
Understanding Viral Infection Mechanisms and Patient Symptoms for the Development of COVID-19 Therapeutics

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Understanding of SARS-CoV-2 and COVID-19
2.1. Classification of SARS-CoV-2
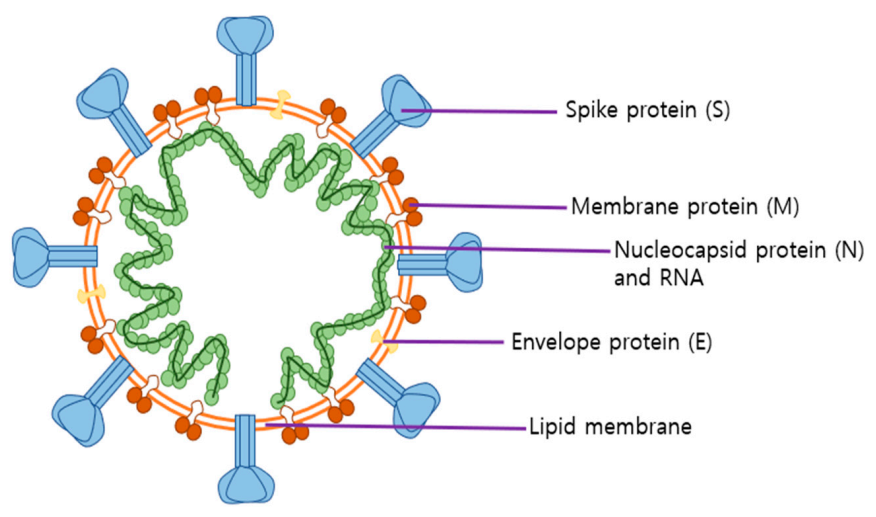
2.2. Roles of SARS-CoV-2 Structural Proteins in Infection
2.3. SARS-CoV-2 Life Cycle and Its Infected Organs
2.4. Role of the Spike Protein and ACE2 in SARS-CoV-2 Entry
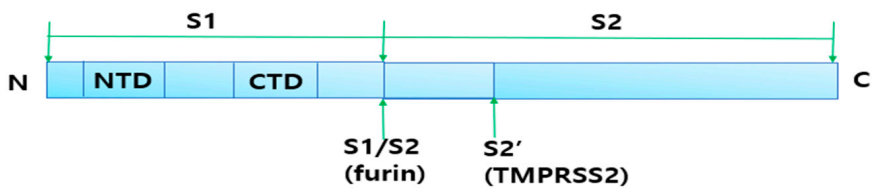
2.4.1. Cleavage of Spike Protein for Its Attachment to ACE2
2.4.2. Role of ACE2 in the Renin-Angiotensin System (RAS)
3. COVID-19 Pathogenesis through Inflammation-Mediated Tissue Damage
3.1. SARS-CoV-2-Induced Inflammation and Cytokine Storm
3.2. Symptoms Seen in COVID-19 Patients
3.3. COVID-19-Induced Tissue Damage and Clinical Presentations
4. Treatment Strategy According to COVID-19 Symptoms
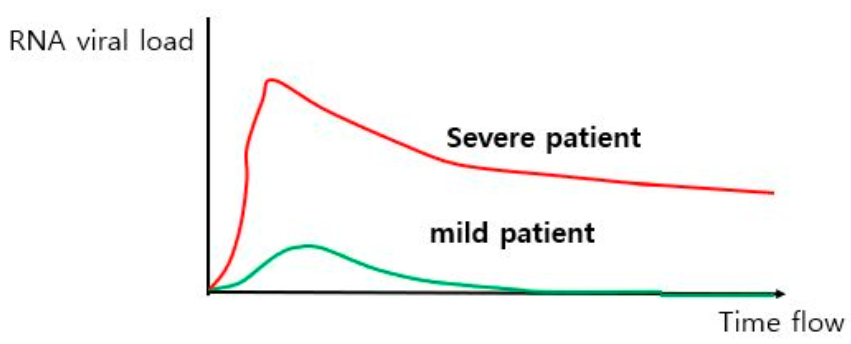
4.1. Differencse in Viral Load between Mild and Severe COVID-19 Patients
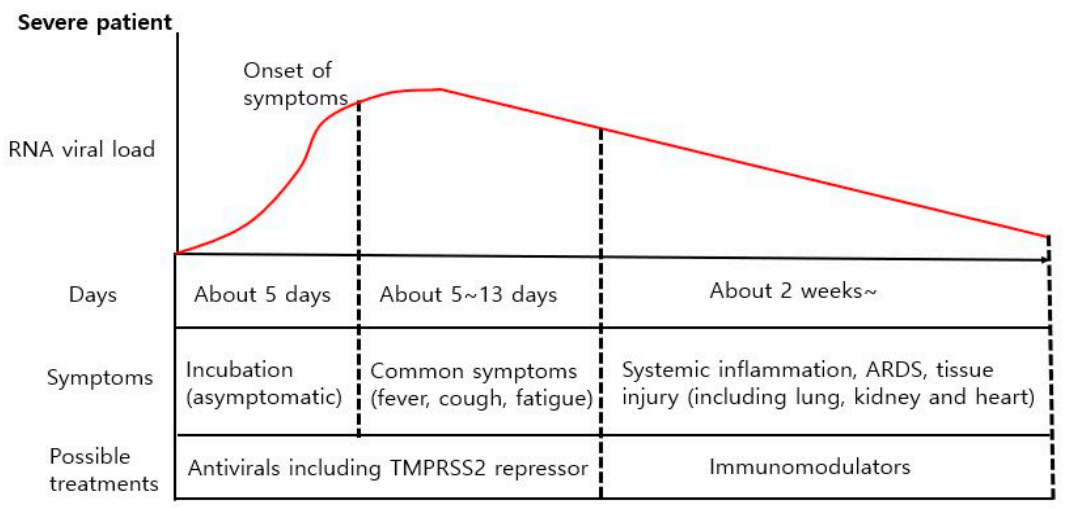
4.2. Different Treatment Strategies for Different COVID-19 Symptoms
5. Current Therapeutics for Treatment of COVID-19
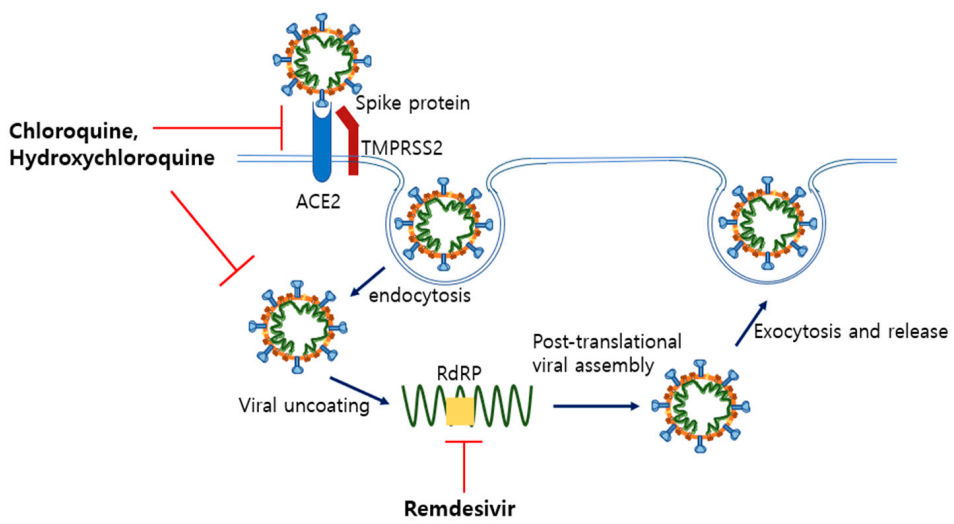
5.1. Remdesivir and Hydroxychloroquine as Anti-Viral Drugs
5.1.1. Remdesivir
5.1.2. Chloroquine and Hydroxychloroquine
5.2. Glucocorticoids (Dexamethasone) as an Anti-Inflammatory Drug
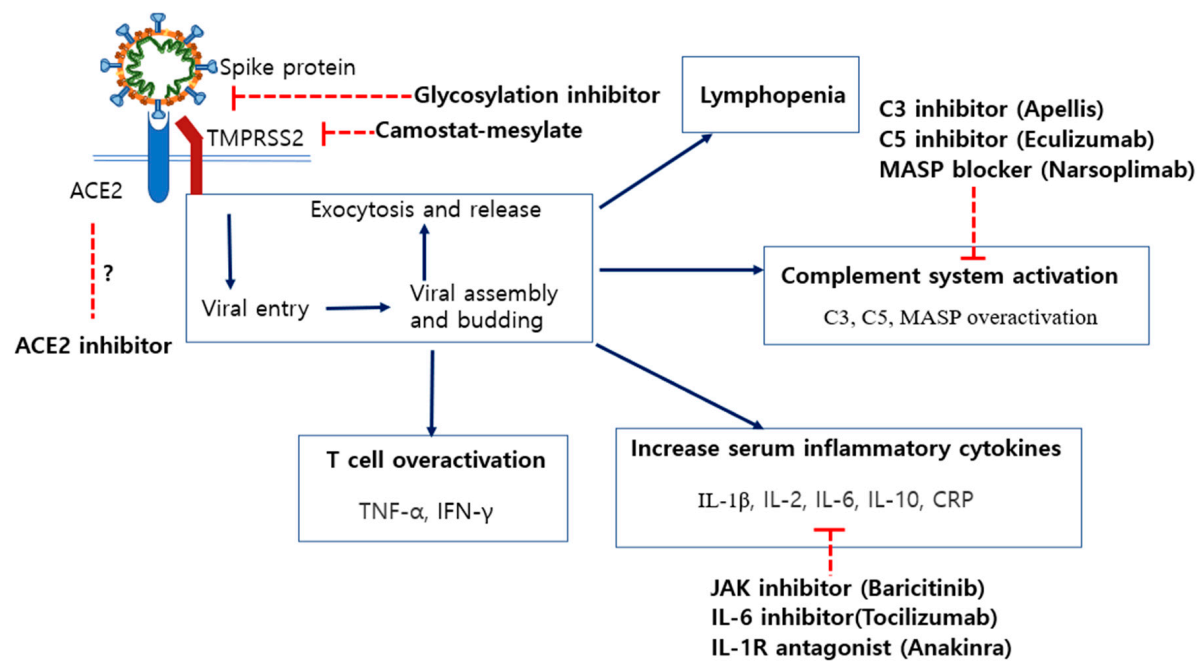
6. Potential Therapeutics for COVID-19 Treatments and Their Disadvantages
6.1. Immunosuppressants for Treatment of Arthritic Diseases
6.1.1. Anakinra, a Recombinant IL-1 Receptor Antagonist Protein
6.1.2. Tocilizumab, a Recombinant Humanized Anti-IL-6 Receptor Antibody
6.1.3. Baricitinib, a Small Molecule That Inhibits Selective Janus Kinase (JAK) 1 and 2
6.2. Inhibition of Specific N-Linked Glycosylation in the S1 Protein
6.3. Modulation of ACE2 Expression by ACE Inhibitors (ACEi) and Angiotensin II Receptor Blockers (ARB)
6.4. Blocking of Spike Protein Cleavage by Inhibiting the Activity of TMPRSS2
6.5. Inhibitors of C3 and C5 Proteins in the Complement System
6.6. Neutrophil Extracellular Trap (NET)-Associated Therapeutics
6.7. Ivermectin
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| ACE | Angiotensin-converting enzyme |
| ARB | Angiotensin II receptor blockers |
| ARDS | Acute respiratory distress syndrome |
| ALI | Acute lung injury |
| ICU | Intensive care unit |
| IL | Interleukin |
| TMPRSS | Transmembrane protease serine subtype |
| RBD | Receptor-binding domain |
| RAS | Renin-angiotensin system |
References
- Pascarella, G.; Strumia, A.; Piliego, C.; Bruno, F.; Del Buono, R.; Costa, F.; Scarlata, S.; Agrò, F.E. COVID-19 diagnosis and management: A comprehensive review. J. Intern. Med. 2020, 288, 192–206. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Le Thanh, T.; Andreadakis, Z.; Kumar, A.; Román, R.G.; Tollefsen, S.; Saville, M.; Mayhew, S. The COVID-19 vaccine development landscape. Nat. Rev. Drug Discov. 2020, 19, 305–306. [Google Scholar] [CrossRef] [PubMed]
- Dotolo, S.; Marabotti, A.; Facchiano, A.; Tagliaferri, R. A review on drug repurposing applicable to COVID-19. Brief. Bioinform. 2020. [Google Scholar] [CrossRef] [PubMed]
- Tanoli, Z.; Seemab, U.; Scherer, A.; Wennerberg, K.; Tang, J.; Vähä-Koskela, M. Exploration of databases and methods supporting drug repurposing: A comprehensive survey. Brief. Bioinform. 2020. [Google Scholar] [CrossRef]
- Wilkinson, G.F.; Pritchard, K. In vitro screening for drug repositioning. J. Biomol. Screen. 2015, 20, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Gorbalenya, A.E.; Baker, S.C.; Baric, R.S.; de Groot, R.J.; Drosten, C.; Gulyaeva, A.A.; Haagmans, B.L.; Lauber, C.; Leontovich, A.M.; Neuman, B.W.; et al. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar]
- Ren, L.L.; Wang, Y.M.; Wu, Z.Q.; Xiang, Z.C.; Guo, L.; Xu, T.; Jiang, Y.Z.; Xiong, Y.; Li, Y.J.; Li, X.W.; et al. Identification of a novel coronavirus causing severe pneumonia in human: A descriptive study. Chin. Med. J. Engl. 2020, 133, 1015–1024. [Google Scholar] [CrossRef]
- Wu, D.; Wu, T.; Liu, Q.; Yang, Z. The SARS-CoV-2 outbreak: What we know. Int. J. Infect. Dis. 2020, 94, 44–48. [Google Scholar] [CrossRef]
- Wei, X.; Li, X.; Cui, J. Evolutionary perspectives on novel coronaviruses identified in pneumonia cases in China. Natl. Sci. Rev. 2020, 7, 239–242. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827. [Google Scholar] [CrossRef]
- Hu, J.; He, C.L.; Gao, Q.; Zhang, G.J.; Cao, X.X.; Long, Q.X.; Deng, H.J.; Huang, L.Y.; Chen, J.; Wang, K.; et al. The D614G mutation of SARS-CoV-2 spike protein enhances viral infectivity and decreases neutralization sensitivity to individual convalescent sera. bioRxiv 2020. [Google Scholar] [CrossRef]
- Plante, J.A.; Liu, Y.; Liu, J.; Xia, H.; Johnson, B.A.; Lokugamage, K.G.; Zhang, X.; Muruato, A.E.; Zou, J.; Fontes-Garfias, C.R.; et al. Spike mutation D614G alters SARS-CoV-2 fitness. Nature 2020, 1–6. [Google Scholar] [CrossRef]
- Weissman, D.; Alameh, M.-G.; de Silva, T.; Collini, P.; Hornsby, H.; Brown, R.; LaBranche, C.C.; Edwards, R.J.; Sutherland, L.; Santra, S.; et al. D614G Spike Mutation Increases SARS CoV-2 Susceptibility to Neutralization. Cell Host Microbe 2021, 29, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Stohlman, S.A.; Baric, R.S.; Nelson, G.N.; Soe, L.H.; Welter, L.M.; Deans, R.J. Specific interaction between coronavirus leader RNA and nucleocapsid protein. J. Virol. 1988, 62, 4288–4295. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Wang, H.; Ji, Y.; Yang, J.; Xu, S.; Huang, X.; Wang, Z.; Qin, L.; Tien, P.; Zhou, X.; et al. The Nucleocapsid Protein of Coronaviruses Acts as a Viral Suppressor of RNA Silencing in Mammalian Cells. J. Virol. 2015, 89, 9029–9043. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Liu, J.; Wang, Q.; Liu, X.; Li, X.; Li, P.; Ma, Q.; Cao, C. The Nucleocapsid Protein of Severe Acute Respiratory Syndrome Coronavirus Inhibits Cell Cytokinesis and Proliferation by Interacting with Translation Elongation Factor 1α. J. Virol. 2008, 82, 6962–6971. [Google Scholar] [CrossRef]
- Nal, B.; Chan, C.; Kien, F.; Siu, L.; Tse, J.; Chu, K.; Kam, J.; Staropoli, S.; Crescenzo-Chaigne, B.; Escriou, N.; et al. Differential maturation and subcellular localization of severe acute respiratory syndrome coronavirus surface proteins S, M and E. J. Gen. Virol. 2005, 86, 1423–1434. [Google Scholar] [CrossRef]
- Schoeman, D.; Fielding, B.C. Coronavirus envelope protein: Current knowledge. Virol. J. 2019, 16, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Alimohamadi, Y.; Taghdir, M.; Sepandi, M. Estimate of the basic reproduction number for COVID-19: A systematic review and meta-Analysis. J. Prev. Med. Public Health 2020, 288, 192–206. [Google Scholar] [CrossRef]
- Liu, Y.; Gayle, A.A.; Wilder-Smith, A.; Rocklöv, J. The reproductive number of COVID-19 is higher compared to SARS coronavirus. J. Travel Med. 2020, 27, 27. [Google Scholar] [CrossRef]
- CDC. Duration of Isolation and Precautions for Adults with COVID-19. Available online: https://www.cdc.gov/coronavirus/2019-ncov/hcp/duration-isolation.html (accessed on 6 February 2021).
- Xiao, F.; Tang, M.; Zheng, X.; Liu, Y.; Li, X.; Shan, H. Evidence for Gastrointestinal Infection of SARS-CoV-2. Gastroenterology 2020, 158, 1831–1833. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Chen, K.; Zou, J.; Han, P.; Hao, J.; Han, Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front. Med. 2020, 14, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Yang, H.; Ji, W.; Wu, W.; Chen, S.; Zhang, W.; Duan, G. Virology, epidemiology, pathogenesis, and control of covid-19. Viruses 2020, 12, 372. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Allen, J.D.; Wrapp, D.; McLellan, J.S.; Crispin, M. Site-specific glycan analysis of the SARS-CoV-2 spike. Science 2020, 369, 330–333. [Google Scholar] [CrossRef]
- Li, W.; Moore, M.J.; Vasllieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greeneugh, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Rabaan, A.A.; Al-Ahmed, S.H.; Haque, S.; Sah, R.; Tiwari, R.; Malik, Y.S.; Dhama, K.; Yatoo, M.I.; Bonilla-Aldana, D.K.; Rodriguez-Morales, A.J. SARS-CoV-2, SARS-CoV, and MERS-COV: A comparative overview. Infez. Med. 2020, 28, 174–184. [Google Scholar]
- Mehdipour, A.R.; Hummer, G. Dual nature of human ACE2 glycosylation in binding to SARS-CoV-2 spike. BioRxiv 2020. [Google Scholar] [CrossRef]
- Glowacka, I.; Bertram, S.; Herzog, P.; Pfefferle, S.; Steffen, I.; Muench, M.O.; Simmons, G.; Hofmann, H.; Kuri, T.; Weber, F.; et al. Differential Downregulation of ACE2 by the Spike Proteins of Severe Acute Respiratory Syndrome Coronavirus and Human Coronavirus NL63. J. Virol. 2010, 84, 1198–1205. [Google Scholar] [CrossRef]
- Silva, A.C.S.; Teixeira, M.M. ACE inhibition, ACE2 and angiotensin-(1-7) axis in kidney and cardiac inflammation and fibrosis. Pharmacol. Res. 2016, 107, 154–162. [Google Scholar] [CrossRef]
- Jia, H. Pulmonary Angiotensin-Converting Enzyme 2 (ACE2) and Inflammatory Lung Disease. Shock 2016, 46, 239–248. [Google Scholar] [CrossRef]
- Wösten-Van Asperen, R.M.; Lutter, R.; Specht, P.A.; Moll, G.N.; Van Woensel, J.B.; Van Der Loos, C.M.; Van Goor, H.; Kamilic, J.; Florquin, S.; Bos, A.P. Acute respiratory distress syndrome leads to reduced ratio of ACE/ACE2 activities and is prevented by angiotensin-(1-7) or an angiotensin II receptor antagonist. J. Pathol. 2011, 225, 618–627. [Google Scholar] [CrossRef] [PubMed]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Sun, J.; Dai, Z.; Deng, H.; Li, X.; Huang, Q.; Wu, Y.; Sun, L.; Xu, Y. Prevalence and severity of corona virus disease 2019 (COVID-19): A systematic review and meta-analysis. J. Clin. Virol. 2020, 127, 104371. [Google Scholar] [CrossRef]
- Yang, X.; Yu, Y.; Xu, J.; Shu, H.; Xia, J.; Liu, H.; Wu, Y.; Zhang, L.; Yu, Z.; Fang, M.; et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: A single-centered, retrospective, observational study. Lancet Respir. Med. 2020, 8, 475–481. [Google Scholar] [CrossRef]
- Zhang, J.; Dong, X.; Cao, Y.; Yuan, Y.D.; Yang, Y.B.; Yan, Y.Q.; Akdis, C.A.; Gao, Y.D. Clinical characteristics of 140 patients infected with SARS-CoV-2 in Wuhan, China. Allergy Eur. J. Allergy Clin. Immunol. 2020, 75, 1730–1741. [Google Scholar] [CrossRef]
- Rico-Mesa, J.S.; White, A.; Anderson, A.S. Outcomes in Patients with COVID-19 Infection Taking ACEI/ARB. Curr. Cardiol. Rep. 2020, 22, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Haimei, M.A. Pathogenesis and Treatment Strategies of COVID-19-Related Hypercoagulant and Thrombotic Complications. Clin. Appl. Thromb. 2020, 26, 1076029620944497. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Z.; Li, C.; Chen, Y.; Gerotziafas, G.; Zhang, Z.; Wan, J.; Liu, P.; Elalamy, I.; Wang, C. Prevention and Treatment of Venous Thromboembolism Associated with Coronavirus Disease 2019 Infection: A Consensus Statement before Guidelines. Thromb. Haemost. 2020, 120, 937. [Google Scholar] [CrossRef]
- Klok, F.A.; Kruip, M.J.H.A.; van der Meer, N.J.M.; Arbous, M.S.; Gommers, D.A.M.P.J.; Kant, K.M.; Kaptein, F.H.J.; van Paassen, J.; Stals, M.A.M.; Huisman, M.V.; et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb. Res. 2020, 191, 145–147. [Google Scholar] [CrossRef] [PubMed]
- NIH. Coronavirus Disease 2019 (COVID-19) Treatment Guidelines. Available online: https://covid19treatmentguidelines.nih.gov/ (accessed on 6 February 2021).
- Henry, B.M.; De Oliveira, M.H.S.; Benoit, S.; Plebani, M.; Lippi, G. Hematologic, biochemical and immune biomarker abnormalities associated with severe illness and mortality in coronavirus disease 2019 (COVID-19): A meta-analysis. Clin. Chem. Lab. Med. 2020, 58, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Cheng, Y.; Wu, Y. Understanding SARS-CoV-2-Mediated Inflammatory Responses: From Mechanisms to Potential Therapeutic Tools. Virol. Sin. 2020, 35, 266–271. [Google Scholar] [CrossRef]
- Li, M.Y.; Li, L.; Zhang, Y.; Wang, X.S. Expression of the SARS-CoV-2 cell receptor gene ACE2 in a wide variety of human tissues. Infect. Dis. Poverty 2020, 9, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Gong, E.; Zhang, B.; Zheng, J.; Gao, Z.; Zhong, Y.; Zou, W.; Zhan, J.; Wang, S.; Xie, Z.; et al. Multiple organ infection and the pathogenesis of SARS. J. Exp. Med. 2005, 202, 415–424. [Google Scholar] [CrossRef]
- Chen, I.Y.; Moriyama, M.; Chang, M.F.; Ichinohe, T. Severe acute respiratory syndrome coronavirus viroporin 3a activates the NLRP3 inflammasome. Front. Microbiol. 2019, 10, 50. [Google Scholar] [CrossRef] [PubMed]
- Yang, M. Cell Pyroptosis, a Potential Pathogenic Mechanism of 2019-nCoV Infection. SSRN Electron. J. 2020, 20, 363–374. [Google Scholar] [CrossRef]
- Tay, M.Z.; Poh, C.M.; Rénia, L.; MacAry, P.A.; Ng, L.F.P. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef]
- Ye, Q.; Wang, B.; Mao, J. Cytokine Storm in COVID-19 and Treatment. J. Infect. 2020, 80, 607–613. [Google Scholar] [CrossRef]
- Li, L.Q.; Huang, T.; Wang, Y.Q.; Wang, Z.P.; Liang, Y.; Huang, T.B.; Zhang, H.Y.; Sun, W.; Wang, Y. COVID-19 patients’ clinical characteristics, discharge rate, and fatality rate of meta-analysis. J. Med. Virol. 2020, 92, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Zisman, L.S. ACE and ACE2: A tale of two enzymes. Eur. Heart J. 2005, 26, 322–324. [Google Scholar] [CrossRef]
- Crackower, M.A.; Sarao, R.; Oliveira-dos-Santos, A.J.; Da Costa, J.; Zhang, L. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature 2002, 417, 822–828. [Google Scholar] [CrossRef]
- Hashimoto, T.; Perlot, T.; Rehman, A.; Trichereau, J.; Ishiguro, H.; Paolino, M.; Sigl, V.; Hanada, T.; Hanada, R.; Lipinski, S.; et al. ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature 2012, 487, 477–481. [Google Scholar] [CrossRef]
- Li, G.; Fan, Y.; Lai, Y.; Han, T.; Li, Z.; Zhou, P.; Pan, P.; Wang, W.; Hu, D.; Liu, X.; et al. Coronavirus infections and immune responses. J. Med. Virol. 2020, 92, 424–432. [Google Scholar] [CrossRef]
- Soy, M.; Keser, G.; Atagündüz, P.; Tabak, F.; Atagündüz, I.; Kayhan, S. Cytokine storm in COVID-19: Pathogenesis and overview of anti-inflammatory agents used in treatment. Clin. Rheumatol. 2020, 39, 2085–2094. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Shi, L.; Wang, Y.; Zhang, J.; Huang, L.; Zhang, C.; Liu, S.; Zhao, P.; Liu, H.; Zhu, L.; et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020, 8, 420–422. [Google Scholar] [CrossRef]
- Madabhavi, I.; Sarkar, M.; Kadakol, N. CoviD-19: A review. Monaldi Arch. Chest Dis. 2020, 90, 248–258. [Google Scholar] [CrossRef]
- Song, P.; Li, W.; Xie, J.; Hou, Y.; You, C. Cytokine storm induced by SARS-CoV-2. Clin. Chim. Acta 2020, 509, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Lian, X.; Su, X.; Wu, W.; Marraro, G.A.; Zeng, Y. From SARS and MERS to COVID-19: A brief summary and comparison of severe acute respiratory infections caused by three highly pathogenic human coronaviruses. Respir. Res. 2020, 21, 1–14. [Google Scholar] [CrossRef]
- WHO. Novel Coronavirus (2019-nCoV) Situation Report-3; WHO: Geneva, Switzerland, 2020. [Google Scholar]
- Baj, J.; Karakuła-Juchnowicz, H.; Teresiński, G.; Buszewicz, G.; Ciesielka, M.; Sitarz, E.; Forma, A.; Karakuła, K.; Flieger, W.; Portincasa, P.; et al. COVID-19: Specific and Non-Specific Clinical Manifestations and Symptoms: The Current State of Knowledge. J. Clin. Med. 2020, 9, 1753. [Google Scholar] [CrossRef] [PubMed]
- Larsen, J.R.; Martin, M.R.; Martin, J.D.; Kuhn, P.; Hicks, J.B. Modeling the Onset of Symptoms of COVID-19. Front. Public Health 2020, 8, 473. [Google Scholar] [CrossRef]
- Del Rio, C.; Collins, L.F.; Malani, P. Long-term Health Consequences of COVID-19. J. Am. Med. Assoc. 2020, 324, 1723. [Google Scholar] [CrossRef]
- Halpin, S.J.; McIvor, C.; Whyatt, G.; Adams, A.; Harvey, O.; McLean, L.; Walshaw, C.; Kemp, S.; Corrado, J.; Singh, R.; et al. Postdischarge symptoms and rehabilitation needs in survivors of COVID-19 infection: A cross-sectional evaluation. J. Med. Virol. 2020, 93, 1013–1022. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Media Statement Knowing the Risks for COVID-19. Available online: http://www.who.int/indonesia/news/detail/08-03-2020-knowing-the-risk-for-covid-19#/ (accessed on 6 February 2021).
- Rodriguez-Morales, A.J.; Cardona-Ospina, J.A.; Gutiérrez-Ocampo, E.; Villamizar-Peña, R.; Holguin-Rivera, Y.; Escalera-Antezana, J.P.; Alvarado-Arnez, L.E.; Bonilla-Aldana, D.K.; Franco-Paredes, C.; Henao-Martinez, A.F.; et al. Clinical, laboratory and imaging features of COVID-19: A systematic review and meta-analysis. Travel Med. Infect. Dis. 2020, 34, 101623. [Google Scholar] [CrossRef]
- Lee, J.Y.; Hong, S.W.; Hyun, M.; Park, J.S.; Lee, J.H.; Suh, Y.S.; Kim, D.H.; Han, S.W.; Cho, C.H.; Kim, H.A. Epidemiological and clinical characteristics of coronavirus disease 2019 in Daegu, South Korea. Int. J. Infect. Dis. 2020, 98, 462–466. [Google Scholar] [CrossRef]
- Verity, R.; Okell, L.C.; Dorigatti, I.; Winskill, P.; Whittaker, C.; Imai, N.; Cuomo-Dannenburg, G.; Thompson, H.; Walker, P.G.T.; Fu, H.; et al. Estimates of the severity of coronavirus disease 2019: A model-based analysis. Lancet Infect. Dis. 2020, 20, 669–677. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. People Who Are at Higher Risk for Severe Illness; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2020.
- Lippi, G.; Simundic, A.M.; Plebani, M. Potential preanalytical and analytical vulnerabilities in the laboratory diagnosis of coronavirus disease 2019 (COVID-19). Clin. Chem. Lab. Med. 2020, 58, 1070–1076. [Google Scholar] [CrossRef]
- Li, X.; Ma, X. Acute respiratory failure in COVID-19: Is it “typical” ARDS? Crit. Care 2020, 24, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef]
- Batah, S.S.; Fabro, A.T. Pulmonary pathology of ARDS in COVID-19: A pathological review for clinicians. Respir. Med. 2021, 176, 106239. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Gupta, A.; Madhavan, M.V.; Sehgal, K.; Nair, N.; Mahajan, S.; Sehrawat, T.S.; Bikdeli, B.; Ahluwalia, N.; Ausiello, J.C.; Wan, E.Y.; et al. Extrapulmonary manifestations of COVID-19. Nat. Med. 2020, 26, 1017–1032. [Google Scholar] [CrossRef] [PubMed]
- Tavazzi, G.; Pellegrini, C.; Maurelli, M.; Belliato, M.; Sciutti, F.; Bottazzi, A.; Sepe, P.A.; Resasco, T.; Camporotondo, R.; Bruno, R.; et al. Myocardial localization of coronavirus in COVID-19 cardiogenic shock. Eur. J. Heart Fail. 2020, 22, 911–915. [Google Scholar] [CrossRef] [PubMed]
- Lindner, D.; Fitzek, A.; Bräuninger, H.; Aleshcheva, G.; Edler, C.; Meissner, K.; Scherschel, K.; Kirchhof, P.; Escher, F.; Schultheiss, H.P.; et al. Association of Cardiac Infection with SARS-CoV-2 in Confirmed COVID-19 Autopsy Cases. JAMA Cardiol. 2020, 5, 1281–1285. [Google Scholar] [CrossRef]
- Walters, T.E.; Kalman, J.M.; Patel, S.K.; Mearns, M.; Velkoska, E.; Burrell, L.M. Angiotensin converting enzyme 2 activity and human atrial fibrillation: Increased plasma angiotensin converting enzyme 2 activity is associated with atrial fibrillation and more advanced left atrial structural remodelling. Europace 2017, 19, 1280–1287. [Google Scholar] [CrossRef] [PubMed]
- Ullah, W.; Saeed, R.; Sarwar, U.; Patel, R.; Fischman, D.L. COVID-19 Complicated by Acute Pulmonary Embolism and Right-Sided Heart Failure. JACC Case Rep. 2020, 2, 1379–1382. [Google Scholar] [CrossRef]
- Desforges, M.; Le Coupanec, A.; Stodola, J.K.; Meessen-Pinard, M.; Talbot, P.J. Human coronaviruses: Viral and cellular factors involved in neuroinvasiveness and neuropathogenesis. Virus Res. 2014, 194, 145–158. [Google Scholar] [CrossRef]
- Sungnak, W.; Huang, N.; Bécavin, C.; Berg, M.; Queen, R.; Litvinukova, M.; Talavera-López, C.; Maatz, H.; Reichart, D.; Sampaziotis, F.; et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 2020, 26, 681–687. [Google Scholar] [CrossRef]
- Ellul, M.A.; Benjamin, L.; Singh, B.; Lant, S.; Michael, B.D.; Easton, A.; Kneen, R.; Defres, S.; Sejvar, J.; Solomon, T. Neurological associations of COVID-19. Lancet Neurol. 2020, 19, 767–783. [Google Scholar] [CrossRef]
- Lee, M.H.; Perl, D.P.; Nair, G.; Li, W.; Maric, D.; Murray, H.; Dodd, S.J.; Koretsky, A.P.; Watts, J.A.; Cheung, V.; et al. Microvascular Injury in the Brains of Patients with Covid-19. N. Engl. J. Med. 2020, 10, 3369. [Google Scholar]
- Liu, Y.; Yan, L.M.; Wan, L.; Xiang, T.X.; Le, A.; Liu, J.M.; Peiris, M.; Poon, L.L.M.; Zhang, W. Viral dynamics in mild and severe cases of COVID-19. Lancet Infect. Dis. 2020, 20, 656–657. [Google Scholar] [CrossRef]
- To, K.K.W.; Tsang, O.T.Y.; Leung, W.S.; Tam, A.R.; Wu, T.C.; Lung, D.C.; Yip, C.C.Y.; Cai, J.P.; Chan, J.M.C.; Chik, T.S.H.; et al. Temporal profiles of viral load in posterior oropharyngeal saliva samples and serum antibody responses during infection by SARS-CoV-2: An observational cohort study. Lancet Infect. Dis. 2020, 20, 565–574. [Google Scholar] [CrossRef]
- Kim, J.Y.; Ko, J.H.; Kim, Y.; Kim, Y.J.; Kim, J.M.; Chung, Y.S.; Kim, H.M.; Han, M.G.; Kim, S.Y.; Chin, B.S. Viral load kinetics of SARS-CoV-2 infection in first two patients in Korea. J. Korean Med. Sci. 2020, 35, 7. [Google Scholar] [CrossRef]
- Pujadas, E.; Chaudhry, F.; McBride, R.; Richter, F.; Zhao, S.; Wajnberg, A.; Nadkarni, G.; Glicksberg, B.S.; Houldsworth, J.; Cordon-Cardo, C. SARS-CoV-2 viral load predicts COVID-19 mortality. Lancet Respir. Med. 2020, 8, e70. [Google Scholar] [CrossRef]
- Zeng, Z.; Yu, H.; Chen, H.; Qi, W.; Chen, L.; Chen, G.; Yan, W.; Chen, T. Longitudinal changes of inflammatory parameters and their correlation with disease severity and outcomes in patients with COVID-19 from Wuhan, China. Crit. Care 2020, 24, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Azkur, A.K.; Akdis, M.; Azkur, D.; Sokolowska, M.; van de Veen, W.; Brüggen, M.C.; O’Mahony, L.; Gao, Y.; Nadeau, K.; Akdis, C.A. Immune response to SARS-CoV-2 and mechanisms of immunopathological changes in COVID-19. Allergy Eur. J. Allergy Clin. Immunol. 2020, 75, 1564–1581. [Google Scholar] [CrossRef]
- Dhama, K.; Khan, S.; Tiwari, R.; Sircar, S.; Bhat, S.; Malik, Y.S.; Singh, K.P.; Chaicumpa, W.; Bonilla-Aldana, D.K.; Rodriguez-Morales, A.J. Coronavirus disease 2019—COVID-19. Clin. Microbiol. Rev. 2020, 33, 1–48. [Google Scholar] [CrossRef]
- Risitano, A.M.; Mastellos, D.C.; Huber-Lang, M.; Yancopoulou, D.; Garlanda, C.; Ciceri, F.; Lambris, J.D. Complement as a target in COVID-19? Nat. Rev. Immunol. 2020, 20, 343–344. [Google Scholar] [CrossRef]
- Li, L.; Huang, Q.; Wang, D.C.; Ingbar, D.H.; Wang, X. Acute lung injury in patients with COVID-19 infection. Clin. Transl. Med. 2020, 10, 20–27. [Google Scholar] [CrossRef]
- Jawaid, A.M. COVID19 inhibitors: A prospective therapeutics. Bioorg. Chem. 2020, 101, 104027. [Google Scholar] [CrossRef] [PubMed]
- Vankadari, N. Arbidol: A potential antiviral drug for the treatment of SARS-CoV-2 by blocking trimerization of the spike glycoprotein. Int. J. Antimicrob. Agents 2020, 56, 105998. [Google Scholar] [CrossRef]
- Graham, R.L.; Donaldson, E.F.; Baric, R.S. A decade after SARS: Strategies for controlling emerging coronaviruses. Nat. Rev. Microbiol. 2013, 11, 836–848. [Google Scholar] [CrossRef]
- Williamson, B.N.; Feldmann, F.; Schwarz, B.; Meade-White, K.; Porter, D.P.; Schulz, J.; van Doremalen, N.; Leighton, I.; Yinda, C.K.; Pérez-Pérez, L.; et al. Clinical benefit of remdesivir in rhesus macaques infected with SARS-CoV-2. Nature 2020, 585, 273–276. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, D.; Du, G.; Du, R.; Zhao, J.; Jin, Y.; Fu, S.; Gao, L.; Cheng, Z.; Lu, Q.; et al. Remdesivir in adults with severe COVID-19: A randomised, double-blind, placebo-controlled, multicentre trial. Lancet 2020, 395, 1569–1578. [Google Scholar] [CrossRef]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the Treatment of Covid-19—Preliminary Report. N. Engl. J. Med. 2020, 383, 1813–1826. [Google Scholar] [CrossRef]
- Goldman, J.D.; Lye, D.C.B.; Hui, D.S.; Marks, K.M.; Bruno, R.; Montejano, R.; Spinner, C.D.; Galli, M.; Ahn, M.-Y.; Nahass, R.G.; et al. Remdesivir for 5 or 10 Days in Patients with Severe Covid-19. N. Engl. J. Med. 2020, 19, 1827–1837. [Google Scholar] [CrossRef]
- Spinner, C.D.; Gottlieb, R.L.; Criner, G.J.; López, J.R.A.; Cattelan, A.M.; Viladomiu, A.S.; Ogbuagu, O.; Malhotra, P.; Mullane, K.M.; Castagna, A.; et al. Effect of Remdesivir vs Standard Care on Clinical Status at 11 Days in Patients with Moderate COVID-19: A Randomized Clinical Trial. J. Am. Med. Assoc. 2020, 324, 1048–1057. [Google Scholar] [CrossRef]
- Slater, A.F.G. Chloroquine: Mechanism of drug action and resistance in plasmodium falciparum. Pharmacol. Ther. 1993, 57, 203–235. [Google Scholar] [CrossRef]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef]
- Gies, V.; Bekaddour, N.; Dieudonné, Y.; Guffroy, A.; Frenger, Q.; Gros, F.; Rodero, M.P.; Herbeuval, J.P.; Korganow, A.S. Beyond Anti-viral Effects of Chloroquine/Hydroxychloroquine. Front. Immunol. 2020, 11, 1409. [Google Scholar] [CrossRef]
- Liu, J.; Cao, R.; Xu, M.; Wang, X.; Zhang, H.; Hu, H.; Li, Y.; Hu, Z.; Zhong, W.; Wang, M. Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discov. 2020, 6, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Sarma, P.; Kaur, H.; Kumar, H.; Mahendru, D.; Avti, P.; Bhattacharyya, A.; Prajapat, M.; Shekhar, N.; Kumar, S.; Singh, R.; et al. Virological and clinical cure in COVID-19 patients treated with hydroxychloroquine: A systematic review and meta-analysis. J. Med. Virol. 2020, 92, 776–785. [Google Scholar] [CrossRef]
- Das, S.; Bhowmick, S.; Tiwari, S.; Sen, S. An Updated Systematic Review of the Therapeutic Role of Hydroxychloroquine in Coronavirus Disease-19 (COVID-19). Clin. Drug Investig. 2020, 40, 591–601. [Google Scholar] [CrossRef]
- Rakedzon, S.; Khoury, Y.; Rozenberg, G.; Neuberger, A. Hydroxychloroquine and Coronavirus Disease 2019: A Systematic Review of a Scientific Failure. Rambam Maimonides Med. J. 2020, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ip, A.; Berry, D.A.; Hansen, E.; Goy, A.H.; Pecora, A.L.; Sinclaire, B.A.; Bednarz, U.; Marafelias, M.; Berry, S.M.; Berry, N.S.; et al. Hydroxychloroquine and Tocilizumab Therapy in COVID-19 Patients-An Observational Study. PLoS ONE 2020, 15, e0237693. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Singh, A.; Singh, R.; Misra, A. Hydroxychloroquine in patients with COVID-19: A Systematic Review and meta-analysis. Diabetes Metab. Syndr. Clin. Res. Rev. 2020, 14, 589–596. [Google Scholar] [CrossRef] [PubMed]
- RECOVERY Collaborative Group. Effect of Hydroxychloroquine in Hospitalized Patients with Covid-19. N. Engl. J. Med. 2020, 383, 2030–2040. [Google Scholar] [CrossRef]
- Abd-Elsalam, S.; Esmail, E.S.; Khalaf, M.; Abdo, E.F.; Medhat, M.A.; El Ghafar, M.S.A.; Ahmed, O.A.; Soliman, S.; Serangawy, G.N.; Alboraie, M. Hydroxychloroquine in the treatment of COVID-19: A multicenter randomized controlled study. Am. J. Trop. Med. Hyg. 2020, 103, 1635–1639. [Google Scholar] [CrossRef]
- Skipper, C.P.; Pastick, K.A.; Engen, N.W.; Bangdiwala, A.S.; Abassi, M.; Lofgren, S.M.; Williams, D.A.; Okafor, E.C.; Pullen, M.F.; Nicol, M.R.; et al. Hydroxychloroquine in Nonhospitalized Adults With Early COVID-19: A Randomized Trial. Ann. Intern. Med. 2020, 383, 2030–2040. [Google Scholar]
- Boulware, D.R.; Pullen, M.F.; Bangdiwala, A.S.; Pastick, K.A.; Lofgren, S.M.; Okafor, E.C.; Skipper, C.P.; Nascene, A.A.; Nicol, M.R.; Abassi, M.; et al. A Randomized Trial of Hydroxychloroquine as Postexposure Prophylaxis for Covid-19. N. Engl. J. Med. 2020, 383, 517–525. [Google Scholar] [CrossRef]
- Mitjà, O.; Corbacho-Monné, M.; Ubals, M.; Alemany, A.; Suñer, C.; Tebé, C.; Tobias, A.; Peñafiel, J.; Ballana, E.; Pérez, C.A.; et al. A Cluster-Randomized Trial of Hydroxychloroquine for Prevention of Covid-19. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Stockman, L.J.; Bellamy, R.; Garner, P. SARS: Systematic review of treatment effects. PLoS Med. 2006, 3, e343. [Google Scholar] [CrossRef]
- Arabi, Y.M.; Mandourah, Y.; Al-Hameed, F.; Sindi, A.A.; Almekhlafi, G.A.; Hussein, M.A.; Jose, J.; Pinto, R.; Al-Omari, A.; Kharaba, A.; et al. Corticosteroid therapy for critically ill patients with middle east respiratory syndrome. Am. J. Respir. Crit. Care Med. 2018, 197, 757–767. [Google Scholar] [CrossRef]
- Lansbury, L.; Rodrigo, C.; Leonardi-Bee, J.; Nguyen-Van-Tam, J.; Lim, W.S. Corticosteroids as adjunctive therapy in the treatment of influenza. Cochrane Database Syst. Rev. 2019. [Google Scholar] [CrossRef]
- Mammen, M.J.; Aryal, K.; Alhazzani, W.; Alexander, P.E. Corticosteroids for patients with acute respiratory distress syndrome: A systematic review and meta-analysis of randomized trials. Polish Arch. Intern. Med. 2020, 130, 276–286. [Google Scholar] [CrossRef]
- RECOVERY Collaborative Group. Dexamethasone in Hospitalized Patients with Covid-19—Preliminary Report. N. Engl. J. Med. 2020, 383, 2030–2040. [Google Scholar] [CrossRef]
- Tomazini, B.M.; Maia, I.S.; Cavalcanti, A.B.; Berwanger, O.; Rosa, R.G.; Veiga, V.C.; Avezum, A.; Lopes, R.D.; Bueno, F.R.; Silva, M.V.A.O.; et al. Effect of Dexamethasone on Days Alive and Ventilator-Free in Patients With Moderate or Severe Acute Respiratory Distress Syndrome and COVID-19: The CoDEX Randomized Clinical Trial. JAMA 2020, 324, 1307–1316. [Google Scholar] [CrossRef]
- Rafiullah, M.; Siddiqui, K. Corticosteroid use in viral pneumonia: Experience so far and the dexamethasone breakthrough in coronavirus disease-2019. J. Comp. Eff. Res. 2020, 9, 1247–1254. [Google Scholar] [CrossRef]
- Jeronimo, C.M.P.; Farias, M.E.L.; Val, F.F.A.; Sampaio, V.S.; Alexandre, M.A.A.; Melo, G.C.; Safe, I.P.; Borba, M.G.S.; Netto, R.L.A.; Maciel, A.B.S.; et al. Methylprednisolone as Adjunctive Therapy for Patients Hospitalized With Coronavirus Disease 2019 (COVID-19; Metcovid): A Randomized, Double-blind, Phase IIb, Placebo-controlled Trial. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Dequin, P.F.; Heming, N.; Meziani, F.; Plantefève, G.; Voiriot, G.; Badié, J.; François, B.; Aubron, C.; Ricard, J.D.; Ehrmann, S.; et al. Effect of Hydrocortisone on 21-Day Mortality or Respiratory Support among Critically Ill Patients with COVID-19: A Randomized Clinical Trial. J. Am. Med. Assoc. 2020, 324, 1298–1306. [Google Scholar] [CrossRef]
- Mertens, M.; Singh, J.A. Anakinra for rheumatoid arthritis. Cochrane Database Syst. Rev. 2009. [Google Scholar] [CrossRef]
- Cavalli, G.; De Luca, G.; Campochiaro, C.; Della-Torre, E.; Ripa, M.; Canetti, D.; Oltolini, C.; Castiglioni, B.; Din, C.T.; Boffini, N.; et al. Interleukin-1 blockade with high-dose anakinra in patients with COVID-19, acute respiratory distress syndrome, and hyperinflammation: A retrospective cohort study. Lancet Rheumatol. 2020, 2, 325–331. [Google Scholar] [CrossRef]
- Huet, T.; Beaussier, H.; Voisin, O.; Jouveshomme, S.; Dauriat, G.; Lazareth, I.; Sacco, E.; Naccache, J.M.; Bézie, Y.; Laplanche, S.; et al. Anakinra for severe forms of COVID-19: A cohort study. Lancet Rheumatol. 2020, 2, 393–400. [Google Scholar] [CrossRef]
- Scott, L.J. Tocilizumab: A Review in Rheumatoid Arthritis. Drugs 2017, 77, 1865–1879. [Google Scholar] [CrossRef]
- Nabil, A.; Uto, K.; Elshemy, M.M.; Soliman, R.; Hassan, A.A.; Ebara, M.; Shiha, G. Current coronavirus (Sars-cov-2) epidemiological, diagnostic and therapeutic approaches: An updated review until june 2020. EXCLI J. 2020, 19, 992–1016. [Google Scholar]
- Michot, J.M.; Albiges, L.; Chaput, N.; Saada, V.; Pommeret, F.; Griscelli, F.; Balleyguier, C.; Besse, B.; Marabelle, A.; Netzer, F.; et al. Tocilizumab, an anti-IL-6 receptor antibody, to treat COVID-19-related respiratory failure: A case report. Ann. Oncol. 2020, 31, 961–964. [Google Scholar] [CrossRef]
- Xu, X.; Han, M.; Li, T.; Sun, W.; Wang, D.; Fu, B.; Zhou, Y.; Zheng, X.; Yang, Y.; Li, X.; et al. Effective treatment of severe COVID-19 patients with tocilizumab. Proc. Natl. Acad. Sci. USA 2020, 117, 10970–10975. [Google Scholar] [CrossRef]
- Antwi-Amoabeng, D.; Kanji, Z.; Ford, B.; Beutler, B.D.; Riddle, M.S.; Siddiqui, F. Clinical outcomes in COVID-19 patients treated with tocilizumab: An individual patient data systematic review. J. Med. Virol. 2020, 92, 2516–2522. [Google Scholar] [CrossRef]
- Lan, S.H.; Lai, C.C.; Huang, H.T.; Chang, S.P.; Lu, L.C.; Hsueh, P.R. Tocilizumab for severe COVID-19: A systematic review and meta-analysis. Int. J. Antimicrob. Agents 2020, 56, 106103. [Google Scholar] [CrossRef]
- Keske, Ş.; Tekin, S.; Sait, B.; İrkören, P.; Kapmaz, M.; Çimen, C.; Uğur, S.; Çelebi, İ.; Bakır, V.O.; Palaoğlu, E.; et al. Appropriate use of tocilizumab in COVID-19 infection. Int. J. Infect. Dis. 2020, 99, 338–343. [Google Scholar] [CrossRef]
- Pawar, A.; Desai, R.J.; Solomon, D.H.; Ortiz, A.J.S.; Gale, S.; Bao, M.; Sarsour, K.; Schneeweiss, S.; Kim, S.C. Risk of serious infections in tocilizumab versus other biologic drugs in patients with rheumatoid arthritis: A multidatabase cohort study. Ann. Rheum. Dis. 2019, 78, 456–464. [Google Scholar] [CrossRef]
- Burmester, G.R.; Pope, J.E. Novel treatment strategies in rheumatoid arthritis. Lancet 2017, 389, 2338–2348. [Google Scholar] [CrossRef]
- Richardson, P.; Griffin, I.; Tucker, C.; Smith, D.; Oechsle, O.; Phelan, A.; Stebbing, J. Baricitinib as potential treatment for 2019-nCoV acute respiratory disease. Lancet 2020, 395, 30–31. [Google Scholar] [CrossRef]
- Cantini, F.; Niccoli, L.; Matarrese, D.; Nicastri, E.; Stobbione, P.; Goletti, D. Baricitinib therapy in COVID-19: A pilot study on safety and clinical impact. J. Infect. 2020, 81, 318–356. [Google Scholar] [CrossRef]
- Favalli, E.G.; Biggioggero, M.; Maioli, G.; Caporali, R. Baricitinib for COVID-19: A suitable treatment? Lancet Infect. Dis. 2020, 20, 1012–1013. [Google Scholar] [CrossRef]
- Ke, Z.; Oton, J.; Qu, K.; Cortese, M.; Zila, V.; McKeane, L.; Nakane, T.; Zivanov, J.; Neufeldt, C.J.; Lu, J.M.; et al. Structures, conformations and distributions of SARS-CoV-2 spike protein trimers on intact virions. bioRxiv 2020. [Google Scholar] [CrossRef]
- Williams, S.J.; Goddard-Borger, E.D. α-glucosidase inhibitors as host-directed antiviral agents with potential for the treatment of COVID-19. Biochem. Soc. Trans. 2020, 48, 1287–1295. [Google Scholar] [CrossRef]
- Zheng, Y.Y.; Ma, Y.T.; Zhang, J.Y.; Xie, X. COVID-19 and the cardiovascular system. Nat. Rev. Cardiol. 2020, 17, 259–260. [Google Scholar] [CrossRef]
- Onweni, C.L.; Zhang, Y.S.; Caulfield, T.; Hopkins, C.E.; Fairweather, D.L.; Freeman, W.D. ACEI/ARB therapy in COVID-19: The double-edged sword of ACE2 and SARS-CoV-2 viral docking. Crit. Care 2020, 24, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Del Río, J.M.; Piqueras-Flores, J.; Nieto-Sandoval, M.; Sierra, P.; Negreira-Caamaño, M.; Águila-Gordo, D.; Mateo-Gómez, C.; Salas-Bravo, D.; Rodríguez-Martínez, M. Comparative analysis between the use of renin-angiotensin system antagonists and clinical outcomes of hospitalized patients with COVID-19 respiratory infection. Med. Clin. 2020, 155, 473–481. [Google Scholar]
- Barochiner, J.; Martinez, R. Use of inhibitors of the renin angiotensin system and COVID-19 prognosis: A systematic review and meta-analysis. medRxiv 2020. [Google Scholar] [CrossRef]
- Messerli, F.H.; Messerli, F.H.; Messerli, F.H.; Siontis, G.C.M.; Rexhaj, E. COVID-19 and Renin Angiotensin Blockers: Current Evidence and Recommendations. Circulation 2020, 25, 2042–2044. [Google Scholar] [CrossRef]
- Shanmugaraj, B.; Siriwattananon, K.; Wangkanont, K.; Phoolcharoen, W. Perspectives on monoclonal antibody therapy as potential therapeutic intervention for Coronavirus disease-19 (COVID-19). Asian Pac. J. Allergy Immunol. 2020, 38, 10–18. [Google Scholar] [PubMed]
- Kim, T.S.; Heinlein, C.; Hackman, R.C.; Nelson, P.S. Phenotypic Analysis of Mice Lacking the Tmprss2-Encoded Protease. Mol. Cell. Biol. 2006, 26, 965–975. [Google Scholar] [CrossRef] [PubMed]
- Iwata-Yoshikawa, N.; Okamura, T.; Shimizu, Y.; Hasegawa, H.; Takeda, M.; Nagata, N. TMPRSS2 Contributes to Virus Spread and Immunopathology in the Airways of Murine Models after Coronavirus Infection. J. Virol. 2019, 93, 6. [Google Scholar] [CrossRef]
- Kawase, M.; Shirato, K.; van der Hoek, L.; Taguchi, F.; Matsuyama, S. Simultaneous Treatment of Human Bronchial Epithelial Cells with Serine and Cysteine Protease Inhibitors Prevents Severe Acute Respiratory Syndrome Coronavirus Entry. J. Virol. 2012, 86, 6537–6545. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Pöhlmann, S. A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol. Cell 2020, 78, 779–784. [Google Scholar] [CrossRef]
- Xia, S.; Lan, Q.; Su, S.; Wang, X.; Xu, W.; Liu, Z.; Zhu, Y.; Wang, Q.; Lu, L.; Jiang, S. The role of furin cleavage site in SARS-CoV-2 spike protein-mediated membrane fusion in the presence or absence of trypsin. Signal Transduct. Target. Ther. 2020, 5, 1–3. [Google Scholar] [CrossRef]
- Papa, G.; Mallery, D.L.; Albecka, A.; Welch, L.; Cattin-Ortolá, J.; Luptak, J.; Paul, D.; McMahon, H.; Goodfellow, I.G.; Carter, A.; et al. Furin cleavage of SARS-CoV-2 Spike promotes but is not essential for infection and cell-cell fusion. bioRxiv 2020, 17, e1009246. [Google Scholar]
- Gao, T.; Hu, M.; Zhang, X.; Li, H.; Zhu, L.; Liu, H.; Dong, Q.; Zhang, Z.; Wang, Z.; Hu, Y.; et al. Highly pathogenic coronavirus N protein aggravates lung injury by MASP-2-mediated complement over-activation. MedRxiv 2020. [Google Scholar] [CrossRef]
- Gralinski, L.E.; Sheahan, T.P.; Morrison, T.E.; Menachery, V.D.; Jensen, K.; Leist, S.R.; Whitmore, A.; Heise, M.T.; Baric, R.S. Complement activation contributes to severe acute respiratory syndrome coronavirus pathogenesis. mBio 2018, 9, e01753-18. [Google Scholar] [CrossRef]
- Magro, C.; Mulvey, J.J.; Berlin, D.; Nuovo, G.; Salvatore, S.; Harp, J.; Baxter-Stoltzfus, A.; Laurence, J. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: A report of five cases. Transl. Res. 2020, 220, 1–13. [Google Scholar] [CrossRef]
- Java, A.; Apicelli, A.J.; Liszewski, M.K.; Coler-Reilly, A.; Atkinson, J.P.; Kim, A.H.; Kulkarni, H.S. The complement system in COVID-19: Friend and foe? JCI Insight 2020, 5, 5. [Google Scholar] [CrossRef]
- De Bont, C.M.; Boelens, W.C.; Pruijn, G.J.M. NETosis, complement, and coagulation: A triangular relationship. Cell. Mol. Immunol. 2019, 16, 19–27. [Google Scholar] [CrossRef]
- Zuo, Y.; Yalavarthi, S.; Shi, H.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.; Weber, A.; Barnes, B.J.; Egeblad, M.; et al. Neutrophil extracellular traps in COVID-19. JCI Insight 2020, 5, e138999. [Google Scholar] [CrossRef]
- Barnes, B.J.; Adrover, J.M.; Baxter-Stoltzfus, A.; Borczuk, A.; Cools-Lartigue, J.; Crawford, J.M.; Daßler-Plenker, J.; Guerci, P.; Huynh, C.; Knight, J.S.; et al. Targeting potential drivers of COVID-19: Neutrophil extracellular traps. J. Exp. Med. 2020, 6, 217. [Google Scholar] [CrossRef]
- Semeraro, F.; Ammollo, C.T.; Morrissey, J.H.; Dale, G.L.; Friese, P.; Esmon, N.L.; Esmon, C.T. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: Involvement of platelet TLR2 and TLR4. Blood 2011, 118, 1952–1961. [Google Scholar] [CrossRef]
- Zuo, Y.; Yalavarthi, S.; Shi, H.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.; Weber, A.; Barnes, B.J.; Egeblad, M.; et al. Neutrophil extracellular traps (NETs) as markers of disease severity in COVID-19. MedRxiv 2020. [Google Scholar] [CrossRef]
- Middleton, E.A.; He, X.Y.; Denorme, F.; Campbell, R.A.; Ng, D.; Salvatore, S.P.; Mostyka, M.; Baxter-Stoltzfus, A.; Borczuk, A.C.; Loda, M.; et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood 2020, 136, 1169–1179. [Google Scholar] [CrossRef]
- Juarez, M.; Schcolnik-Cabrera, A.; Dueñas-Gonzalez, A. The multitargeted drug ivermectin: From an antiparasitic agent to a repositioned cancer drug. Am. J. Cancer Res. 2018, 8, 317–331. [Google Scholar]
- Yang, S.N.Y.; Atkinson, S.C.; Wang, C.; Lee, A.; Bogoyevitch, M.A.; Borg, N.A.; Jans, D.A. The broad spectrum antiviral ivermectin targets the host nuclear transport importin α/β1 heterodimer. Antivir. Res. 2020, 177, 104760. [Google Scholar] [CrossRef]
- Caly, L.; Druce, J.D.; Catton, M.G.; Jans, D.A.; Wagstaff, K.M. The FDA-approved drug ivermectin inhibits the replication of SARS-CoV-2 in vitro. Antivir. Res. 2020, 178, 104787. [Google Scholar] [CrossRef] [PubMed]
- Chaccour, C.; Hammann, F.; Ramón-García, S.; Rabinovich, N.R. Ivermectin and COVID-19: Keeping rigor in times of urgency. Am. J. Trop. Med. Hyg. 2020, 102, 1156–1157. [Google Scholar] [CrossRef] [PubMed]
- Rajter, J.C.; Sherman, M.S.; Fatteh, N.; Vogel, F.; Sacks, J.; Rajter, J.-J. Use of Ivermectin Is Associated With Lower Mortality in Hospitalized Patients With Coronavirus Disease 2019. Chest 2020, 159, 85–92. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, H.M.; Moon, S.Y.; Yang, H.I.; Kim, K.S. Understanding Viral Infection Mechanisms and Patient Symptoms for the Development of COVID-19 Therapeutics. Int. J. Mol. Sci. 2021, 22, 1737. https://doi.org/10.3390/ijms22041737
Choi HM, Moon SY, Yang HI, Kim KS. Understanding Viral Infection Mechanisms and Patient Symptoms for the Development of COVID-19 Therapeutics. International Journal of Molecular Sciences. 2021; 22(4):1737. https://doi.org/10.3390/ijms22041737
Chicago/Turabian StyleChoi, Hyung Muk, Soo Youn Moon, Hyung In Yang, and Kyoung Soo Kim. 2021. "Understanding Viral Infection Mechanisms and Patient Symptoms for the Development of COVID-19 Therapeutics" International Journal of Molecular Sciences 22, no. 4: 1737. https://doi.org/10.3390/ijms22041737
APA StyleChoi, H. M., Moon, S. Y., Yang, H. I., & Kim, K. S. (2021). Understanding Viral Infection Mechanisms and Patient Symptoms for the Development of COVID-19 Therapeutics. International Journal of Molecular Sciences, 22(4), 1737. https://doi.org/10.3390/ijms22041737