
Endoplasmic Reticulum Stress Signaling and the Pathogenesis of Hepatocarcinoma

Abstract
1. Introduction
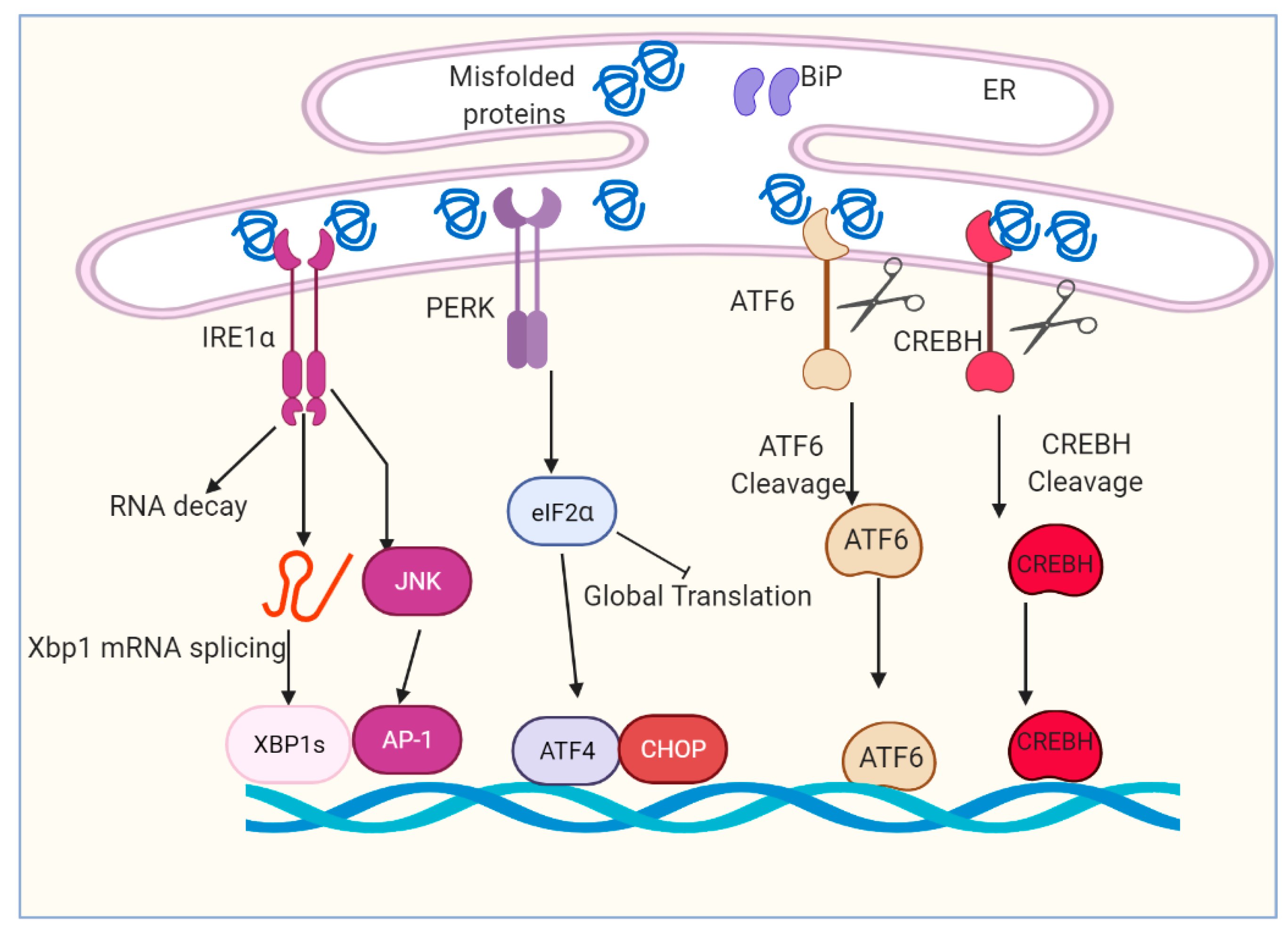
2. ER Stress and UPR
2.1. Serine/Threonine-Protein Kinase/Endoribonuclease IRE1α and Transcription Factor Xbp1s
2.2. The Kinase PERK and Transcription Factor ATF4 and CHOP
2.3. Transcription Factor ATF6α
2.4. Transcription Factor CREBH
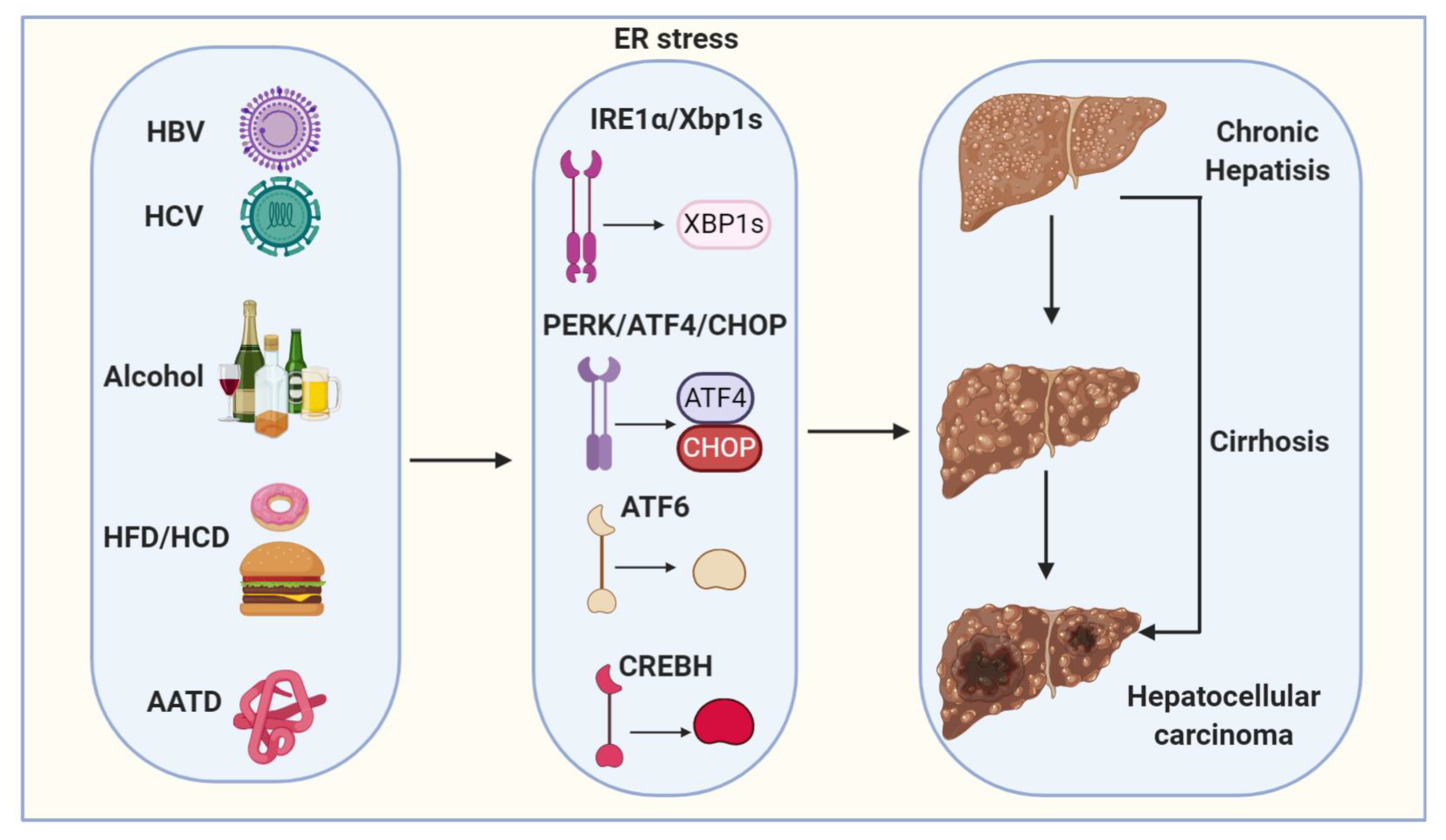
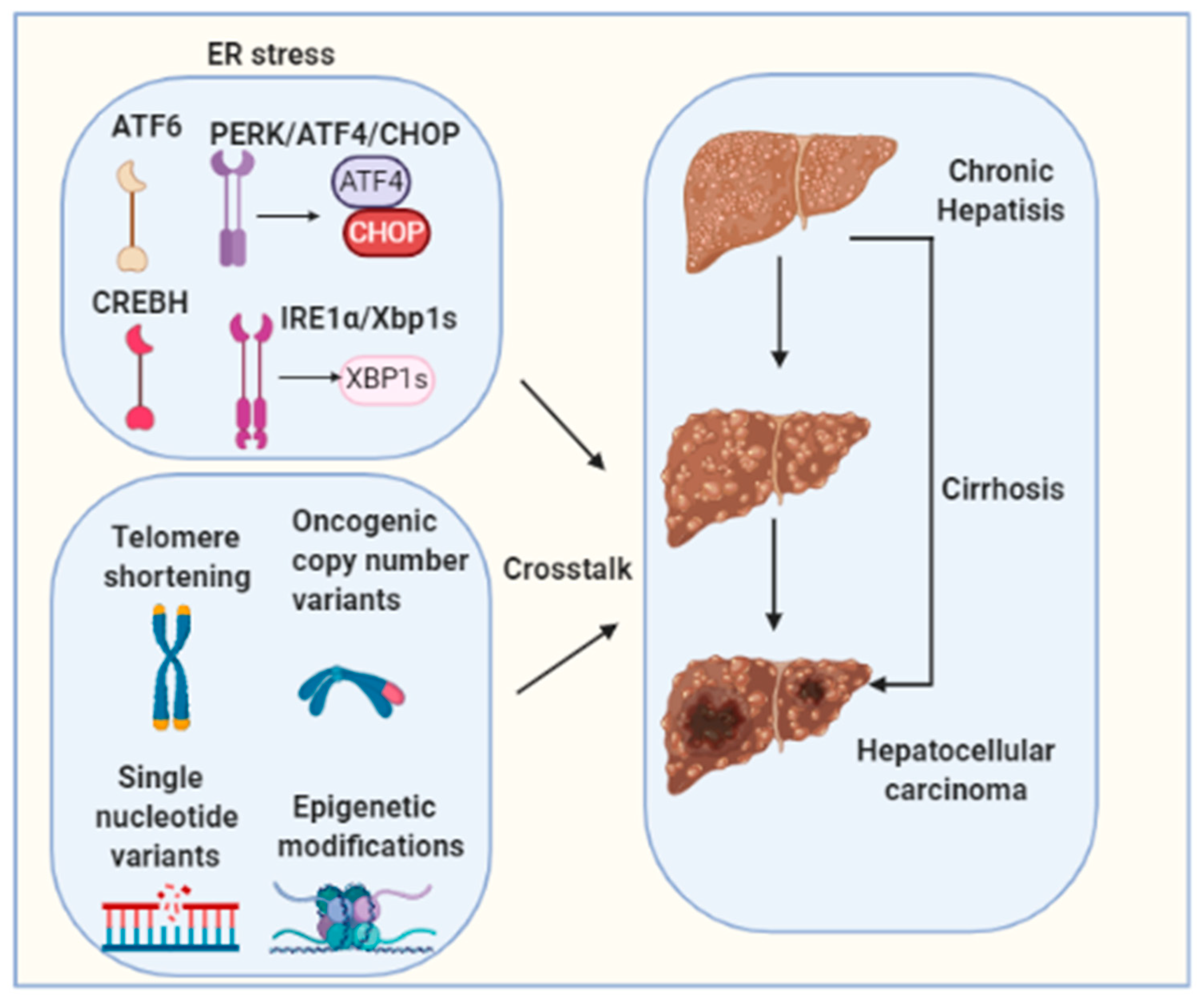
3. Pathogenesis of Hepatocellular Carcinoma
3.1. Viral Hepatitis
3.2. Alcoholic Fatty Liver Disease (ALD)
3.3. Non-Alcoholic Fatty Liver Disease (NAFLD) and Nonalcoholic Steatohepatitis (NASH)
3.4. Alpha1-Antitrypsin Deficiency (AATD)
4. Targeting ER Stress for HCC Treatment
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Halperin, L.; Jung, J.; Michalak, M. The many functions of the endoplasmic reticulum chaperones and folding enzymes. IUBMB Life 2014, 66, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Kaufman, R.J. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 2016, 529, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell. Mol. Life Sci. 2016, 73, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Beretta, M.; Santos, C.X.; Molenaar, C.; Hafstad, A.D.; Miller, C.C.; Revazian, A.; Betteridge, K.; Schröder, K.; Streckfuß-Bömeke, K.; Doroshow, J.H.; et al. Nox4 regulates InsP(3) receptor-dependent Ca(2+) release into mitochondria to promote cell survival. EMBO J. 2020, 39, e103530. [Google Scholar] [CrossRef] [PubMed]
- Sasako, T.; Ohsugi, M.; Kubota, N.; Itoh, S.; Okazaki, Y.; Terai, A.; Kubota, T.; Yamashita, S.; Nakatsukasa, K.; Kamura, T.; et al. Hepatic Sdf2l1 controls feeding-induced ER stress and regulates metabolism. Nat. Commun. 2019, 10, 947. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, K. Endoplasmic Reticulum Stress-Associated Lipid Droplet Formation and Type II Diabetes. Biochem. Res. Int. 2012, 2012, 247275. [Google Scholar] [CrossRef] [PubMed]
- Song, M.J.; Malhi, H. The unfolded protein response and hepatic lipid metabolism in non alcoholic fatty liver disease. Pharmacol. Ther. 2019, 203, 107401. [Google Scholar] [CrossRef]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef]
- Dandekar, A.; Mendez, R.; Zhang, K. Cross talk between ER stress, oxidative stress, and inflammation in health and disease. Methods Mol. Biol. 2015, 1292, 205–214. [Google Scholar] [PubMed]
- Zhang, K. Endoplasmic reticulum stress response and transcriptional reprogramming. Front. Genet. 2014, 5, 460. [Google Scholar] [PubMed]
- Sabath, N.; Levy-Adam, F.; Younis, A.; Rozales, K.; Meller, A.; Hadar, S.; Soueid-Baumgarten, S.; Shalgi, R. Cellular proteostasis decline in human senescence. Proc. Natl. Acad. Sci. USA 2020, 117, 31902–31913. [Google Scholar] [CrossRef] [PubMed]
- Platko, K.; Lebeau, P.F.; Byun, J.H.; Poon, S.V.; Day, E.A.; MacDonald, M.E.; Holzapfel, N.; Mejia-Benitez, A.; Maclean, K.N.; Krepinsky, J.C.; et al. GDF10 blocks hepatic PPARγ activation to protect against diet-induced liver injury. Mol. Metab. 2019, 27, 62–74. [Google Scholar] [CrossRef]
- Wei, J.; Yuan, Y.; Chen, L.; Xu, Y.; Zhang, Y.; Wang, Y.; Yang, Y.; Peek, C.B.; Diebold, L.; Yang, Y.; et al. ER-associated ubiquitin ligase HRD1 programs liver metabolism by targeting multiple metabolic enzymes. Nat. Commun. 2018, 9, 3659. [Google Scholar] [CrossRef]
- Ji, C. New Insights into the Pathogenesis of Alcohol-Induced ER Stress and Liver Diseases. Int. J. Hepatol. 2014, 2014, 513787. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Green, R.M. Endoplasmic reticulum stress and liver diseases. Liver Res. 2019, 3, 55–64. [Google Scholar] [CrossRef]
- Lebeaupin, C.; Vallée, D.; Hazari, Y.; Hetz, C.; Chevet, E.; Bailly-Maitre, B. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J. Hepatol. 2018, 69, 927–947. [Google Scholar] [CrossRef]
- Mohan, S.; Brown, L.; Ayyappan, P. Endoplasmic reticulum stress: A master regulator of metabolic syndrome. Eur. J. Pharmacol. 2019, 860, 172553. [Google Scholar] [CrossRef]
- Maiers, J.L.; Malhi, H. Endoplasmic Reticulum Stress in Metabolic Liver Diseases and Hepatic Fibrosis. Semin. Liver Dis. 2019, 39, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Zhang, C.; Zhang, K. Measurement of ER stress response and inflammation in the mouse model of nonalcoholic fatty liver disease. Methods Enzymol. 2011, 489, 329–348. [Google Scholar] [PubMed]
- Malhi, H.; Kaufman, R.J. Endoplasmic reticulum stress in liver disease. J. Hepatol. 2011, 54, 795–809. [Google Scholar] [CrossRef]
- Kapuy, O.; Márton, M.; Bánhegyi, G.; Vinod, P.K. Multiple system-level feedback loops control life-and-death decisions in endoplasmic reticulum stress. FEBS Lett. 2020, 594, 1112–1123. [Google Scholar] [CrossRef]
- Xu, C.; Bailly-Maitre, B.; Reed, J.C. Endoplasmic reticulum stress: Cell life and death decisions. J. Clin. Investig. 2005, 115, 2656–2664. [Google Scholar] [CrossRef]
- Yu, J.; Li, T.; Liu, Y.; Wang, X.; Zhang, J.; Wang, X.; Shi, G.; Lou, J.; Wang, L.; Wang, C.C.; et al. Phosphorylation switches protein disulfide isomerase activity to maintain proteostasis and attenuate ER stress. EMBO J. 2020, 39, e103841. [Google Scholar] [CrossRef]
- Wang, S.; Kaufman, R.J. The impact of the unfolded protein response on human disease. J. Cell. Biol. 2012, 197, 857–867. [Google Scholar] [CrossRef] [PubMed]
- Schröder, M.; Kaufman, R.J. ER stress and the unfolded protein response. Mutat. Res. 2005, 569, 29–63. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Fang, D. Endoplasmic reticulum-associated degradation and beyond: The multitasking roles for HRD1 in immune regulation and autoimmunity. J. Autoimmun. 2020, 109, 102423. [Google Scholar] [CrossRef] [PubMed]
- Rainbolt, T.K.; Frydman, J. Dynamics and clustering of IRE1α during ER stress. Proc. Natl. Acad. Sci. USA 2020, 117, 3352–3354. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Brandizzi, F. IRE1: ER stress sensor and cell fate executor. Trends Cell Biol. 2013, 23, 547–555. [Google Scholar] [CrossRef]
- Rozpedek, W.; Pytel, D.; Mucha, B.; Leszczynska, H.; Diehl, J.A.; Majsterek, I. The Role of the PERK/eIF2α/ATF4/CHOP Signaling Pathway in Tumor Progression During Endoplasmic Reticulum Stress. Curr. Mol. Med. 2016, 16, 533–544. [Google Scholar] [CrossRef]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Davé, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef]
- Zhang, K.; Shen, X.; Wu, J.; Sakaki, K.; Saunders, T.; Rutkowski, D.T.; Back, S.H.; Kaufman, R.J. Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell 2006, 124, 587–599. [Google Scholar] [CrossRef] [PubMed]
- Belyy, V.; Tran, N.H.; Walter, P. Quantitative microscopy reveals dynamics and fate of clustered IRE1α. Proc. Natl. Acad. Sci. USA 2020, 117, 1533–1542. [Google Scholar] [CrossRef]
- Dufey, E.; Bravo-San Pedro, J.M.; Eggers, C.; González-Quiroz, M.; Urra, H.; Sagredo, A.I.; Sepulveda, D.; Pihán, P.; Carreras-Sureda, A.; Hazari, Y.; et al. Genotoxic stress triggers the activation of IRE1α-dependent RNA decay to modulate the DNA damage response. Nat. Commun. 2020, 11, 2401. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.B.; Snyder, J.T.; Alonso, L.C. Atf6α impacts cell number by influencing survival, death and proliferation. Mol. Metab. 2019, 27s, S69–S80. [Google Scholar] [CrossRef] [PubMed]
- Duvigneau, J.C.; Luís, A.; Gorman, A.M.; Samali, A.; Kaltenecker, D.; Moriggl, R.; Kozlov, A.V. Crosstalk between inflammatory mediators and endoplasmic reticulum stress in liver diseases. Cytokine 2019, 124, 154577. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Huang, L.; Gong, J.; Shi, C.; Wang, Z.; Ye, B.; Xuan, A.; He, X.; Long, D.; Zhu, X.; et al. NF-κB pathway link with ER stress-induced autophagy and apoptosis in cervical tumor cells. Cell Death Discov. 2017, 3, 17059. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Trillo-Tinoco, J.; Sierra, R.A.; Anadon, C.; Dai, W.; Mohamed, E.; Cen, L.; Costich, T.L.; Magliocco, A.; Marchion, D.; et al. ER stress-induced mediator C/EBP homologous protein thwarts effector T cell activity in tumors through T-bet repression. Nat. Commun. 2019, 10, 1280. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, D.; Neuman, S.D.; Yang, A.; Lough, L.; Brown, B.; Bashirullah, A.; Cardozo, T.; Ryoo, H.D. Translational induction of ATF4 during integrated stress response requires noncanonical initiation factors eIF2D and DENR. Nat. Commun. 2020, 11, 4677. [Google Scholar] [CrossRef] [PubMed]
- Kasetti, R.B.; Patel, P.D.; Maddineni, P.; Patil, S.; Kiehlbauch, C.; Millar, J.C.; Searby, C.C.; Raghunathan, V.; Sheffield, V.C.; Zode, G.S. ATF4 leads to glaucoma by promoting protein synthesis and ER client protein load. Nat. Commun. 2020, 11, 5594. [Google Scholar] [CrossRef] [PubMed]
- Schulze, R.J.; Schott, M.B.; Casey, C.A.; Tuma, P.L.; McNiven, M.A. The cell biology of the hepatocyte: A membrane trafficking machine. J. Cell. Biol. 2019, 218, 2096–2112. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Wang, Z.V.; Tao, C.; Gao, N.; Holland, W.L.; Ferdous, A.; Repa, J.J.; Liang, G.; Ye, J.; Lehrman, M.A.; et al. The Xbp1s/GalE axis links ER stress to postprandial hepatic metabolism. J. Clin. Investig. 2013, 123, 455–468. [Google Scholar] [CrossRef] [PubMed]
- Maillo, C.; Martín, J.; Sebastián, D.; Hernández-Alvarez, M.; García-Rocha, M.; Reina, O.; Zorzano, A.; Fernandez, M.; Méndez, R. Circadian- and UPR-dependent control of CPEB4 mediates a translational response to counteract hepatic steatosis under ER stress. Nat. Cell Biol. 2017, 19, 94–105. [Google Scholar] [CrossRef]
- Wei, J.; Chen, L.; Li, F.; Yuan, Y.; Wang, Y.; Xia, W.; Zhang, Y.; Xu, Y.; Yang, Z.; Gao, B.; et al. HRD1-ERAD controls production of the hepatokine FGF21 through CREBH polyubiquitination. EMBO J. 2018, 37. [Google Scholar] [CrossRef]
- Chen, L.; Wei, J.; Zhu, H.; Pan, H.; Fang, D. Energy supplementation rescues growth restriction and female infertility of mice with hepatic HRD1 ablation. Am. J. Transl. Res. 2020, 12, 2018–2027. [Google Scholar] [PubMed]
- Sun, S.; Shi, G.; Sha, H.; Ji, Y.; Han, X.; Shu, X.; Ma, H.; Inoue, T.; Gao, B.; Kim, H.; et al. IRE1α is an endogenous substrate of endoplasmic-reticulum-associated degradation. Nat. Cell Biol. 2015, 17, 1546–1555. [Google Scholar] [CrossRef]
- Sepulveda, D.; Rojas-Rivera, D.; Rodríguez, D.A.; Groenendyk, J.; Köhler, A.; Lebeaupin, C.; Ito, S.; Urra, H.; Carreras-Sureda, A.; Hazari, Y.; et al. Interactome Screening Identifies the ER Luminal Chaperone Hsp47 as a Regulator of the Unfolded Protein Response Transducer IRE1α. Mol. Cell 2018, 69, 238–252.e7. [Google Scholar] [CrossRef]
- Ito, S.; Nagata, K. Roles of the endoplasmic reticulum-resident, collagen-specific molecular chaperone Hsp47 in vertebrate cells and human disease. J. Biol. Chem. 2019, 294, 2133–2141. [Google Scholar] [CrossRef]
- Lu, Y.; Liang, F.X.; Wang, X. A synthetic biology approach identifies the mammalian UPR RNA ligase RtcB. Mol. Cell 2014, 55, 758–770. [Google Scholar] [CrossRef]
- Xu, Y.; Melo-Cardenas, J.; Zhang, Y.; Gau, I.; Wei, J.; Montauti, E.; Zhang, Y.; Gao, B.; Jin, H.; Sun, Z.; et al. The E3 ligase Hrd1 stabilizes Tregs by antagonizing inflammatory cytokine-induced ER stress response. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Wong, H.N.; Song, B.; Miller, C.N.; Scheuner, D.; Kaufman, R.J. The unfolded protein response sensor IRE1alpha is required at 2 distinct steps in B cell lymphopoiesis. J. Clin. Investig. 2005, 115, 268–281. [Google Scholar] [CrossRef]
- Reimold, A.M.; Etkin, A.; Clauss, I.; Perkins, A.; Friend, D.S.; Zhang, J.; Horton, H.F.; Scott, A.; Orkin, S.H.; Byrne, M.C.; et al. An essential role in liver development for transcription factor XBP-1. Genes Dev. 2000, 14, 152–157. [Google Scholar]
- Lee, A.H.; Scapa, E.F.; Cohen, D.E.; Glimcher, L.H. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science 2008, 320, 1492–1496. [Google Scholar] [CrossRef] [PubMed]
- So, J.S.; Hur, K.Y.; Tarrio, M.; Ruda, V.; Frank-Kamenetsky, M.; Fitzgerald, K.; Koteliansky, V.; Lichtman, A.H.; Iwawaki, T.; Glimcher, L.H.; et al. Silencing of lipid metabolism genes through IRE1α-mediated mRNA decay lowers plasma lipids in mice. Cell Metab. 2012, 16, 487–499. [Google Scholar] [CrossRef]
- McQuiston, A.; Diehl, J.A. Recent insights into PERK-dependent signaling from the stressed endoplasmic reticulum. F1000Research 2017, 6, 1897. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Li, J.; Sha, B. The ER stress sensor PERK luminal domain functions as a molecular chaperone to interact with misfolded proteins. Acta Crystallogr. Sect. D Struct. Biol. 2016, 72 Pt 12, 1290–1297. [Google Scholar] [CrossRef]
- Wang, P.; Li, J.; Tao, J.; Sha, B. The luminal domain of the ER stress sensor protein PERK binds misfolded proteins and thereby triggers PERK oligomerization. J. Biol. Chem. 2018, 293, 4110–4121. [Google Scholar] [CrossRef]
- B’Chir, W.; Maurin, A.C.; Carraro, V.; Averous, J.; Jousse, C.; Muranishi, Y.; Parry, L.; Stepien, G.; Fafournoux, P.; Bruhat, A. The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucl. Acids Res. 2013, 41, 7683–7699. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M.; et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell. Biol. 2013, 15, 481–490. [Google Scholar] [CrossRef]
- Dey, S.; Sayers, C.M.; Verginadis, I.I.; Lehman, S.L.; Cheng, Y.; Cerniglia, G.J.; Tuttle, S.W.; Feldman, M.D.; Zhang, P.J.; Fuchs, S.Y.; et al. ATF4-dependent induction of heme oxygenase 1 prevents anoikis and promotes metastasis. J. Clin. Investig. 2015, 125, 2592–2608. [Google Scholar] [CrossRef]
- Novoa, I.; Zeng, H.; Harding, H.P.; Ron, D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J. Cell. Biol. 2001, 153, 1011–1022. [Google Scholar] [CrossRef]
- Harding, H.P.; Zeng, H.; Zhang, Y.; Jungries, R.; Chung, P.; Plesken, H.; Sabatini, D.D.; Ron, D. Diabetes mellitus and exocrine pancreatic dysfunction in perk-/- mice reveals a role for translational control in secretory cell survival. Mol. Cell 2001, 7, 1153–1163. [Google Scholar] [CrossRef]
- Zhang, P.; McGrath, B.; Li, S.; Frank, A.; Zambito, F.; Reinert, J.; Gannon, M.; Ma, K.; McNaughton, K.; Cavener, D.R. The PERK eukaryotic initiation factor 2 alpha kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Mol. Cell. Biol. 2002, 22, 3864–3874. [Google Scholar] [CrossRef]
- Fusakio, M.E.; Willy, J.A.; Wang, Y.; Mirek, E.T.; Al Baghdadi, R.J.; Adams, C.M.; Anthony, T.G.; Wek, R.C. Transcription factor ATF4 directs basal and stress-induced gene expression in the unfolded protein response and cholesterol metabolism in the liver. Mol. Biol. Cell 2016, 27, 1536–1551. [Google Scholar] [CrossRef] [PubMed]
- Jo, H.; Choe, S.S.; Shin, K.C.; Jang, H.; Lee, J.H.; Seong, J.K.; Back, S.H.; Kim, J.B. Endoplasmic reticulum stress induces hepatic steatosis via increased expression of the hepatic very low-density lipoprotein receptor. Hepatology 2013, 57, 1366–1377. [Google Scholar] [CrossRef]
- Yeh, K.Y.; Lai, C.Y.; Lin, C.Y.; Hsu, C.C.; Lo, C.P.; Her, G.M. ATF4 overexpression induces early onset of hyperlipidaemia and hepatic steatosis and enhances adipogenesis in zebrafish. Sci. Rep. 2017, 7, 16362. [Google Scholar] [CrossRef] [PubMed]
- Malhi, H.; Kropp, E.M.; Clavo, V.F.; Kobrossi, C.R.; Han, J.; Mauer, A.S.; Yong, J.; Kaufman, R.J. C/EBP homologous protein-induced macrophage apoptosis protects mice from steatohepatitis. J. Biol. Chem. 2013, 288, 18624–18642. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Gao, J.; Ishigaki, Y.; Kondo, K.; Sawada, S.; Izumi, T.; Uno, K.; Kaneko, K.; Tsukita, S.; Takahashi, K.; et al. ER Stress Protein CHOP Mediates Insulin Resistance by Modulating Adipose Tissue Macrophage Polarity. Cell. Rep. 2017, 18, 2045–2057. [Google Scholar] [CrossRef]
- Yamamoto, K.; Sato, T.; Matsui, T.; Sato, M.; Okada, T.; Yoshida, H.; Harada, A.; Mori, K. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev. Cell 2007, 13, 365–376. [Google Scholar] [CrossRef]
- Forouhan, M.; Mori, K.; Boot-Handford, R.P. Paradoxical roles of ATF6α and ATF6β in modulating disease severity caused by mutations in collagen X. Matrix Biol. J. Int. Soc. Matrix Biol. 2018, 70, 50–71. [Google Scholar] [CrossRef]
- Wu, J.; Rutkowski, D.T.; Dubois, M.; Swathirajan, J.; Saunders, T.; Wang, J.; Song, B.; Yau, G.D.; Kaufman, R.J. ATF6alpha optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev. Cell 2007, 13, 351–364. [Google Scholar] [CrossRef]
- Shen, J.; Snapp, E.L.; Lippincott-Schwartz, J.; Prywes, R. Stable binding of ATF6 to BiP in the endoplasmic reticulum stress response. Mol. Cell. Biol. 2005, 25, 921–932. [Google Scholar] [CrossRef]
- Kaneko, M.; Yasui, S.; Niinuma, Y.; Arai, K.; Omura, T.; Okuma, Y.; Nomura, Y. A different pathway in the endoplasmic reticulum stress-induced expression of human HRD1 and SEL1 genes. FEBS Lett. 2007, 581, 5355–5360. [Google Scholar] [CrossRef]
- Yamamoto, K.; Takahara, K.; Oyadomari, S.; Okada, T.; Sato, T.; Harada, A.; Mori, K. Induction of liver steatosis and lipid droplet formation in ATF6alpha-knockout mice burdened with pharmacological endoplasmic reticulum stress. Mol. Biol. Cell 2010, 21, 2975–2986. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, F.; Gong, Q.; Cui, A.; Zhuo, S.; Hu, Z.; Han, Y.; Gao, J.; Sun, Y.; Liu, Z.; et al. Hepatic ATF6 Increases Fatty Acid Oxidation to Attenuate Hepatic Steatosis in Mice Through Peroxisome Proliferator-Activated Receptor α. Diabetes 2016, 65, 1904–1915. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Lu, M.; Mori, K.; Luo, S.; Lee, A.S.; Zhu, Y.; Shyy, J.Y. ATF6 modulates SREBP2-mediated lipogenesis. EMBO J. 2004, 23, 950–958. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Vera, L.; Fischer, W.H.; Montminy, M. The CREB coactivator CRTC2 links hepatic ER stress and fasting gluconeogenesis. Nature 2009, 460, 534–537. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Williams, D.; Qiu, Y.; Song, Z.; Yang, Z.; Kimler, V.; Goldberg, A.; Zhang, R.; Yang, Z.; Chen, X.; et al. Regulation of hepatic autophagy by stress-sensing transcription factor CREBH. FASEB J. Off. Publ. Federat. Am. Soc. Exp. Biol. 2019, 33, 7896–7914. [Google Scholar] [CrossRef]
- Zheng, Z.; Kim, H.; Qiu, Y.; Chen, X.; Mendez, R.; Dandekar, A.; Zhang, X.; Zhang, C.; Liu, A.C.; Yin, L.; et al. CREBH Couples Circadian Clock With Hepatic Lipid Metabolism. Diabetes 2016, 65, 3369–3383. [Google Scholar] [CrossRef]
- Song, Y.; Zhao, M.; Cheng, X.; Shen, J.; Khound, R.; Zhang, K.; Su, Q. CREBH mediates metabolic inflammation to hepatic VLDL overproduction and hyperlipoproteinemia. J. Mol. Med. 2017, 95, 839–849. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, G.; Zheng, Z.; Maddipati, K.R.; Zhang, X.; Dyson, G.; Williams, P.; Duncan, S.A.; Kaufman, R.J.; Zhang, K. Endoplasmic reticulum-tethered transcription factor cAMP responsive element-binding protein, hepatocyte specific, regulates hepatic lipogenesis, fatty acid oxidation, and lipolysis upon metabolic stress in mice. Hepatology 2012, 55, 1070–1082. [Google Scholar] [CrossRef]
- Lee, J.H.; Giannikopoulos, P.; Duncan, S.A.; Wang, J.; Johansen, C.T.; Brown, J.D.; Plutzky, J.; Hegele, R.A.; Glimcher, L.H.; Lee, A.H. The transcription factor cyclic AMP-responsive element-binding protein H regulates triglyceride metabolism. Nat. Med. 2011, 17, 812–815. [Google Scholar] [CrossRef]
- Xu, X.; Park, J.G.; So, J.S.; Hur, K.Y.; Lee, A.H. Transcriptional regulation of apolipoprotein A-IV by the transcription factor CREBH. J. Lipid Res. 2014, 55, 850–859. [Google Scholar] [CrossRef]
- Kim, H.; Mendez, R.; Zheng, Z.; Chang, L.; Cai, J.; Zhang, R.; Zhang, K. Liver-enriched transcription factor CREBH interacts with peroxisome proliferator-activated receptor α to regulate metabolic hormone FGF21. Endocrinology 2014, 155, 769–782. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Mendez, R.; Chen, X.; Fang, D.; Zhang, K. Lysine Acetylation of CREBH Regulates Fasting-Induced Hepatic Lipid Metabolism. Mol. Cell. Biol. 2015, 35, 4121–4134. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Reeves, H.L.; Kotsiliti, E.; Govaere, O.; Heikenwalder, M. From NASH to HCC: Current concepts and future challenges. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 411–428. [Google Scholar] [CrossRef] [PubMed]
- McGlynn, K.A.; London, W.T. The global epidemiology of hepatocellular carcinoma: Present and future. Clin. Liver Dis. 2011, 15, 223–243, vii–x. [Google Scholar] [CrossRef] [PubMed]
- Nordenstedt, H.; White, D.L.; El-Serag, H.B. The changing pattern of epidemiology in hepatocellular carcinoma. Digest. Liver Dis. Off. J. Ital. Soc. Gastroenterol. Ital. Assoc. Study Liver 2010, 42 (Suppl. 3), S206–S214. [Google Scholar] [CrossRef]
- Golabi, P.; Fazel, S.; Otgonsuren, M.; Sayiner, M.; Locklear, C.T.; Younossi, Z.M. Mortality assessment of patients with hepatocellular carcinoma according to underlying disease and treatment modalities. Medicine 2017, 96, e5904. [Google Scholar] [CrossRef]
- Colagrande, S.; Inghilesi, A.L.; Aburas, S.; Taliani, G.G.; Nardi, C.; Marra, F. Challenges of advanced hepatocellular carcinoma. World J. Gastroenterol. 2016, 22, 7645–7659. [Google Scholar] [CrossRef] [PubMed]
- Hamed, M.A.; Ali, S.A. Non-viral factors contributing to hepatocellular carcinoma. World J. Hepatol. 2013, 5, 311–322. [Google Scholar] [CrossRef]
- El-Serag, H.B. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology 2012, 142, 1264–1273.e1. [Google Scholar] [CrossRef] [PubMed]
- Petruzziello, A. Epidemiology of Hepatitis B Virus (HBV) and Hepatitis C Virus (HCV) Related Hepatocellular Carcinoma. Open Virol. J. 2018, 12, 26–32. [Google Scholar] [CrossRef]
- Lee, H.W.; Lee, J.S.; Ahn, S.H. Hepatitis B Virus Cure: Targets and Future Therapies. Int. J. Mol. Sci. 2020, 22, 213. [Google Scholar] [CrossRef]
- Kim, S.Y.; Kyaw, Y.Y.; Cheong, J. Functional interaction of endoplasmic reticulum stress and hepatitis B virus in the pathogenesis of liver diseases. World J. Gastroenterol. 2017, 23, 7657–7665. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.C.; Wu, H.C.; Chen, C.F.; Fausto, N.; Lei, H.Y.; Su, I.J. Different types of ground glass hepatocytes in chronic hepatitis B virus infection contain specific pre-S mutants that may induce endoplasmic reticulum stress. Am. J. Pathol. 2003, 163, 2441–2449. [Google Scholar] [CrossRef]
- Wang, C.; Teng, Z.; Zhu, Y.; Zhao, A.Z.; Sun, C. Associations between pre-S deletion mutation of hepatitis B virus and risk of hepatocellular carcinoma in the Asian population: A meta-analysis. Med. Sci. Monitor Int. Med. J. Exp. Clin. Res. 2015, 21, 1072–1077. [Google Scholar]
- Choi, Y.M.; Lee, S.Y.; Kim, B.J. Naturally Occurring Hepatitis B Virus Mutations Leading to Endoplasmic Reticulum Stress and Their Contribution to the Progression of Hepatocellular Carcinoma. Int. J. Mol. Sci. 2019, 20, 597. [Google Scholar] [CrossRef]
- Cho, H.K.; Cheong, K.J.; Kim, H.Y.; Cheong, J. Endoplasmic reticulum stress induced by hepatitis B virus X protein enhances cyclo-oxygenase 2 expression via activating transcription factor 4. Biochem. J. 2011, 435, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Gao, B.; Ye, L.; Han, X.; Wang, W.; Kong, L.; Fang, X.; Zeng, Y.; Zheng, H.; Li, S.; et al. Hepatitis B virus X protein (HBx) activates ATF6 and IRE1-XBP1 pathways of unfolded protein response. Virus Res. 2007, 124, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.K.; Kim, S.Y.; Kyaw, Y.Y.; Win, A.A.; Koo, S.H.; Kim, H.H.; Cheong, J. HBx induces the proliferation of hepatocellular carcinoma cells via AP1 over-expressed as a result of ER stress. Biochem. J. 2015, 466, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Chusri, P.; Kumthip, K.; Hong, J.; Zhu, C.; Duan, X.; Jilg, N.; Fusco, D.N.; Brisac, C.; Schaefer, E.A.; Cai, D.; et al. HCV induces transforming growth factor β1 through activation of endoplasmic reticulum stress and the unfolded protein response. Sci. Rep. 2016, 6, 22487. [Google Scholar] [CrossRef]
- Goto, K.; Roca Suarez, A.A.; Wrensch, F.; Baumert, T.F.; Lupberger, J. Hepatitis C Virus and Hepatocellular Carcinoma: When the Host Loses Its Grip. Int. J. Mol. Sci. 2020, 21, 57. [Google Scholar] [CrossRef]
- Tardif, K.D.; Mori, K.; Kaufman, R.J.; Siddiqui, A. Hepatitis C virus suppresses the IRE1-XBP1 pathway of the unfolded protein response. J. Biol. Chem. 2004, 279, 17158–17164. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Kang, R.; Huang, H.; Xi, X.; Wang, B.; Wang, J.; Zhao, Z. Hepatitis C virus core protein activates autophagy through EIF2AK3 and ATF6 UPR pathway-mediated MAP1LC3B and ATG12 expression. Autophagy 2014, 10, 766–784. [Google Scholar] [CrossRef] [PubMed]
- Chida, T.; Ito, M.; Nakashima, K.; Kanegae, Y.; Aoshima, T.; Takabayashi, S.; Kawata, K.; Nakagawa, Y.; Yamamoto, M.; Shimano, H.; et al. Critical role of CREBH-mediated induction of transforming growth factor β2 by hepatitis C virus infection in fibrogenic responses in hepatic stellate cells. Hepatology 2017, 66, 1430–1443. [Google Scholar] [CrossRef]
- Ji, C.; Mehrian-Shai, R.; Chan, C.; Hsu, Y.H.; Kaplowitz, N. Role of CHOP in hepatic apoptosis in the murine model of intragastric ethanol feeding. Alcohol. Clin. Exp. Res. 2005, 29, 1496–1503. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.; Chen, Y.; Wang, J.; Hao, L.; Huang, C.; Griffiths, A.; Sun, Z.; Zhou, Z.; Song, Z. ER stress-induced upregulation of NNMT contributes to alcohol-related fatty liver development. J. Hepatol. 2020, 73, 783–793. [Google Scholar] [CrossRef]
- Howarth, D.L.; Lindtner, C.; Vacaru, A.M.; Sachidanandam, R.; Tsedensodnom, O.; Vasilkova, T.; Buettner, C.; Sadler, K.C. Activating transcription factor 6 is necessary and sufficient for alcoholic fatty liver disease in zebrafish. PLoS Genet. 2014, 10, e1004335. [Google Scholar] [CrossRef]
- Chanda, D.; Kim, Y.H.; Li, T.; Misra, J.; Kim, D.K.; Kim, J.R.; Kwon, J.; Jeong, W.I.; Ahn, S.H.; Park, T.S.; et al. Hepatic cannabinoid receptor type 1 mediates alcohol-induced regulation of bile acid enzyme genes expression via CREBH. PLoS ONE 2013, 8, e68845. [Google Scholar] [CrossRef]
- Wang, J.M.; Qiu, Y.; Yang, Z.; Kim, H.; Qian, Q.; Sun, Q.; Zhang, C.; Yin, L.; Fang, D.; Back, S.H.; et al. IRE1α prevents hepatic steatosis by processing and promoting the degradation of select microRNAs. Sci. Signal. 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Meng, Q.; Xiao, F.; Chen, S.; Du, Y.; Yu, J.; Wang, C.; Guo, F. ATF4 deficiency protects mice from high-carbohydrate-diet-induced liver steatosis. Biochem. J. 2011, 438, 283–289. [Google Scholar] [CrossRef]
- Wang, C.; Li, H.; Meng, Q.; Du, Y.; Xiao, F.; Zhang, Q.; Yu, J.; Li, K.; Chen, S.; Huang, Z.; et al. ATF4 deficiency protects hepatocytes from oxidative stress via inhibiting CYP2E1 expression. J. Cell. Mol. Med. 2014, 18, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Tam, A.B.; Mercado, E.L.; Hoffmann, A.; Niwa, M. ER stress activates NF-κB by integrating functions of basal IKK activity, IRE1 and PERK. PLoS ONE 2012, 7, e45078. [Google Scholar] [CrossRef]
- Attanasio, S.; Ferriero, R.; Gernoux, G.; De Cegli, R.; Carissimo, A.; Nusco, E.; Campione, S.; Teckman, J.; Mueller, C.; Piccolo, P.; et al. CHOP and c-JUN up-regulate the mutant Z α(1)-antitrypsin, exacerbating its aggregation and liver proteotoxicity. J. Biol. Chem. 2020, 295, 13213–13223. [Google Scholar] [CrossRef] [PubMed]
- Testino, G.; Leone, S.; Borro, P. Alcohol and hepatocellular carcinoma: A review and a point of view. World J. Gastroenterol. 2014, 20, 15943–15954. [Google Scholar] [CrossRef] [PubMed]
- McKillop, I.H.; Schrum, L.W.; Thompson, K.J. Role of alcohol in the development and progression of hepatocellular carcinoma. Hepatic Oncol. 2016, 3, 29–43. [Google Scholar] [CrossRef] [PubMed]
- Morgan, T.R.; Mandayam, S.; Jamal, M.M. Alcohol and hepatocellular carcinoma. Gastroenterology 2004, 127 (Suppl. 1), S87–S96. [Google Scholar] [CrossRef]
- Edenberg, H.J. The genetics of alcohol metabolism: Role of alcohol dehydrogenase and aldehyde dehydrogenase variants. Alcohol. Res. Health J. Natl. Inst. Alcohol. Abuse Alcohol. 2007, 30, 5–13. [Google Scholar]
- Zakhari, S. Overview: How is alcohol metabolized by the body? Alcohol. Res. Health J. Natl. Inst. Alcohol. Abuse Alcohol. 2006, 29, 245–254. [Google Scholar]
- Waris, G.; Ahsan, H. Reactive oxygen species: Role in the development of cancer and various chronic conditions. J. Carcinogenes. 2006, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Nowak, A.J.; Relja, B. The Impact of Acute or Chronic Alcohol Intake on the NF-κB Signaling Pathway in Alcohol-Related Liver Disease. Int. J. Mol. Sci. 2020, 21, 9407. [Google Scholar] [CrossRef]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef] [PubMed]
- Osna, N.A.; Donohue, T.M., Jr.; Kharbanda, K.K. Alcoholic Liver Disease: Pathogenesis and Current Management. Alcohol. Res. Curr. Rev. 2017, 38, 147–161. [Google Scholar]
- Lugea, A.; Tischler, D.; Nguyen, J.; Gong, J.; Gukovsky, I.; French, S.W.; Gorelick, F.S.; Pandol, S.J. Adaptive unfolded protein response attenuates alcohol-induced pancreatic damage. Gastroenterology 2011, 140, 987–997. [Google Scholar] [CrossRef]
- Li, K.; Xiao, Y.; Yu, J.; Xia, T.; Liu, B.; Guo, Y.; Deng, J.; Chen, S.; Wang, C.; Guo, F. Liver-specific Gene Inactivation of the Transcription Factor ATF4 Alleviates Alcoholic Liver Steatosis in Mice. J. Biol. Chem. 2016, 291, 18536–18546. [Google Scholar] [CrossRef] [PubMed]
- Dhamija, E.; Paul, S.B.; Kedia, S. Non-alcoholic fatty liver disease associated with hepatocellular carcinoma: An increasing concern. Ind. J. Med. Res. 2019, 149, 9–17. [Google Scholar]
- Bence, K.K.; Birnbaum, M.J. Metabolic drivers of non-alcoholic fatty liver disease. Mol. Metab. 2020, 101143. [Google Scholar] [CrossRef]
- Berardo, C.; Di Pasqua, L.G.; Cagna, M.; Richelmi, P.; Vairetti, M.; Ferrigno, A. Nonalcoholic Fatty Liver Disease and Non-Alcoholic Steatohepatitis: Current Issues and Future Perspectives in Preclinical and Clinical Research. Int. J. Mol. Sci. 2020, 21, 9646. [Google Scholar] [CrossRef] [PubMed]
- Kanwal, F.; Kramer, J.R.; Mapakshi, S.; Natarajan, Y.; Chayanupatkul, M.; Richardson, P.A.; Li, L.; Desiderio, R.; Thrift, A.P.; Asch, S.M.; et al. Risk of Hepatocellular Cancer in Patients With Non-Alcoholic Fatty Liver Disease. Gastroenterology 2018, 155, 1828–1837.e2. [Google Scholar] [CrossRef]
- Henkel, A.; Green, R.M. The unfolded protein response in fatty liver disease. Semin. Liver Dis. 2013, 33, 321–329. [Google Scholar] [PubMed]
- Liu, X.; Henkel, A.S.; LeCuyer, B.E.; Schipma, M.J.; Anderson, K.A.; Green, R.M. Hepatocyte X-box binding protein 1 deficiency increases liver injury in mice fed a high-fat/sugar diet. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G965–G974. [Google Scholar] [CrossRef] [PubMed]
- Farrell, G.C.; van Rooyen, D.; Gan, L.; Chitturi, S. NASH is an Inflammatory Disorder: Pathogenic, Prognostic and Therapeutic Implications. Gut Liver 2012, 6, 149–171. [Google Scholar] [CrossRef]
- Luedde, T.; Schwabe, R.F. NF-κB in the liver--linking injury, fibrosis and hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 108–118. [Google Scholar] [CrossRef]
- He, G.; Yu, G.Y.; Temkin, V.; Ogata, H.; Kuntzen, C.; Sakurai, T.; Sieghart, W.; Peck-Radosavljevic, M.; Leffert, H.L.; Karin, M. Hepatocyte IKKbeta/NF-kappaB inhibits tumor promotion and progression by preventing oxidative stress-driven STAT3 activation. Cancer Cell 2010, 17, 286–297. [Google Scholar] [CrossRef] [PubMed]
- Luedde, T.; Beraza, N.; Kotsikoris, V.; van Loo, G.; Nenci, A.; De Vos, R.; Roskams, T.; Trautwein, C.; Pasparakis, M. Deletion of NEMO/IKKgamma in liver parenchymal cells causes steatohepatitis and hepatocellular carcinoma. Cancer Cell 2007, 11, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, Y.; Oikawa, F.; Mizuno, S.; Ohno, H.; Yagishita, Y.; Satoh, A.; Osaki, Y.; Takei, K.; Kikuchi, T.; Han, S.I.; et al. Hyperlipidemia and hepatitis in liver-specific CREB3L3 knockout mice generated using a one-step CRISPR/Cas9 system. Sci. Rep. 2016, 6, 27857. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, E.L.; Khan, Z. Liver Disease in Alpha-1 Antitrypsin Deficiency: Current Approaches and Future Directions. Curr. Pathobiol. Rep. 2017, 5, 243–252. [Google Scholar] [CrossRef]
- Feldman, A.; Sokol, R.J. Alpha-1-Antitrypsin Deficiency: An Important Cause of Pediatric Liver Disease. Lung Health Profess. Magaz. 2013, 4, 8–11. [Google Scholar]
- Blanco, I.; Bueno, P.; Diego, I.; Pérez-Holanda, S.; Lara, B.; Casas-Maldonado, F.; Esquinas, C.; Miravitlles, M. Alpha-1 antitrypsin Pi*SZ genotype: Estimated prevalence and number of SZ subjects worldwide. Int. J. Chronic Obstruct. Pulm. Dis. 2017, 12, 1683–1694. [Google Scholar] [CrossRef]
- Tanash, H.A.; Piitulainen, E. Liver disease in adults with severe alpha-1-antitrypsin deficiency. J. Gastroenterol. 2019, 54, 541–548. [Google Scholar] [CrossRef]
- Strnad, P.; Buch, S.; Hamesch, K.; Fischer, J.; Rosendahl, J.; Schmelz, R.; Brueckner, S.; Brosch, M.; Heimes, C.V.; Woditsch, V.; et al. Heterozygous carriage of the alpha1-antitrypsin Pi*Z variant increases the risk to develop liver cirrhosis. Gut 2019, 68, 1099–1107. [Google Scholar] [CrossRef]
- Berg, N.O.; Eriksson, S. Liver disease in adults with alpha-1 -antitrypsin deficiency. N. Engl. J. Med. 1972, 287, 1264–1267. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, S. Alpha 1-antitrypsin deficiency and liver cirrhosis in adults. An analysis of 35 Swedish autopsied cases. Acta Med. Scand. 1987, 221, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Topic, A.; Ljujic, M.; Radojkovic, D. Alpha-1-antitrypsin in pathogenesis of hepatocellular carcinoma. Hepat. Mon. 2012, 12, e7042. [Google Scholar] [CrossRef] [PubMed]
- Perlmutter, D.H. α1-antitrypsin Deficiency: A Misfolded Secretory Protein Variant with Unique Effects on the Endoplasmic Reticulum. Endoplasmic Reticulum Stress Dis. 2016, 3, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Hidvegi, T.; Schmidt, B.Z.; Hale, P.; Perlmutter, D.H. Accumulation of mutant alpha1-antitrypsin Z in the endoplasmic reticulum activates caspases-4 and -12, NFkappaB, and BAP31 but not the unfolded protein response. J. Biol. Chem. 2005, 280, 39002–39015. [Google Scholar] [CrossRef] [PubMed]
- Ordóñez, A.; Snapp, E.L.; Tan, L.; Miranda, E.; Marciniak, S.J.; Lomas, D.A. Endoplasmic reticulum polymers impair luminal protein mobility and sensitize to cellular stress in alpha1-antitrypsin deficiency. Hepatology 2013, 57, 2049–2060. [Google Scholar] [CrossRef] [PubMed]
- Ponzetto, A.; Perez-Perez, G.I.; Figura, N. Alpha1-antitrypsin deficiency and c-JUN. Hepatology 2017, 66, 677. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Garcia-Carbonell, R.; Yamachika, S.; Zhao, P.; Dhar, D.; Loomba, R.; Kaufman, R.J.; Saltiel, A.R.; Karin, M. ER Stress Drives Lipogenesis and Steatohepatitis via Caspase-2 Activation of S1P. Cell 2018, 175, 133–145.e15. [Google Scholar] [CrossRef]
- Pavlović, N.; Calitz, C.; Thanapirom, K.; Mazza, G.; Rombouts, K.; Gerwins, P.; Heindryckx, F. Inhibiting IRE1α-endonuclease activity decreases tumor burden in a mouse model for hepatocellular carcinoma. eLife 2020, 9. [Google Scholar] [CrossRef]
- Wu, S.; Du, R.; Gao, C.; Kang, J.; Wen, J.; Sun, T. The role of XBP1s in the metastasis and prognosis of hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2018, 500, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Vandewynckel, Y.P.; Laukens, D.; Bogaerts, E.; Paridaens, A.; Van den Bussche, A.; Verhelst, X.; Van Steenkiste, C.; Descamps, B.; Vanhove, C.; Libbrecht, L.; et al. Modulation of the unfolded protein response impedes tumor cell adaptation to proteotoxic stress: A PERK for hepatocellular carcinoma therapy. Hepatol. Int. 2015, 9, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Lu, Q.; Liu, J.; Fan, L.; Wang, Y.; Wei, W.; Wang, H.; Sun, G. Melatonin Increases the Sensitivity of Hepatocellular Carcinoma to Sorafenib through the PERK-ATF4-Beclin1 Pathway. Int. J. Biol. Sci. 2019, 15, 1905–1920. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Umemura, A.; Taniguchi, K.; Font-Burgada, J.; Dhar, D.; Ogata, H.; Zhong, Z.; Valasek, M.A.; Seki, E.; Hidalgo, J.; et al. ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell 2014, 26, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Pafili, K.; Roden, M. Nonalcoholic fatty liver disease (NAFLD) from pathogenesis to treatment concepts in humans. Mol. Metab. 2020, 101122. [Google Scholar] [CrossRef] [PubMed]
- Alqahtani, A.; Khan, Z.; Alloghbi, A.; Said Ahmed, T.S.; Ashraf, M.; Hammouda, D.M. Hepatocellular Carcinoma: Molecular Mechanisms and Targeted Therapies. Medicina 2019, 55, 526. [Google Scholar] [CrossRef]
- Ringelhan, M.; Pfister, D.; O’Connor, T.; Pikarsky, E.; Heikenwalder, M. The immunology of hepatocellular carcinoma. Nat. Immunol. 2018, 19, 222–232. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Risk Factor | UPR Pathway Activation | Cellular and Molecular Mechanisms of Liver Injury | Reference |
|---|---|---|---|
| Hepatitis B virus | PERK-ATF4 | Oxidative stress and apoptosis | [97] |
| ATF6 cleavage | Proliferation of hepatocellular carcinoma cells | [98,99] | |
| IRE1α-XBP1s | |||
| Hepatitis C virus | IRE1α-XBP1s | Misfolded proteins are more stable | [103] |
| IRE1α-JNK | TGFβ1 expression and proliferation | [101,105] | |
| CREBH cleavage | |||
| PERK pathway | Autophagy induction | [104] | |
| ATF6 cleavage | |||
| Alcohol | PERK-ATF4-CHOP | Apoptosis | [106] |
| NNMT expression | [107] | ||
| ATF6 cleavage | Lipogenesis and steatosis | [108] | |
| CREBH cleavage | Perturbation of bile acid homeostasis | [109] | |
| fatty liver (NAFLD/NASH) | IRE1α-XBP1s | Hepatic steatosis and insulin resistance | [110] |
| miRNA expression and liver injury | |||
| PERK-ATF4 | SCD1 and CYP2E1 expression; liver steatosis | [111,112] | |
| ATF6 cleavage | Hepatic steatosis and insulin resistance | [74] | |
| CREBH cleavage | Nonalcoholic steatohepatitis | [80] | |
| IREα—JNKs and NF-κB | Hepatic injury and fibrosis | [113] | |
| Alpha1-antitrypsin deficiency (AATD) | CHOP | Upregulates A1AT expression and apoptosis | [114] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, J.; Fang, D. Endoplasmic Reticulum Stress Signaling and the Pathogenesis of Hepatocarcinoma. Int. J. Mol. Sci. 2021, 22, 1799. https://doi.org/10.3390/ijms22041799
Wei J, Fang D. Endoplasmic Reticulum Stress Signaling and the Pathogenesis of Hepatocarcinoma. International Journal of Molecular Sciences. 2021; 22(4):1799. https://doi.org/10.3390/ijms22041799
Chicago/Turabian StyleWei, Juncheng, and Deyu Fang. 2021. "Endoplasmic Reticulum Stress Signaling and the Pathogenesis of Hepatocarcinoma" International Journal of Molecular Sciences 22, no. 4: 1799. https://doi.org/10.3390/ijms22041799
APA StyleWei, J., & Fang, D. (2021). Endoplasmic Reticulum Stress Signaling and the Pathogenesis of Hepatocarcinoma. International Journal of Molecular Sciences, 22(4), 1799. https://doi.org/10.3390/ijms22041799