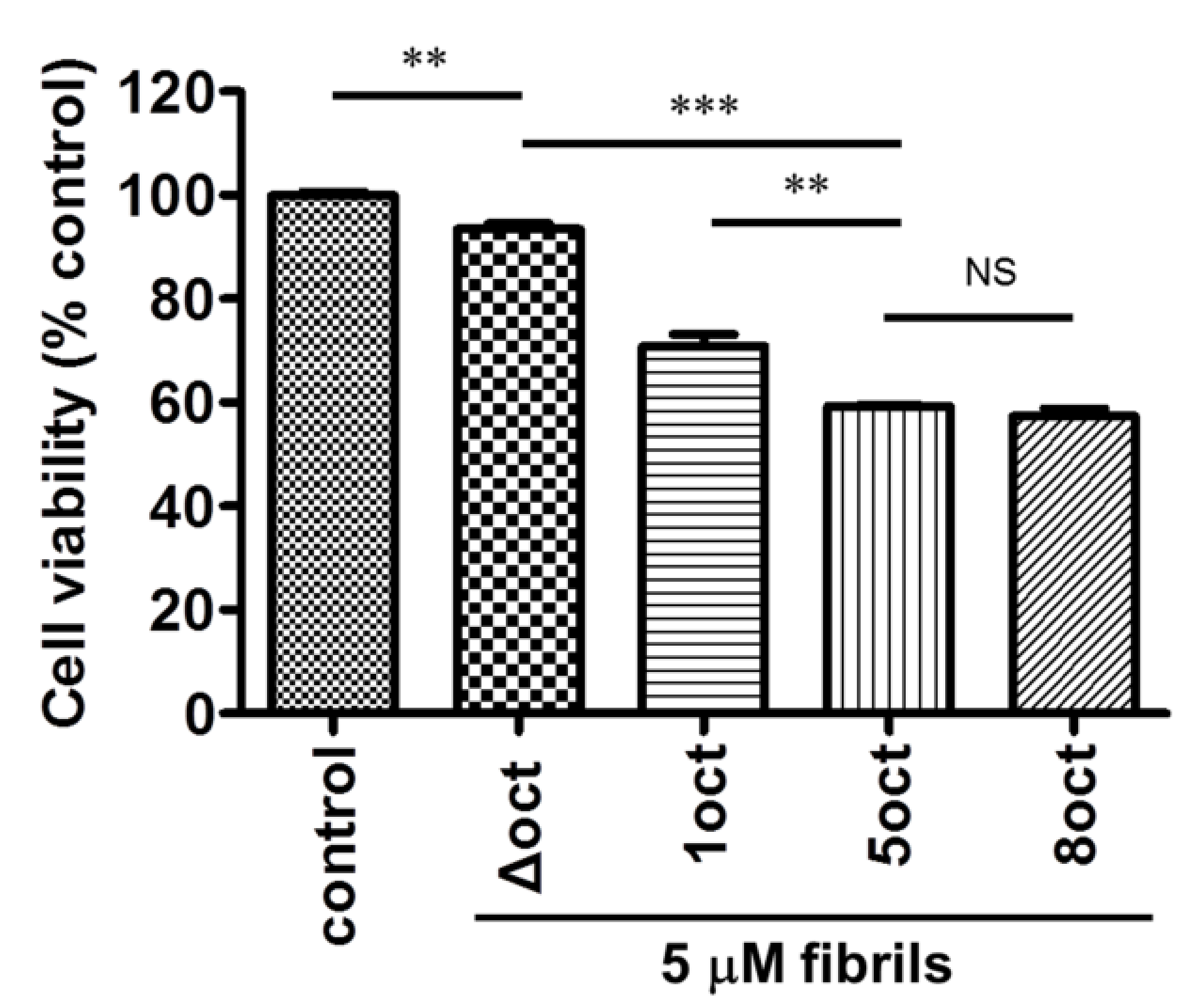
2.4. The Number of Octapeptide Repeats Affects the Cytotoxicity of the Amyloid Fibrils
3. Discussion
4. Materials and Methods
4.1. Plasmid Preparations of PrP-Δoct, PrP-1oct and PrP-8oct
4.2. Expression and Purification of PrP-Δoct, PrP-1oct, PrP-5oct and PrP-8oct
4.3. Circular Dichroism Spectroscopy (CD)
4.4. Kinetics of Fibril Conversion from PrP Variants
4.5. Transmissible Electron Microscopy (TEM)
4.6. PK-Digestion Assay
4.7. Cell Culture and Viability Assay
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Griffith, J.S. Self-replication and scrapie. Nature 1967, 215, 1043–1044. [Google Scholar] [CrossRef]
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef]
- Prusiner, S.B. Prion biology and diseases—Laughing cannibals, mad cows, and scientific heresy. Med. Res. Rev. 1996, 16, 487–505. [Google Scholar] [CrossRef]
- Palmer, M.S.; Mahal, S.P.; Campbell, T.A.; Hill, A.F.; Sidle, K.C.; Laplanche, J.-L.; Collinge, J. Deletions in the prion protein gene are not associated with CJD. Hum. Mol. Genet. 1993, 2, 541–544. [Google Scholar] [CrossRef]
- Reder, A.T.; Mednick, A.S.; Brown, P.; Spire, J.P.; Van Cauter, E.; Wollmann, R.L.; Cervenakova, L.; Goldfarb, L.G.; Garay, A.; Ovsiew, F.; et al. Clinical and genetic studies of fatal familial insomnia. Neurology 1995, 45, 1068–1075. [Google Scholar] [CrossRef]
- Perry, R.T.; Go, R.C.; Harrell, L.E.; Acton, R.T. SSCP analysis and sequencing of the human prion protein gene (PRNP) detects two different 24 bp deletions in an atypical Alzheimer’s disease family. Am. J. Med. Genet. 1995, 60, 12–18. [Google Scholar] [CrossRef]
- Zahn, R.; Von Schroetter, C.; Wüthrich, K. Human prion proteins expressed in Escherichia coli and purified by high-affinity column refolding. FEBS Lett. 1997, 417, 400–404. [Google Scholar] [CrossRef]
- Pan, K.M.; Baldwin, M.; Nguyen, J.; Gasset, M.; Serban, A.; Groth, D.; Mehlhorn, I.; Huang, Z.; Fletterick, R.J.; Cohen, F.E.; et al. Conversion of α-helices into β-sheets features in the formation of the scrapie prion proteins. Proc. Natl. Acad. Sci. USA 1993, 90, 10962–10966. [Google Scholar] [CrossRef]
- Martins, S.M.; Frosoni, D.J.; Martinez, A.M.B.; De Felice, F.G.; Ferreira, S.T. Formation of soluble oligomers and amyloid fibrils with physical properties of the scrapie isoform of the prion protein from the C-terminal domain of recombinant murine prion protein mPrP-(121–231). J. Biol. Chem. 2006, 281, 26121–26128. [Google Scholar] [CrossRef] [PubMed]
- Millhauser, G.L. Copper binding in the prion protein. Acc. Chem. Res. 2004, 37, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Whittal, R.M.; Ball, H.L.; Cohen, F.E.; Burlingame, A.L.; Prusiner, S.B.; Baldwin, M.A. Copper binding to octarepeat peptides of the prion protein monitored by mass spectrometry. Protein Sci. 2000, 9, 332–343. [Google Scholar] [CrossRef] [PubMed]
- Wopfner, F.; Weidenhöfer, G.; Schneider, R.; Von Brunn, A.; Gilch, S.; Schwarz, T.F.; Werner, T.; Schätzl, H.M. Analysis of 27 mammalian and 9 avian PrPs reveals high conservation of flexible regions of the prion protein. J. Mol. Biol. 1999, 289, 1163–1178. [Google Scholar] [CrossRef]
- Croes, E.; Theuns, J.; Houwing-Duistermaat, J.; Dermaut, B.; Sleegers, K.; Roks, G.; Broeck, M.V.D.; Van Harten, B.; Van Swieten, J.C.; Cruts, M.; et al. Octapeptide repeat insertions in the prion protein gene and early onset dementi. J. Neurol. Neurosurg. Psychiatry 2004, 75, 1166–1170. [Google Scholar] [CrossRef] [PubMed]
- Beck, J.; Poulter, M.; Campbell, T.A.; Adamson, G.; Uphill, J.B.; Guerreiro, R.; Jackson, G.S.; Stevens, J.C.; Manji, H.; Collinge, J.; et al. PRNP allelic series from 19 years of prion protein gene sequencing at the MRC Prion Unit. Hum. Mutat. 2010, 31, E1551–E1563. [Google Scholar] [CrossRef]
- Legname, G.; Nguyen, H.-O.B.; Peretz, D.; Cohen, F.E.; DeArmond, S.J.; Prusiner, S.B. Continuum of prion protein structures enciphers a multitude of prion isolate-specified phenotypes. Proc. Natl. Acad. Sci. USA 2006, 103, 19105–19110. [Google Scholar] [CrossRef] [PubMed]
- Caughey, B.; Lansbury, P.T. Protofibrils, pores, fibrils, and neurodegeneration: Separating the responsible protein aggregates from the innocent bystanders. Annu. Rev. Neurosci. 2003, 26, 267–298. [Google Scholar] [CrossRef]
- Makarava, N.; Baskakov, I.V. The same primary structure of the prion protein yields two distinct self-propagating states. J. Biol. Chem. 2008, 283, 15988–15996. [Google Scholar] [CrossRef] [PubMed]
- Priola, S.A.; Chesebro, B. Abnormal properties of prion protein with insertional mutations in different cell types. J. Biol. Chem. 1998, 273, 11980–11985. [Google Scholar] [CrossRef]
- Swietnicki, W.; Morillas, M.; Chen, S.G.; Gambetti, P.; Surewicz, W.K. Aggregation and fibrillization of the recombinant human prion protein huPrP90-231. Biochemistry 2000, 39, 424–431. [Google Scholar] [CrossRef]
- Legname, G.; Baskakov, I.V.; Nguyen, H.-O.B.; Riesner, D.; Cohen, F.E.; DeArmond, S.J.; Prusiner, S.B. Synthetic mammalian prions. Science 2004, 305, 673–676. [Google Scholar] [CrossRef] [PubMed]
- Bocharova, O.V.; Breydo, L.; Salnikov, V.V.; Gill, A.C.; Baskakov, I.V. Synthetic prions generated in vitro are similar to a newly identified subpopulation of PrPSc from sporadic Creutzfeldt-Jakob disease. Protein Sci. 2005, 14, 1222–1232. [Google Scholar] [CrossRef]
- Leliveld, S.R.; Stitz, L.; Korth, C. Expansion of the octarepeat domain alters the misfolding pathway but not the folding pathway of the prion protein. Biochemistry 2008, 47, 6267–6278. [Google Scholar] [CrossRef]
- De Simone, A.; Zagari, A.; Derreumaux, P. Structural and hydration properties of the partially unfolded states of the prion protein. Biophys. J. 2007, 93, 1284–1292. [Google Scholar] [CrossRef]
- Viles, J.H.; Donne, D.; Kroon, G.; Prusiner, S.B.; Cohen, F.E.; Dyson, H.J.; Wright, P.E. Local structural plasticity of the prion protein. Analysis of NMR relaxation dynamics. Biochemistry 2001, 40, 2743–2753. [Google Scholar] [CrossRef]
- Morrissey, M.P.; Shakhnovich, E.I. Evidence for the role of PrPC helix 1 in the hydrophilic seeding of prion aggregates. Proc. Natl. Acad. Sci. USA 1999, 96, 11293–11298. [Google Scholar] [CrossRef] [PubMed]
- Heise, H.; Hoyer, W.; Becker, S.; Andronesi, O.C.; Riedel, D.; Baldus, M. Molecular-level secondary structure, polymorphism, and dynamics of full-length alpha-synuclein fibrils studied by solid-state NMR. Proc. Natl. Acad. Sci. USA 2005, 102, 15871–15876. [Google Scholar] [CrossRef] [PubMed]
- Peelaerts, W.; Bousset, L.; Van Der Perren, A.; Moskalyuk, A.; Pulizzi, R.; Giugliano, M.; Haute, C.V.D.; Melki, R.; Baekelandt, V. alpha-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 2005, 522, 340–344. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Savtchenko, R.; Ostapchenko, V.G.; Makarava, N.; Baskakov, I.V. Molecular structure of amyloid fibrils controls the relationship between fibrillar size and toxicity. PLoS ONE 2011, 6, e20244. [Google Scholar] [CrossRef]
- Scherzinger, E.; Lurz, R.; Turmaine, M.; Mangiarini, L.; Hollenbach, B.; Hasenbank, R.; Bates, G.P.; Davies, S.W.; Lehrach, H.; E Wanker, E. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell 1997, 90, 549–558. [Google Scholar] [CrossRef]
- Vital, C.; Gray, F.; Vital, A.; Parchi, P.; Capellari, S.; Petersen, R.B.; Ferrer, X.; Jarnier, D.; Julien, J.; Gambetti, P. Prion encephalopathy with insertion of octapeptide repeats: The number of repeats determines the type of cerebellar deposits. Neuropathol. Appl. Neurobiol. 1998, 24, 125–130. [Google Scholar] [CrossRef]
- Hsu, C.T.; Ting, C.Y.; Ting, C.J.; Chen, T.Y.; Lin, C.P.; Whang-Peng, J.; Hwang, J. Vaccination against gonadotropin-releasing hormone (GnRH) using toxin receptor-binding domain-conjugated GnRH repeats. Cancer Res. 2000, 60, 3701–3705. [Google Scholar]
- Bocharova, O.V.; Breydo, L.; Parfenov, A.S.; Salnikov, V.V.; Baskakov, I.V. In vitro conversion of full-length mammalian prion protein produces amyloid form with physical properties of PrPSc. J. Mol. Biol. 2005, 346, 645–659. [Google Scholar] [CrossRef]
- Luo, J.-C.; Wang, S.-C.; Jian, W.-B.; Chen, C.-H.; Tang, J.-L.; Lee, C.-I. Formation of amyloid fibrils from β-amylase. FEBS Lett. 2012, 586, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.-J.; Yu, K.-H.; Wu, J.-R.; Lee, C.-F.; Jheng, C.-P.; Chen, H.-R.; Lee, C.-I. Liberation of GPI-anchored prion from phospholipids accelerates amyloidogenic conversion. Int. J. Mol. Sci. 2013, 14, 17943–17957. [Google Scholar] [CrossRef] [PubMed]
- Jen, H.-I.; Lin, Z.-Y.; Guo, J.-X.; Lee, C.-I. The effects of divalent cation-chelated prion fibrils on the immune response of EOC 13.31 microglia cells. Cells 2020, 9, 2285. [Google Scholar] [CrossRef] [PubMed]
- Whitmore, L.; Wallace, B.A. Protein secondary structure analyses from circular dichroism spectroscopy: Methods and reference databases. Biopolymers 2007, 89, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Whitmore, L.; Wallace, B.A. DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 2004, 32, W668–W673. [Google Scholar] [CrossRef]
- Makarava, N.; Lee, C.-I.; Ostapchenko, V.G.; Baskakov, I.V. Highly promiscuous nature of prion polymerization. J. Biol. Chem. 2007, 282, 36704–36713. [Google Scholar] [CrossRef]
- Schagger, H. Tricine-SDS-PAGE. Nat. Protoc. 2006, 1, 16–22. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, K.-H.; Huang, M.-Y.; Lee, Y.-R.; Lin, Y.-K.; Chen, H.-R.; Lee, C.-I. The Effect of Octapeptide Repeats on Prion Folding and Misfolding. Int. J. Mol. Sci. 2021, 22, 1800. https://doi.org/10.3390/ijms22041800
Yu K-H, Huang M-Y, Lee Y-R, Lin Y-K, Chen H-R, Lee C-I. The Effect of Octapeptide Repeats on Prion Folding and Misfolding. International Journal of Molecular Sciences. 2021; 22(4):1800. https://doi.org/10.3390/ijms22041800
Chicago/Turabian StyleYu, Kun-Hua, Mei-Yu Huang, Yi-Ru Lee, Yu-Kie Lin, Hau-Ren Chen, and Cheng-I Lee. 2021. "The Effect of Octapeptide Repeats on Prion Folding and Misfolding" International Journal of Molecular Sciences 22, no. 4: 1800. https://doi.org/10.3390/ijms22041800
APA StyleYu, K.-H., Huang, M.-Y., Lee, Y.-R., Lin, Y.-K., Chen, H.-R., & Lee, C.-I. (2021). The Effect of Octapeptide Repeats on Prion Folding and Misfolding. International Journal of Molecular Sciences, 22(4), 1800. https://doi.org/10.3390/ijms22041800